
Abstract
Rechargeable non-aqueous lithium-oxygen batteries with a large theoretical capacity are emerging as a high-energy electrochemical device for sustainable energy strategy. Despite many efforts made to understand the fundamental Li-O2 electrochemistry, the kinetic process of cathodic reactions, associated with the formation and decomposition of a solid Li2O2 phase during charging and discharging, remains debate. Here we report direct visualization of the charge/discharge reactions on a gold cathode in a non-aqueous lithium-oxygen micro-battery using liquid-cell aberration-corrected scanning transmission electron microscopy (STEM) combining with synchronized electrochemical measurements. The real-time and real-space characterization by time-resolved STEM reveals the electrochemical correspondence of discharge/charge overpotentials to the nucleation, growth and decomposition of Li2O2 at a constant current density. The nano-scale operando observations would enrich our knowledge on the underlying reaction mechanisms of lithium-oxygen batteries during round-trip discharging and charging and shed lights on the strategies in improving the performances of lithium-oxygen batteries by tailoring the cathodic reactions.
Similar content being viewed by others

Introduction
Lithium-oxygen (Li-O2) batteries have recently attracted enormous research attention as a new generation of high energy storage devices for all electric vehicles and other high-energy-demanded applications because of the high theoretical capacity1. However, the practical implementation of Li-O2 batteries is facing substantial challenges, particularly, in the development of high performance cathodes and stable electrolytes for high round-trip efficiency, long lifetimes and high rate capability1,2,3,4. Unlike intercalation reactions in lithium-ion batteries, the electrochemistry of Li-O2 batteries is based on a reversible reaction of 2Li + O2 ⇔ Li2O2. The formation and decomposition of the solid Li2O2 phase during the discharge-recharge cycles via oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are the critical processes which determine the overall battery performances2,3,4,5,6,7,8,9,10,11. Despite many efforts made to understand the fundamental Li-O2 electrochemistry in recent years, the evolution of Li2O2 on cathodes remains debate with several critical questions which include but are not limited to9,10,11,12,13,14,15,16,17 (1) is the formation of Li2O2 by a non-Faradaic disproportionation reaction of intermediate LiO2 or by a direct electrochemical reduction of O2 to Li2O2? (2) how can the continuous formation of insulating Li2O2 on the cathode surface keep a constant or near constant discharge potential? (3) does the decomposition of Li2O2 take place at Li2O2/electrolyte interfaces or the Li2O2/electrode interfaces? The investigations of cathodic reactions, associated with the formation and decomposition of Li2O2, have been conducted extensively by differential electrochemical mass spectrometry11, nuclear magnetic resonance13, surface enhanced Raman spectroscopy14, X-ray diffraction15, ab initio calculations16, ex situ transmission electron microscopy (TEM) and scanning electron microscopy (SEM)17, and so on. However, the direct correspondence of Li2O2 nucleation, growth and decomposition to electrochemical discharge/charge in the liquid-state Li-O2 system has not been well established owing to the lack of operando characterization with real-space visualization. The rationale of the basic cathodic reaction kinetic process, related to those questions, is still in dispute. Therefore, the direct observations of the nucleation, growth and decomposition of Li2O2, accompanying with simultaneous electrochemical measurements of discharging/charging response, is highly desired to answer those fundamental questions and thus the reaction kinetics of the Li-O2 batteries.
TEM with a high spatial resolution has been demonstrated as a powerful tool for in situ characterization of electrochemical and chemical reactions12,18,19,20. In particular, the recently-developed micro-fabricated electrochemical cells (EC) provide a unique advantage for imaging electrochemical reactions while electrochemical measurements can be performed simultaneously. For lithium ion batteries, in situ EC TEM and scanning TEM (STEM) have been used to observe lithiation and delithiation dynamics in silicon nanowires and LiFePO4 nanoparticle electrodes21,22. Although in situ TEM has been utilized to study the formation and/or decomposition of Li2O2 in Li-O2 batteries12,23,24, electrochemical processes of these studies were performed at a constant voltage and thus cannot provide the information on the electrochemical correspondence of the nucleation, growth and decomposition of Li2O2 to discharge/charge overpotentials. Moreover, the weak contrast of Li2O2 under TEM mode, especially interfered by liquid surroundings in EC, limits the spatial resolution for detecting the formation/decomposition of light Li2O2 under non-equilibrium high-rate charge/discharge conditions. In contrast, the state-of-the-art spherical aberration corrected STEM with enhanced contrast by a high angle annular dark field (HAADF) detector is a promising technique for characterizing basic electrochemical reactions in liquid-state at a high spatial resolution of ~1 nm19. In this study we employ in situ EC HAADF-STEM to investigate the nucleation, growth and decomposition of Li2O2 in a basic liquid-state Li-O2 system during discharging and charging at a constant current density, together with synchronized electrochemical measurements.
Figure 1a shows the schematic diagram of the Li-O2 micro-battery for in situ STEM observations. A micro-channel was sandwiched between two micro-chips with ~30 nm thick electron-transparent Si3N4 membranes to form a flowing liquid cell. The top chip was patterned with two Au pads as the electrodes for electrochemical measurements. One Au pad loaded with LiFePO4 nanoparticles was used as the counter electrode. The formation and decomposition of Li2O2 during discharge-charge cycles were recorded on the other Au pad which acted as the working electrode (Fig. 1b). 1.0 M LiClO4 in dimethylsulfoxide (DMSO) saturated with oxygen was used as the electrolyte and fed into the cell at a low flowing rate of 1 μL/s through an external liquid pump. The flowing electrolyte keeps near constant oxygen and Li ion concentrations during electrochemical measurements and STEM observations at a very low flow velocity (smaller than 0.05 nm/s) of the electrolyte in the vicinity of the Au cathode, calculated by the finite element analysis (Fig. S2). The influence of the flowing electrolyte at the low velocity on the cathodic reaction kinetics is ignorable as verified by invisible mechnical motion of small Li2O2 nanoparticles. The assembled liquid Li-O2 micro-cell was loaded into a TEM holder and connected to an electrochemical workstation. To achieve high-quality images, a Cs-corrected STEM mode with a mass-sensitive HAADF detector was employed for the operando observations. The spatial resolution of the Cs-corrected STEM liquid cell was estimated to be ~1 nm19,25. Additionally, HAADF-STEM images were taken at a relatively lower resolution of 512 by 512 pixels and a short scanning time of 5 μs/pixel to avoid electron beam irradiation and sample drift and to capture more frames during observations. Under the imaging condition, the electron dosage is as low as 0.53 e−/Å2 with low magnification of 50k, which is several orders of magnitude lower than that of the typical high-resolution Cs-corrected STEM (~105 e−/Å2)26,27 (Table S1). The experimental parameters of the low-dose STEM can be found in Supplementary Materials. To test the electrochemical performance of the Li-O2 micro-battery inside TEM, the cyclic voltammetry (CV) measurements were performed as shown in Fig. 1c. The CV curves well trace the reversible cathodic reactions and are consistent with those of conventional Li-O2 batteries28,29, demonstrating that the electrochemical functions of our Li-O2 micro-battery are essentially identical to these of conventional coin batteries although their apparent configurations look different. A time-capacity dependent potential evolution curve during galvanostatic discharge and charge at a constant current density of 1.3 μA/cm2 was recorded during HAADF-STEM observations (Fig. 1d). In spite of the slightly high charge/discharge overpotentials due to large series resistance and complex configuration of the liquid cell setup12,30, the characteristic voltammetric profile of the micro-battery is consistent with those of conventional Au based Li-O2 batteries6,31,32. Moreover, the galvanostatic charge/discharge profiles do not show obvious changes when the electron beam for STEM imaging turns on and off (Fig. S3). Moreover, we have verified that the electrolyte and reaction product Li2O2 are stable under the low-dose STEM mode in the experimental time scale (Supplementary Materials). Therefore, the influence of electron beam irradiation on the cathodic reactions during our operando observations should not be observable. Under the applied galvanostatic discharge-charge cycles, the detailed electrochemical reactions on the Au cathodes of the Li-O2 micro-battery can be monitored simultaneously by STEM.

(a) Schematic diagram of the Li-O2 micro-battery setup for operando STEM characterization of cathodic reactions during charging and discharging. A top microchip is patterned with 120 nm wide Au electrode. (b) Schematic reactions in the micro-battery with the formation and decomposition of solid Li2O2 on the Au cathode during discharge-charge cycles. LiFePO4 nanoparticles are loaded on the Au anode as the Li+ source. (c) Three cyclic voltammetry cycles with voltage range from 1–5 V of the micro-battery measured in TEM liquid holder. (d) The synchronized galvanostatic discharge-charge curves of the micro-battery at the current of 100 nA with the cut-off capacity of ~1.67 nAh. The electrolyte is 1 M LiClO4 in DMSO.
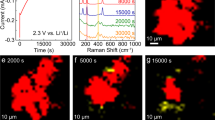
The time sequential HAADF-STEM images during discharging are presented in Fig. 2a–h. To enhance the weak contrast of the reaction products of light Li2O2, color correction with blue, white and red is used. For reference, the original grey HAADF-STEM images are shown in Fig. S15. The regions with a bright white contrast represent the Li2O2 phase while the very bright rim of the gold electrode in Fig. 2a may be from the edge effect of the electrode because it keeps unchanging with discharging and charging. During the first 10 seconds of the discharging reaction, a diffuse layer with continuous bright contrast emerges, as marked by white dashed line in Fig. 2b. Although discrete Li2O2 particles cannot be identified from the diffuse layer by HAADF-STEM, correspondingly, the discharge overpotential reaches the discharge plateau with continuous increase of capacity. It is known that the solubility of Li2O2 in DMSO is very low7. The absence of the Li2O2 phase on the electrode surface and diffuse layer suggests that the discharge reaction is carried out by the one-electron reduction of O2 with the formation of lithium superoxide (LiO2) intermediates which have high solubility in DMSO. Since the contrast of HAADF images arises from the high-angle electron scattering, the white contrast of the diffuse layer evidences the enhanced electron scattering from the electrolyte in the vicinity of charged electrode. Thus, the enhanced contrast of the diffuse layer most likely originates from the charged or polarized molecules of the electrolyte in electric fields, which may have higher structure ordering by polarized molecule alignment or higher density from reduced inter-molecule distances. This assumption is supported by the fact that the diffuse layer can also be observed around the Au electrodes with a pure DMSO electrolyte without the additives of O2 and Li ions (Fig. S5). The thickness of the diffuse layer gradually increases with discharge time and reaches the maximum of about 80–90 nm before the precipitation of solid Li2O2 phase (Fig. S4). When discharging for about 16 seconds, nanoparticles gradually become visible within the diffuse zone and preferentially start from the topmost surface of the gold electrode. Significantly, the precipitation of isolated nanoparticles in the electrolyte without direct contact with the electrode can also be observed, as marked by arrows in Fig. 2c and d. These observations indicate that the formation of solid Li2O2 phase is most likely by a non-Faradaic disproportionation reaction lagging behind the one-electron ORR. The earlier precipitation of Li2O2 in the vicinity of Au electrode surfaces is also in agreement with the reaction mechanism as the formation of Li2O2 by the disproportionation reaction is expected to be controlled by LiO2 diffusion. With continuously discharging, the thickness of the Li2O2 layer keeps increasing from ~90 nm to ~160 nm mainly by the formation of new nanoparticles from the diffuse layer (Fig. 2d–g). The newly-formed nanoparticles often connect with pre-existed ones and form a network like porous structure. After ~60 seconds, the discharge products forms a ~160 nm thick Li2O2 layer on the Au cathode, which is composed of loosely interconnected nanoparticles with open pore channels (Fig. 2h).

Time sequential HAADF-STEM images during a discharge-charge cycle. The blue-white-red colors are used to enhance the weak Z-contrast of the reaction products of light Li2O2. The regions with a bright white contrast represent the Li2O2 phase. (a,b) A diffuse layer with weak contrast in the vicinity of the charged electrode. (c–h) Nucleation and growth of Li2O2 nanoparticles and the formation of a network structural Li2O2 film during discharging. (i and j) The preferential dissolution of Li2O2 at the electrode/Li2O2 interface. (k–o) The decomposition of Li2O2 via the continuous dissolution of Li2O2 particles at electrolyte/Li2O2 interfaces during charging. (p) A diffuse layer around the gold electrode after the complete dissolution of Li2O2 particles. (scale bar: 100 nm, E0 = 200 keV, magnification: 50 k, dwell time: 5 μs/pixel, image size: 512 × 512 pixels, electron dose: 0.53 e−/Å2).
The formation of Li2O2 has been suggested to follow a two-step reaction, which includes the one-electron reduction of O2 to form the lithium superoxide (LiO2) intermediates (Eq. 1) and subsequently either a non-Faradaic disproportionation reaction (Eq. 2) or a second reduction reaction (Eq. 3) to form the final Li2O2 discharge product5,14:
The operando STEM observations provide compelling evidence that the formation of Li2O2 is through a hybrid electrochemical and chemical process, i.e, the one-electron reduction of O2 to LiO2 by Eq. 1 and followed by the non-Faradaic disproportionation of LiO2 with Eq. 2 under the current experimental conditions. The hybrid mechanism allows Li2O2 nanoparticles to directly precipitate from electrolyte, rather than layer-by-layer growth on the electrode surface. The operando observation is also consistent with the reaction mechanism proposed by Bruce et al. The high-donor-number solvent DMSO with a high solubility of LiO2 facilitates the formation of Li2O2 through a solution pathway28. It has been reported that the disproportionation of LiO2 to Li2O2 has a half-life of tens of seconds14, close to the time scale of the discharge in our in situ STEM experiments. Therefore, the direct second electron reduction pathway by charge transfer (Eq. 3) appears unfavorable at a low constant discharging current. As shown in the time sequential HAADF-STEM images, the formation of Li2O2 is dominated by the nucleation process in the initial stage. The soluble LiO2 produced at the electrode surface diffuses into the electrolyte and forms Li2O2 by the disproportionation reaction (Eq. 2). When the concentration of Li2O2 exceeds the solubility limit, crystallites precipitate from the electrolyte as the initial Li2O2 nuclei and gradually grow up during continuous discharging. The smallest Li2O2 precipitates, which can be identified by the STEM observations, are about 3–4 nm (Fig. S6), which could be the upper bound of the critical size of Li2O2 precipitates. The formation of the network structure with interconnected Li2O2 particles may result from heterogeneous nucleation of Li2O2 nanoparticles during which the pre-existed particles act as the nucleation sites to reduce the nucleation barrier of newly-formed Li2O2 particles, or attachment growth of the nanoparticles. The open channels in the network structure enable the gold electrode to contact with electrolyte. In this way the LiO2 intermediates can be continuously generated from the electrode by ORR at a nearly constant overpotential until the electrode is completely passivated by a solid Li2O2 film which blocks the contact between the gold electrode and electrolyte (Fig. S13)33. The crystal structure and chemistry of the Li2O2 phase formed during the operando observations are verified by separate selected area electron diffraction (SAED) and electron energy loss spectroscopy (EELS) analysis at a low acceleration voltage of 120 kV (Fig. S12).
The formation kinetics of Li2O2 was investigated by measuring the evolution of the size and number of Li2O2 particles during discharging. As shown in Fig. 3a and b, the average particle size via discharge time is parabolic while the particle number quickly increases within first 20 seconds and then remains nearly constant, indicating that the formation of solid Li2O2 phase is via a nucleation-growth process. From the initial discharge plateau to the end of cut-off capacity, the mean particle size increases from ~10 to ~35 nm within ~60 seconds at the average growth rate of 0.4 nm/s. The low growth rate is in line with the low current density used in this study. We also estimated the total volume changes of Li2O2 with discharge time by assuming the Li2O2 nanoparticles have an approximate spherical shape (Fig. 3c). Since the liquid layer is very thin (~120 nm), the density of Li2O2 particles is not very high and the contrast difference between the solid Li2O2 particles and electrolyte is large, most Li2O2 nanoparticles can be counted during the in situ observations, although a small number of nanoparticles may be missing because of the overlap along the electron beam direction in the early stage of discharging. Interestingly, except the first 10 second corresponding to the formation of the diffuse layer, the Li2O2 volume has a linear relation with discharge time and thereby capacity, which is consistent with the smooth discharge curve at a constant current density (Fig. 1d). After the initial stage of Li2O2 formation, the thickness of the Li2O2 layer keeps nearly unchanged during continuous discharging (Fig. 3d) and the increase of the charge capacity is by the growth of individual Li2O2 nanoparticles. The nearly constant Li2O2 layer thickness could be associated with the relatively stable concentration gradient of LiO2 around the electrode because the contour of the Li2O2 layer shows the similarity with the local curvature of the Au electrode. Since Li2O2 is almost insoluble in the non-aqueous DMSO electrolyte7, Li2O2 precipitates once it forms by disproportionation of LiO2. The growth of Li2O2 particles is more likely controlled by the discharge reaction rate, namely LiO2 formation rate and discharge current density, after a stable LiO2 concentration gradient is formed in the vicinity of the electrode. Both the linear relation of Li2O2 volume vs. time and the parabolic curve between Li2O2 particle size and time are in good agreement with the galvanostatic discharge mode which has a constant LiO2 formation rate and thus Li2O2 formation rate (Li2O2 volume per unit time) under a near equilibrium condition.

The evolution of Li2O2 particles during charging and discharging. (a) Parabolic relation between average particle size and discharge/charge time. (b) Relation between particle number and discharge/charge time. (c) Total volume changes of Li2O2 with discharge/charge time. Note that the volume has a linear relation with time. (d) The plot of Li2O2 layer thickness with discharge/charge time.
To investigate the decomposition reaction of Li2O2, galvanostatic charging was conducted after the Li2O2 formation (Fig. 1d). The corresponding evolution of Li2O2 particles during charging is shown in Fig. 2i–p. Within the first 10 seconds, noticeable shrinkage of the Li2O2 nanoparticles takes place at the Au electrode/Li2O2 interface (Fig. 2i,j), indicating that the preferential decomposition of Li2O2 takes place from those directly contacting with the electrode. The process is accompanied by a quick increase of the charge potential (Fig. 1d) with the gradual loss of the direct contact between Li2O2 nanoparticles and the electrode. This observation is consistent with previous in situ TEM studies12,23 and evidences that the decomposition of Li2O2 starts from the electrode surface at lower charging potentials. The kinetics of Li2O2 oxidation is apparently limited by the charge transport between the electrode and indirectly contacted Li2O2 particles. With further charging, noticeable dissolution of Li2O2 particles that reside away from the gold electrode (up to 160 nm) can be observed at high charge potentials (Fig. 2k–n). This phenomenon is different from the previous observations that the decomposition of Li2O2 is carried out by the collapse of Li2O2 films to keep direct contact between Li2O2 and electrode for interfacial oxidation operated at constant voltages and high current densities23. In our experiment, the charge and discharge are conducted at a constant current density of 1.3 μA/cm2, similar to the conventional battery testing, and the charge reaction is under a near electrochemical equilibrium state with a relatively lower reaction rate in comparison with that of ref.23. The Li2O2 oxidation involves charge transfer, i.e.
during which considerable electron transport in the porous Li2O2 film is required for the decomposition of solid Li2O2 via OER during charging. It is known that bulk Li2O2 is insulator and electrons cannot tunnel into Li2O2 deeper than ∼5–10 nm34. However, if the Li2O2 phase formed by ORR during discharging is an insulator, it is difficult to explain the operando observations of continuous oxidation of Li2O2 nanoparticles far away from the electrode. Thus, one possible explanation is that the Li2O2 nanoparticles formed by the solution precipitation may be electrically conductive, or at least Li2O2 particle surfaces are metallic as theoretically predicted by Siegel et al.16,35. As the reaction product layer consists of inter-connected Li2O2 particles, the particle surfaces and the interfaces could provide conductive paths for the decomposition of Li2O2 at Li2O2/electrolyte interfaces. However, after partial dissolution, many Li2O2 particles become isolated with the loss of the surface conductive pathway (Fig. 2k). Significantly, the operando observation reveals that the subsequent charging proceeds via continuous dissolution of the isolated Li2O2 particles while the charge potential smoothly increases (Fig. 1d), showing similar behavior in the final charging stage of conventional Li-O2 batteries. Therefore, the isolated residual Li2O2 nanoparticles are not “flotsam” but contribute to the charge recovery by OER. Except the hypothesis of the conductive Li2O2, the operando observations can also be interpreted by a hybrid chemical and electrochemical process, i.e, the dissolution of Li2O2 by the reverse non-Faradaic disproportionation reaction of Eq. 2: Li2O2 + O2 → 2LiO2, and OER by the one-electron oxidation of LiO2 by reverse Eq. 1: LiO2 →Li+ + O2 + e−. In this case, the charging kinetics is controlled by the non-Faradaic dissolution of Li2O2, which depends on the solubility of both LiO2 and oxygen in the electrolyte. Although recent observations of a redox mediator assisted Li2O2 decomposition provides an additional clue on the existence of non-Faradaic disproportionation reaction36, confirmation of this hypothesis still needs more electrochemical and theoretical evidences. After charging for 58 seconds, all the Li2O2 particles completely disappear but leave behind a diffuse layer around the Au electrode (Fig. 2p), similar to the initial stage of discharging. The much stronger intensity of the diffuse layer in Fig. 2p than that in Fig. 2a suggests that the formation and distribution of the diffuse layer are associated with the applied potentials of the electrode. It is worth noting that after Li2O2 nanoparticles completely dissolve at 58 seconds (Fig. 2p) the charging continues until 60 seconds (Fig. 1d). The short charging time gap could be caused by that the remained Li2O2 nanoparticles are too small to be seen from the low magnification STEM images, the oxidation process of Li2O2 takes place at other location out of the observation area, or the dissolved LiO2 in the electrolyte contributes to the continuous charging by the one-electron oxidation reaction. The measurements of Li2O2 evolution during charging reveal that the structural evolution of these particles is almost the inverse process of Li2O2 formation in discharging with the nearly symmetric plots shown in Fig. 3, although the reaction kinetics and reaction pathways for discharging and charging are very different. On the basis of the operando observations (Movie S1), the ORR and OER processes under round-trip discharging/charging cycles at a constant current density are schematically illustrated in Fig. S16.
We noticed that Luo and his co-workers very recently reported in situ observations of cathodic reactions in a solid-state Li-O2 battery using environmental transmission electron microscopy24. This work reveals new insights into oxygen reduction and oxygen evolution reactions in the solid-state Li-O2 system. However, it is fundamentally different from our work on the cathodic reactions in the liquid-state Li-O2 battery. Electrochemically, the utilization of different electrolytes (liquid versus solid) has rendered them as essentially different systems in the following aspects: (1) the chemistry of the discharge products is different between the two observations. In the study of Luo et al., a mixture of Li2O2 and Li2O is formed as the discharge product while in our liquid-state Li-O2 system the final products of discharge are only solid Li2O2. (2) The morphology of discharge products is different. Luo et al. reported the formation of hollow spherical particles with Li2O outer-shell and Li2O2 inner-shell surfaces. In the liquid system, the discharge products of Li2O2 are solid spherical particles. (3) More importantly, the reaction pathways are different. For the solid-state Li-O2 battery, the ORR on CNTs initially produces solid LiO2 covered by Li2O. The LiO2 subsequently disproportionates into Li2O2 and O2 through a solid-state transformation reaction. While, in our Li-O2 micro-battery with the liquid electrolyte, the LiO2 firstly produced is soluble in the electrolyte (in the forms of solvated Li+ and O2−), and upon the disproportionation of LiO2, Li2O2 is formed by nucleation and growth in the liquid electrolyte. In addition to the above differences, there are several unique advantages of our liquid-state Li-O2 experimental setup: (1) our liquid-cell Li-O2 micro-battery directly accommodates a liquid-state aprotic electrolyte of 1.0 M LiClO4 in DMSO with saturated oxygen and is operated under ambient pressure. This environment is almost identical to that of a practical non-aqueous Li-O2 battery. In contrast, the environmental TEM used by Luo et al. employed a constant O2 pressure of 0.1 mbar. Although it was claimed to be oxygen-rich environment, the pressure is far below the atmospheric pressure of a practical solid-state Li-O2 battery. (2) In our operando experiments, the charging and discharging were conducted at galvanostatic mode with a constant current density of 1.3 μA/cm2, similar to the coin battery testing. In contrast, in situ study in the solid-state cells by Luo et al. was discharged or charged by applying constant potential biasing and therefore the electrode reactions occurred at a far-from-equilibrium state. Therefore, our experimental setup better resembles the condition of practical batteries and hence the observed phenomena in our operando study would be more translatable for normal real-world batteries. Therefore, these two works investigate quite different systems and, we believe, both are of great scientific significance and would contribute to better understandings of the Li-O2 electrochemistry. In particular, the comparison between these two studies would provide insights to how the state of electrolytes affects the fundamental electrochemical reactions and the operation of Li-O2 batteries.
In summary, we have developed a liquid-state Li-O2 micro-battery for operando characterization of fundamental cathodic reactions under aberration-corrected STEM (STEM) synchronized with electrochemical measurements. The direct observations of Li2O2 nucleation, growth and decomposition at known discharge/charge overpotentials shine lights on the underlying mechanisms of cathodic reactions at a constant current density. The formation of porous Li2O2 layer through solution precipitation by the non-Faradaic disproportionation reaction of intermediate LiO2 is critical to keep a constant or near constant discharge potential for a large capacity. The dissolution of resultant Li2O2 nanoparticles during charging is mainly contributed by the Li2O2/electrode interfacial oxidation at the early stage of the charging reaction. We propose that the hybrid chemical and electrochemical process would be responsible for the oxidation of Li2O2 particles away from the electrode surfaces. Inspired by the operando observations, we have developed a full performance nanoporous graphene based Li-O2 battery by utilizing stable high-donor-number organic solvent DMSO as the electrolyte to enhance the solution mechanism of Li2O2 formation, the large surface area of nanoporous graphene for multiple heterogeneous nucleation/growth of small and porous Li2O2 nanoparticles, and an oxidation redox mediator to effectively reduce the charge overpotential by improving the charge transfer between insulating Li2O2 and electrode37.
References
Bruce, P. G., Freunberger, S. A., Hardwick, L. J. & Tarascon, J.-M. Li-O2 and Li-S batteries with high energy storage. Nature Mater. 11, 19–29 (2012).
Luntz, A. C. & McCloskey, B. D. Nonaqueous Li-air Batteries: A Status Report. Chem. Rev. 114, 11721–11750 (2014).
Black, R., Adams, B. & Nazar, L. F. Non-aqueous and hybrid Li-O2 batteries. Adv. Energy Mater. 2, 801–815 (2012).
Thotiyl, M. M. O., Freunberger, S. A., Peng, Z. Q. & Bruce, P. G. The carbon electrode in nonaqueous Li-O2 cells. J Am Chem Soc 135, 494–500 (2013).
Lu, Y.-C. & Shao-Horn, Y. Probing the reaction kinetics of the charge reactions of nonaqueous Li-O2 batteries. J Phys Chem Lett 4, 93–99 (2013).
Peng, Z. Q., Freunberger, S. A., Chen, Y. H. & Bruce, P. G. A reversible and higher-rate Li-O2 battery. Science 337, 563–566 (2012).
Imanishi, N., Luntz, A. C. & Bruce, P. G. The Lithium Air Battery (Springer 2014).
Mitchell, R. R., Gallant, B. M., Thompson, C. V. & Shao-Horn, Y. All-carbon-nanofiber electrodes for high-energy rechargeable Li-O2 batteries. Energy Environ. Sci. 4, 2952–2958 (2011).
Aurbach, D., McCloskey, B. D., Nazar, L. F. & Bruce, P. G. Advances in understanding mechanisms underpinning lithium-air batteries. Nat. Energy. https://doi.org/10.1038/nenergy.2016.128.
Black, R., Lee, J., Adams, B., Mims, C. A. & Nazar, L. F. The role of catalysts and peroxide oxidation in lithium-oxygen batteries. Angew. Chem. Int. Ed. 52, 392–396 (2013).
McCloskey, B. D. et al. Twin problems of interfacial carbonate formation in non-aqueous Li-O2 batteries. J Phys Chem Lett 3, 997–1001 (2012).
Zhong, L. et al. In situ transmission electron microscopy observations of electrochemical oxidation of Li2O2. Nano Lett. 13, 2209–2214 (2013).
Leskes, M. et al. Direct detection of discharge products in lithium-oxygen batteries by solid-state NMR spectroscopy. Angew. Chem., Int. Ed. 51, 8560–8563 (2012).
Peng, Z. Q. et al. Oxygen reactions in a non-aqueous Li+ electrolyte. Angew. Chem. Int. Ed. 50, 6351–6355 (2011).
Lim, H., Yilmaz, E. & Byon, H. R. Real-time XRD studies of Li-O2 electrochemical reaction in nonaqueous lithium–oxygen battery. J. Phys. Chem. Lett. 3, 3210–3215 (2012).
Radin, M. D., Rodriguez, J. F., Tian, F. & Siegel, D. J. Lithium peroxide surfaces are metallic, while lithium oxide surfaces are not. J. Am. Chem. Soc. 134, 1093–1103 (2012).
Mitchell, R. R., Gallant, B. M., Shao-Horn, Y. & Thompson, C. V. Mechanisms of morphological evolution of Li2O2 particles during electrochemical growth. J. Phys. Chem. Lett. 4, 1060–1064 (2013).
Fujita, T. et al. Atomic origins of the high catalytic activity of nanoporous gold. Nature Mater. 11, 775–780 (2012).
de Jonge, N. & Ross, F. M. Electron microscopy of specimens in liquid. Nature Nanotech. 6, 695–704 (2011).
Liu, X. H. et al. In situ TEM experiments of electrochemical lithiation and delithiation of individual nanostructures. Adv. Energy Mater. 2, 722–741 (2012).
Gu, M. et al. Demonstration of an electrochemical liquid cell for operando transmission electron microscopy observation of the lithiation/delithiation behavior of Si nanowire battery anodes. Nano Lett. 13, 6106–6112 (2013).
Holtz, M. E. et al. Nanoscale imaging of lithium ion distribution during in situ operation of battery electrode and electrolyte. Nano Lett. 14, 1453–1459 (2014).
Kushima, A. et al. Charging/discharging nanomorphology asymmetry and rate-dependent capacity degradation in Li-oxygen battery. Nano Lett. 15, 8260–8265 (2015).
Luo, L. L. et al. Revealing the reaction mechanisms of Li-O2 batteries using environmental transmission electron microscopy. Nat. Nanotech. 12, 535–539 (2017).
Ishizuka, K. A practical approach for STEM image simulation based on the FFT multislice method. Ultramicroscopy 90, 71–83 (2001).
Vogt, T., Dahmen, W. & Binev, P. Modeling Nanoscale Imaging in Electron Microscopy. (Springer, Ottawa, Canada, 2012).
Buban, J. P. et al. High-resolution low-dose scanning transmission electron microscopy. J. Electron Microsc. 59, 103–112 (2010).
Johnson, L. et al. The role of LiO2 solubility in O2 reduction in aprotic solvents and its consequences for Li-O2 batteries. Nature Chem. 6, 1091–1099 (2014).
Han, J. H. et al. Effect of chemical doping on cathodic performance of bicontinuous nanoporous graphene for Li-O2 batteries. Adv. Energy Mater. 6, 1501870 (2016).
Liu, Y., Hudak, N. S., Huber, D. L., Limmer, S. J., Sullivan, J. P. & Huang, J. Y. Nano Lett. 11, 4188–4194 (2011).
Chen, Y. H., Freunberger, S. A., Peng, Z. Q., Fontaine, O. & Bruce, P. G. Charging a Li-O2 battery using a redox mediator. Nature Chem. 5, 489–494 (2013).
Guo, X. W. et al. Hierarchical nanoporosity enhanced reversible capacity of bicontinuous nanoporous metal based Li-O2 battery. Sci. Rep. 6, 33466 (2016).
Lau, S. & Archer, L. A. Nucleation and growth of lithium peroxide in the Li-O2 battery. Nano Lett. 15, 5995–6002 (2015).
Viswanathan, V. et al. Electrical conductivity in Li2O2 and its role in determining capacity limitations in non-aqueous Li-O2 batteries. J. Chem. Phys. 135, 214704 (2011).
Radin, M. D. & Siegel, D. J. Charge transport in lithium peroxide: relevance for rechargeable metal-air batteries. Energy Environ. Sci. 6, 2370–2379 (2013).
Yang, C. C. et al. Direct observations of the formation and redox-mediator-assisted decomposition of Li2O2 in a liquid-cell Li-O2 microbattery by scanning transmission electron microscopy. Adv. Mater. 1702752 (2017).
Han, J. H. et al Full performance nanoporous graphene based Li-O2 batteries through solution phase oxygen reduction and redox-additive mediated Li2O2 oxidation. Adv. Energy Mater. 7, 1601933 (2017).
Acknowledgements
This work is sponsored by JST-CREST “Phase Interface Science for Highly Efficient Energy Utilization”, JST, Japan; World Premier International (WPI) Research Center Initiative for Atoms, Molecules and Materials, MEXT, Japan; Shanghai Pujiang Program (17PJ1403700); and National Natural Science Foundation of China (11704245).
Author information
Authors and Affiliations
Contributions
M.C. conceived and supervised the project. P.L., J.H., X.G. and C.Y. perform the operando STEM experiments. Y.I. helped the liquid cell assemble. P. L., A.H. and T.F. conducted the STEM data analysis. S.N. did the FEA calculations. P.L., J.H. and M.C. analysed the data and wrote the manuscript. All authors contributed to the extensive discussions of the results.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, P., Han, J., Guo, X. et al. Operando characterization of cathodic reactions in a liquid-state lithium-oxygen micro-battery by scanning transmission electron microscopy. Sci Rep 8, 3134 (2018). https://doi.org/10.1038/s41598-018-21503-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21503-w
This article is cited by
-
Addressing Transport Issues in Non-Aqueous Li–air Batteries to Achieving High Electrochemical Performance
Electrochemical Energy Reviews (2023)
-
Monitoring chemical reactions in liquid media using electron microscopy
Nature Reviews Chemistry (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
