
Abstract
Substantial impacts on climate have been documented for soot‒sulfuric acid (H2SO4) interactions in terms of optical and hygroscopic properties of soot aerosols. However, the influence of H2SO4 on heterogeneous chemistry on soot remains unexplored. Additionally, oxidation rate coefficients for polycyclic aromatic hydrocarbons intrinsic to the atmospheric particles evaluated in laboratory experiments seem to overestimate their degradation in ambient atmosphere, possibly due to matrix effects which are hitherto not mimicked in laboratory experiments. For the first time, our kinetics study reports significant influence of H2SO4 coating on heterogeneous ozonation of benzo(a)pyrene (BaP) deposited on model soot, representative to atmospheric particles. The approximate specific surface area of model soot (5 m2g−1) was estimated as a measure of the availability of surface molecules to a typical gaseous atmospheric oxidant. Heterogeneous bimolecular reaction kinetics and Raman spectroscopy studies suggested plausible reasons for decreased BaP ozonation rate in presence of H2SO4: 1. decreased partitioning of O3 on soot surface and 2. shielding of BaP molecules to gaseous O3 by acid-BaP reaction or O3 oxidation products.
Similar content being viewed by others

Introduction
Synchronous to the rapid pace of urbanization and amplified energy demand, atmospheric pollution with organic toxicants such as polycyclic aromatic hydrocarbons (PAHs) intrinsic to carbonaceous particles or soot, is rapidly enhancing1. Emission of soot2 and PAHs occur from diverse sources which are mainly anthropogenic, for example, incomplete combustion of fossil fuels and biomass in transport, residential, agriculture and commercial sectors. Soot particles absorb and scatter solar radiation influencing the earth’s radiative budget3,4 and the co-emitted organic toxicants pose severe threats to human health5. In fact, laboratory studies have shown that toxicity of the soot surface composition was enhanced by 1.5–2 times upon heterogeneous ozone oxidation6. Freshly emitted soot aggregates are composed of hydrophobic spherules which undergo aging by adsorption or condensation of either directly H2SO4 vapor or gaseous SO2 which eventually ends at H2SO4 and leads to a highly hygroscopic coating on the soot particles7. However, Donaldson and coworkers recently argued for the formation of hygroscopic sulfurous acid (H2SO3) from triplet SO2 as it is difficult for ground state SO2 to cross the high activation barrier of the endothermic reaction8. Nevertheless, the soot particles with hygroscopic coating might act as an effective nucleus to form small water clusters and subsequently grow in size by rapid condensation of more water molecules9. Indeed, H2SO4 coating on propane soot particles enhanced their hygroscopic size and the particles could act as cloud condensation nuclei (CCN) at 80% relative humidity condition7. Additionally, heterogeneous oxidation by hydroxyl radicals, ozone and nitrogen oxides can also aid to enhanced hydrophilicity of the aerosol surface by increasing oxygen- and nitrogen-containing functional groups in the surface adsorbed organic species10. Despite considerable studies on atmospheric aging of soot and subsequent alteration of their physicochemical properties such as morphology, hygroscopicity and optical properties, kinetic studies on heterogeneous oxidation of organics on soot surface are limited and the subject is still poorly understood11. Notably, the studies of Poschl and coworkers reported a non-linear Langmuir type dependence of benzo(a)pyrene (BaP, a 5-ring PAH; PAHs are ubiquitous organic pollutants) ozonation rate with gaseous ozone12. Later, studies involving 2–5 ring PAHs associated with wide range of substrates including solid carboxylic acid aerosols13, dry sodium chloride aerosols13, ammonium sulfate particles14, pyrex glass15, thin film of carboxylic acids16, water17 and 1-octanol17 reported substrate specific but similar non-linear relationships between ozonation rate constant and gaseous ozone concentration, which is consistent with the Langmuir-Hinshelwood model involving an initial rapid equilibration of gaseous ozone on substrate surface, followed by heterogeneous oxidation of PAHs13. So far laboratory studies are focused on the ozonation of PAHs adsorbed on single component surface but in the real-world scenario, the surface composition of atmospheric particles is complex, consisting of both organic and inorganic components.
The current work is the experimental manifestation of a hitherto assumption that shielding effect extends pollutants’ lifetime in the real environment. From the mechanistic consideration of the Langmuir-Hinshelwood kinetic model, we investigated whether a hygroscopic coating of H2SO4 can influence the ozonation kinetics of BaP deposited on laboratory generated soot particles. The approximate specific surface area of the soot particles was estimated not by the accessibility of soot surface but by the availability of surface-bound BaP molecules to gaseous ozone. In addition, the soot samples were characterized by Raman spectroscopy.
Results
To increase reproducibility of the experiments, the organic and inorganic impurities from the collected soot particles generated in controlled combustion of kerosene were removed chemically and thermally (see Supplementary information, pageS3). The resulting soot particles were termed as cleaned soot. The cleaned soot particles coated with BaP were termed as sootBaP and the sootBaP particles coated with H2SO4were termed as \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\).
Sample characterization by Raman Spectroscopy
In order to have a better understanding of the surface properties of the cleaned soot, sootBaP and \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\) samples at the molecular level, we have performed Raman measurements. Figure 1(a‒c) provide the Raman spectra of cleaned soot, sootBaP and \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\) respectively. The Raman spectrum of cleaned soot (Fig. 1a) is well fitted with four Lorentzian shaped bands centered at 1133, 1318, 1502 and 1589 cm−1. The bands are generated due to different structural and compositional types in the soot sample18,19. The mode at ~1589 cm−1 (G-band) is the graphitic lattice vibration with E2g symmetry. The band at ~1318 cm−1 corresponds to disordered graphite lattice with A1g symmetry mode. The observed band at ~1502 cm−1 represents the amorphous fraction of the soot sample. The peak at ~1133 cm−1 corresponds to C–C and C=C bond stretching vibration of polyene like structure with A1g symmetry. Nine bands are identified in the deconvoluted Raman spectra of sootBaP (Fig. 1b) prior to the exposure to gaseous ozone. Apart from the peaks due to carbonaceous soot (vide supra), we observed additional six modes at 425 cm−1, 606 cm−1, 668 cm−1, 803 cm−1, 963 cm−1 and 1219 cm−1, which are attributed to the different types of vibrations of BaP molecules20. The origins of these vibrational bands are summarized in Table 1. Prior to the exposure to gaseous ozone, twelve bands are identified in the deconvoluted Raman spectra of \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\)(Fig. 1c); details of the band assignments are given in Table 1. Again, apart from the peaks due to carbonaceous soot and BaP, the modes at 888 cm−1 and 1054 cm−1 are identified as the vibrational bands of H2SO4 21.

Characterization of soot samples prior to heterogeneous ozonation reaction. Curve fitting results for the typical first order Raman spectra of (1a) cleaned soot (based on four bands); (1b) cleaned soot coated with benzo(a)pyrene (BaP) (based on nine bands); (1c) cleaned soot first coated with BaP followed by coated with H2SO4 (based on twelve bands). The peak positions of soot, BaP and H2SO4 are indicated in the graphs.
Determination of specific surface area of soot
Surface coverage is the fraction of total number of surface active sites of an adsorbent occupied by adsorbate molecules. Previous studies have shown that substrate surface coverage significantly influence the reaction kinetics. For example, Alebic-Juretic and coworkers showed ozonation rate enhancement by a factor of 2.36 at sub-monolayer surface coverage of BaP on non-activated silica gel22. Similar observations have been reported for other substrates, such as spark discharge soot particles12 and azelaic acid aerosols13. Therefore, the monolayer surface coverage of BaP on the sootBaP samples was determined by using the technique utilized by Ray and coworkers for artificial snow samples23. Analogous to those authors, the Langmuir concentration (\({c}_{{\rm{BaP}}}^{{\rm{L}}}\)), i.e, the concentration corresponding to the monolayer coverage of BaP on soot surface was evaluated.
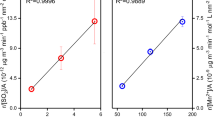
The BaP ozontion kinetics was studied by quantifying the BaP degradation in sootBaP samples with a series of BaP surface loads at ~8 ppm ozone concentration with increasing ozone exposure time (Fig. S2). Figure 2 shows the plot of observed pseudo-first order rate constants, \({k}_{{\rm{obs}}}^{{\rm{I}}}\) values (from Supplementary Fig. S2) against the corresponding BaP surface loads. The \({k}_{{\rm{obs}}}^{{\rm{I}}}\) values are independent at lower BaP surface loads (sub-monolayer), whereas decreasing \({k}_{{\rm{obs}}}^{{\rm{I}}}\) values are observed at higher BaP surface loads (above monolayer). The Langmuir concentration \({c}_{{\rm{BaP}}}^{{\rm{L}}}\) = \(8.6\,\times \,{10}^{-6}{\text{moles}g}^{-1}\) was estimated from the intersection point of two linear least-squares-fits with clearly different slopes22,24.

Determination of Langmuir concentration from kinetic experimental data. BaP ozonation kinetics on sootBaP samples as a function of \({c}_{{\rm{B}}{\rm{a}}{\rm{P}}}^{0}\) inside a quartz glass reactor at room temperature and pressure; \({c}_{{{\rm{O}}}_{3}}\)= 2 × 1014 molecules cm−3; the evaluated \({k}_{{\rm{obs}}}^{{\rm{I}}}\,\)values were corrected for losses due to evaporation and O2 oxidation; error bars represent the standard deviation.
Alebic-Juretic and coworkers explained their observation of ozonation rate enhancement at sub-monolayer BaP surface load by postulating rapid desorption of BaP oxidation products from the surface and chemical activation of BaP by the acidic silica gel surface22. On the contrary, Poschl and coworkers argued that most of the possible BaP oxidation products are not volatile enough for rapid desorption from silica gel surface12. Furthermore, these authors suggested that instead of rate enhancement at sub-monolayer BaP coverage, the reaction rates were reduced at above-monolayer surface loads due to burial effects by multiple layers of BaP and its oxidation products. Indeed, the gaseous ozone molecules are unable to reach the underlying BaP molecules at above monolayer surface load of BaP whereas at sub-monolayer condition the \({k}_{{\rm{obs}}}^{{\rm{I}}}\) values remain independent of the BaP surface loads, resulting in two distinct slopes in Fig. 2.
Additionally, the \({c}_{{\rm{BaP}}}^{{\rm{L}}}\) value was utilized to calculate the specific surface area (SSA) which is the gas accessible area of unit mass of a solid23. Since the accessibility of surface adsorbed BaP molecules to the gaseous ozone was considered instead of the soot surface, the SSA evaluated in this work represents a relative value. Therefore we termed it approximate specific surface area (ASSA). The ASSA of the soot particles was estimated to be 5 m2g−1, using equation (1) 23,24:
where, \({c}_{{\rm{BaP}}}^{{\rm{L}}}\) is the Langmuir concentration, NA is Avogadro’s number and ABaP is the molecular cross-sectional area of BaP15 assumed to be 1 nm2. Numerous studies have measured the specific surface area (SSA) of soot from various sources mainly by using the standard Brunauer–Emmett–Teller (BET) model for N2 adsorption isotherm, where N2 molecules undergo physical adsorption on soot surface25. In fact, the SSA determined by the BET technique is a measure of the gas accessible surface area of a solid. The SSA of soot particles generated from a wide range of sources ranged between 0.1–500 m2 g−1 26. Although the magnitude of ASSA of our model soot (~5 m2 g−1) is found small compared to spark discharge soot (395 m2 g−1)12,26, however, comparable values are observed for other types of soot which are more representative of the real-world atmosphere, such as wood stove27 (1.0 m2 g−1), bus exhaust27 (1.9 m2 g−1), marine vessel exhaust27 (12 m2 g−1) and soot of black smoke from ceramic furnace flue gas28 (15 m2 g−1). Also BET-Kr adsorption isotherm has been used to estimate the SSA of aviation kerosene soot (43 m2 g−1)29.
Heterogeneous ozonation kinetics of BaP in sootBaP and \({\bf{s}}{\bf{o}}{\bf{o}}{{\bf{t}}}_{{\bf{B}}{\bf{a}}{\bf{P}}+{{\bf{H}}}_{2}{\bf{S}}{{\bf{O}}}_{4}}\) samples
The ozonation kinetics of sub-monolayer concentration of BaP (\({c}_{{\rm{B}}{\rm{a}}{\rm{P}}}^{0}\) = \(3.12\,\times \,{10}^{-6}{\text{moles}g}^{-1}\), corresponding to ~0.4 times monolayer) adsorbed on sootBaP and \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\) samples, were studied by exposing to a range of ozone concentrations, \({c}_{{{\rm{O}}}_{3}}\)=8─360 ppm (see Supplementary Fig. S3). The evaluated \({k}_{{\rm{obs}}}^{{\rm{I}}}\) values from Fig. S3a and b were then plotted against the corresponding ozone concentrations in Fig. 3. The non-linear relationship between \({k}_{{\rm{obs}}}^{{\rm{I}}}\) and \({c}_{{{\rm{O}}}_{3}}\) indicates heterogeneous reaction on both sootBaP and \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\) samples. Figure 3 also demonstrates that initially \({k}_{{\rm{obs}}}^{{\rm{I}}}\) values are increasing linearly with increasing \({c}_{{{\rm{O}}}_{3}}\) but become independent and leveled off at higher \({c}_{{{\rm{O}}}_{3}}\) implying the Langmuir-Hinshelwood (LH) type of kinetics. The LH model30 demonstrates heterogeneous bimolecular reaction involving rapid initial equilibrium partitioning of O3 at the air-soot interface prior to heterogeneous reaction between BaP and O3 31. The experimental results of Shiraiwa and coworkers demonstrated possible involvement of long-lived reactive oxygen intermediates in the LH mechanism32. Therefore the plots in Fig. 3 were fitted with modified LH equation, equation (2)15:

Bimolecular heterogeneous reaction on soot surface. Pseudo first-order rate constants (\({k}_{{\rm{obs}}}^{{\rm{I}}}\)) as a function of gaseous ozone concentration for the reaction of BaP (\({c}_{{\rm{Bap}}}^{0}\) = \(3.12\,\times \,{10}^{-6}{\text{moles}g}^{-1}\)) and ozone on clean and H2SO4 coated soot inside the reactor at room temperature and pressure; error bars represent the standard deviation. The \({{\rm{K}}}_{{{\rm{O}}}_{3}}\,\)and k maxvalues were evaluated by fitting the plots with Langmuir ─ Hinshelwood equation.
where, k max is the maximum pseudo-first order rate constant at saturated surface concentration of ozone, \({{\rm{K}}}_{{{\rm{O}}}_{3}}\) is the adsorption equilibrium constant of ozone, and [O3] is the gas phase ozone concentration. The partitioning of gaseous ozone was observed to be higher by more than 1 order of magnitude on azelaic acid aerosol (\({{\rm{K}}}_{{{\rm{O}}}_{3}}\) = ~ \(1.2\,\times \,{10}^{-15}\) cm3) and by 3 orders of magnitudes on spark discharge soot (\({{\rm{K}}}_{{{\rm{O}}}_{3}}\)= \(2.8\,\times {10}^{-13}\) cm3)12 than that on NaCl surface (\({{\rm{K}}}_{{{\rm{O}}}_{3}}\)< \(1.2\,\times {10}^{-16}\) cm3)13, implying that ozone has higher affinity for non-polar surfaces. In good agreement with the previous results, we observed that ozone partitioning was ~ 7 times higher on non-polar sootBaP surface (\({{\rm{K}}}_{{{\rm{O}}}_{3}}\) = \(1.2\,\times \,{10}^{-13}\) cm3) than that on relatively polar surface of the \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\,\)samples (\({{\rm{K}}}_{{{\rm{O}}}_{3}}\) = \(1.8\times {10}^{-14}\) cm3) due to presence of H2SO4. Thus our results support Zhang and coworkers who concluded that uptake of H2SO4 on model soot surface, converted the hydrophobic soot into hydrophilic aerosol33. Also the evaluated \({{\rm{K}}}_{{{\rm{O}}}_{3}}\,\)value of our model soot varied within a factor of 2.5 from the \({{\rm{K}}}_{{{\rm{O}}}_{3}}\) value evaluated by Poschl and coworkers12. Previous studies have shown that \({{\rm{K}}}_{{{\rm{O}}}_{3}}\) may vary by 3 orders of magnitude but the typical range of k max falls within (\({10}^{-3}-{10}^{-2}){{\rm{s}}}^{-1}\) and varies by a factor of 3. The k max for sootBaP(k max = \(9.12\,\times {10}^{-3}{{\rm{s}}}^{-1}\)) and \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\) (k max = \(1.85\times {10}^{-3}{{\rm{s}}}^{-1}\,)\) samples are within this range. This is indicative of a similar rate-determining step for BaP ozonation on model soot surface, possibly involving formation of reactive oxygen intermediates from ozone, as postulated by Shiraiwa and co-workers32. The k max for sootBaP samples was found to be ~5 times higher than \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\,\)samples. Indeed, the possibility of slow bulk reaction on \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\) samples is most unlikely because the nonlinear shape of the plot of \({k}_{{\rm{obs}}}^{{\rm{I}}}\) as a function of \({c}_{{{\rm{O}}}_{3}}\) (Fig. 3) is consistent with LH mechanism indicative of a heterogeneous bimolecular reaction13. According to LH model, limited numbers of active sites are available for air-soot surface partitioning of gaseous ozone. A saturated condition is developed at some ozone concentration when all the active surface sites are occupied. Beyond this saturation point, the ozonation rate constant should become independent of the ozone concentration. This is clearly illustrated in Fig. 3, where the k max forms a plateau at high ozone concentrations.
Mmereki and co-workers studied anthracene (3-ring PAH) ozonation kinetics and observed that in presence of near-monolayer coating of a series of n-carboxylic acid films, the k max of the reaction between gaseous ozone and surface adsorbed anthracene on air-aqueous interface decreased significantly in comparison to uncoated water surface16. The authors postulated the formation of organic acid and anthracene complex at the ozone attacking positions of anthracene molecules. Consequently both ozone and organic acids are in competition for the same reaction site resulting in reduction of ozonation rate. This reasoning can be assumed to be partly effective to explain our observations. If H2SO4─BaP complex formation were the only reason for slower ozonation rate in our study then we would not have observed >70% BaP recovery in prolonged ozone exposure of \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\) samples (Supplementary Fig. S4). Moreover Henning and coworkers reported that H2SO4 possibly consumes PAHs to produce lower molecular weight products34. Additionally, the degree of disorder in soot which is measured from the intensity ratio of defect (D) and ideal graphite (G) bands35, i.e., ID/IGin cleaned soot, sootBaP and \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\) are estimated to be 1.79, 4.8 and 0.51 respectively. The ID/IG ratio also corresponds to the aromatic/olefinic ration of a sample36,37. Thus relative to the ID/IG value of cleaned soot (1.79), the higher ID/IG value for sootBaP (4.8) clearly indicates the presence of BaP on the soot surface, as D bands arise from larger aromatic compounds38. Interestingly, ID/IG ratio is reduced significantly when H2SO4 was coated on sootBaP. Indeed, our Raman spectroscopic measurements (Fig. 1) shows that the ID/IG value i.e., aromatic/olefinic ratio is significantly reduced for \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\)(ID/IG = 0.51) when the sootBaP (ID/IG = 4.8) samples were coated with H2SO4. Therefore possibly the H2SO4–BaP complex and/or products formed from the reaction between H2SO4 and BaP in \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\) samples, hinder the BaP molecules to gaseous ozone resulting in the slow reaction rate compared to that in sootBaP samples. Essentially, extensive investigations are required further to decipher the reasons of slowing down of BaP ozonation rate in presence of H2SO4, inconspicuously.
In this experimental study we have focused on a hitherto unexplored topic of whether the hygroscopic coating on soot aerosol surface influences the heterogeneous oxidation kinetics and explored its consequence. We have shown in this paper for the first time that H2SO4 coating markedly influences the oxidation kinetics of soot surface adsorbed BaP by gaseous ozone. The maximum first order rate constants (k max) of BaP ozonation reaction on sootBaP and \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\,\)samples were found to be \(9.12\,\times {10}^{-3}{{\rm{s}}}^{-1}\) and \(1.85\,\times \,{10}^{-3}{{\rm{s}}}^{-1}\) respectively. Thus k max was reduced by nearly 5 times in presence of H2SO4 coating. In immediate effect, the half-life of soot surface adsorbed BaP is enhanced, as calculated for sootBaP (t 1/2 ~17 minutes) and \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\)(t 1/2 ~2 hours) samples at atmospherically relevant ozone concentration of 100 ppb15. If this is a general phenomenon then the lifetime enhancement would possibly result in long range transport of soot bound chemicals causing pollution to a pristine area14. On the other hand, particles in ambient air affect the atmosphere directly by scattering or absorbing solar radiation as well as indirectly by forming cloud condensation nuclei (CCN), affecting the microphysical properties of cloud4,7. Therefore, at ambient conditions the aged soot particles can also act as CCN and be subsequently removed by rain, resulting in soil or water pollution39. Ageing of soot particles can also bring changes in cloud albedo (Twomey effect) and precipitation pattern34. It is believed that the experimental rate coefficient values are possibly overestimations compared to the real atmospheric values40, because in real atmosphere, particle associated PAHs are shielded from atmospheric oxidants which have not yet been mimicked in the experiments. BaP apparently undergoes faster chemical degradation compared to that in real atmosphere. The H2SO4 coating in the \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\,\)samples however, reflects a multiphase condition to the adsorbed BaP molecules and thus our study is undoubtedly a successful attempt in approaching the real atmospheric conditions. Furthermore, development of a simple soot preparation method from kerosene and subsequent characterization of the soot samples by Raman spectroscopy and validation by evaluating their approximate specific surface area (~5 m2g−1) followed by utilization of the soot samples for kinetics experiments are also novel aspects of this study. Further environmental implications of this work are currently being characterized using models and will be reported in due course.
Methods
Soot particles were generated under controlled combustion of kerosene in a set up similar to that of a typical wick lamp (Fig. S1a). Length of the wick was adjusted to maintain a stable and medium flame. Ultra high purity air (flow rate was adjusted at 0.3 \({{\rm{L}}\min }^{-1}\) for maximum soot collection), carrying the soot particles, was bubbled through 200 mL n-hexane, where the particles were deposited. The collected particles were further washed by dichloromethane (DCM) followed by heated for 5 hrs inside a furnace at 400 °C to ensure maximum removal of the organic impurities. These cleaned soot particles were used as the substrate for heterogeneous ozonation of BaP. The sootBaP samples were prepared by soaking the cleaned soot into different concentrations of BaP in DCM, followed by blowing off DCM under gentle stream of nitrogen gas. The \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\) samples were prepared by soaking 10 g sootBaP particles into 10 mL ~1 μM H2SO4 solution followed by drying off the particles by heating at 180 °C. An experimental set up was developed to study the heterogeneous BaP ozonation kinetics (see Supplementary Fig. S1). In brief, ozone was generated by flowing ultra-high purity grade O2 gas through non-thermal plasma generated inside a homemade dielectric barrier discharge (DBD) reactor. The sootBaP and \({{\rm{soot}}}_{{\rm{BaP}}+{{\rm{H}}}_{2}{{\rm{SO}}}_{4}}\,\)samples were simultaneously exposed to gaseous O3 inside a quartz glass tube reactor. The ozonation kinetics was determined by evaluating the BaP degradation with O3 exposure time. The initial and unreacted BaP concentrations were measured using high performance liquid chromatography (HPLC, Shimadzu Prominance), equipped with a C-18 reversed-phase chromatographic column (SupelcosilTM LC-PAH, 15 cm × 4.6 mm, 5 µm) and a fluorescence detector (Shimadzu RF 10AXL). Raman measurements were performed in backscattering geometry using LabRAM HR (Jobin Yvon) spectrometer equipped with a Peltier-cooled charge-coupled-device (CCD) detector. An air cooled argon ion laser with a wavelength of 488 nm was used as the excitation light source. Raman spectra of all samples have been recorded in the frequency range of 50–2000 cm−1 under similar experimental conditions.
References
Jacobson, M. Z. Control of fossil-fuel particulate black carbon and organic matter, possibly the most effective method of slowing global warming. J. Geophys. Res. Atmos. 107, ACH 16-11–ACH 16-22, https://doi.org/10.1029/2001JD001376 (2002).
Bond, T. C. et al. Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res. Atmos. 118, 5380–5552 (2013).
Jacobson, M. Z. Strong radiative heating due to the mixing state of black carbon in atmospheric aerosols. Nature 409, 695–697 (2001).
Ramanathan, V. & Carmichael, G. Global and regional climate changes due to black carbon. Nature Geosci. 1, 221–227 (2008).
Poschl, U. & Shiraiwa, M. Multiphase chemistry at the atmosphere-biosphere interface influencing climate and public health in the anthropocene. Chem. Rev. 115, 4440–4475 (2015).
Holder, A. L., Carter, B. J., Goth–Goldstein, R., Lucas, D. & Koshland, C. P. Increased cytotoxicity of oxidized flame soot. Atmos. Pollut. Res. 3, 25–31 (2012).
Zhang, R. et al. Variability in morphology, hygroscopicity, and optical properties of soot aerosols during atmospheric processing. Proc. Natl. Acad. Sci. USA 105, 10291–10296 (2008).
Donaldson, D. J., Kroll, J. A. & Vaida, V. Gas-phase hydrolysis of triplet SO2: A possible direct route to atmospheric acid formation. Sci. Rep. 6, 30000 (2016).
Khalizov, A. F., Xue, H., Wang, L., Zheng, J. & Zhang, R. Enhanced Light Absorption and Scattering by Carbon Soot Aerosol Internally Mixed with Sulfuric Acid. J. Phys. Chem. A 113, 1066–1074 (2009).
McMeeking, G. R., Good, N., Petters, M. D., McFiggans, G. & Coe, H. Influences on the fraction of hydrophobic and hydrophilic black carbon in the atmosphere. Atmos. Chem. Phys. 11, 5099–5112 (2011).
George, C., Ammann, M., D’Anna, B., Donaldson, D. J. & Nizkorodov, S. A. Heterogeneous Photochemistry in the Atmosphere. Chem. Rev. 115, 4218–4258 (2015).
Pöschl, U., Letzel, T., Schauer, C. & Niessner, R. Interaction of Ozone and Water Vapor with Spark Discharge Soot Aerosol Particles Coated with Benzo[a]pyrene: O3 and H2O Adsorption, Benzo[a]pyrene Degradation, and Atmospheric Implications. J. Phys. Chem. A 105, 4029–4041 (2001).
Kwamena, N.-O. A., Thornton, J. A. & Abbatt, J. P. D. Kinetics of Surface-Bound Benzo[a]pyrene and Ozone on Solid Organic and Salt Aerosols. J. Phys. Chem. A 108, 11626–11634 (2004).
Zhou, S., Lee, A. K. Y., McWhinney, R. D. & Abbatt, J. P. D. Burial Effects of Organic Coatings on the Heterogeneous Reactivity of Particle-Borne Benzo[a]pyrene (BaP) toward Ozone. J. Phys. Chem. A 116, 7050–7056 (2012).
Kwamena, N.-O. A., Earp, M. E., Young, C. J. & Abbatt, J. P. D. Kinetic and Product Yield Study of the Heterogeneous Gas−Surface Reaction of Anthracene and Ozone. J. Phys. Chem. A 110, 3638–3646 (2006).
Mmereki, B. T., Donaldson, D. J., Gilman, J. B., Eliason, T. L. & Vaida, V. Kinetics and products of the reaction of gas-phase ozone with anthracene adsorbed at the air–aqueous interface. Atmos. Environ. 38, 6091–6103 (2004).
Mmereki, B. T. & Donaldson, D. J. Direct Observation of the Kinetics of an Atmospherically Important Reaction at the Air−Aqueous Interface. J. Phys. Chem. A 107, 11038–11042 (2003).
Han, C., Liu, Y., Ma, J. & He, H. Effect of soot microstructure on its ozonization reactivity. J. Chem. Phys. 137, 084507 (2012).
Sadezky, A., Muckenhuber, H., Grothe, H., Niessner, R. & Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 43, 1731–1742 (2005).
Bao, L. et al. Surface enhanced Raman spectroscopic detection of polycyclic aromatic hydrocarbons (PAHs) using a gold nanoparticles-modified alginate gel network. Analyst 137, 4010–4015 (2012).
Lund Myhre, C. E., Christensen, D. H., Nicolaisen, F. M. & Nielsen, C. J. Spectroscopic Study of Aqueous H2SO4 at Different Temperatures and Compositions: Variations in Dissociation and Optical Properties. J. Phys. Chem. A 107, 1979–1991, https://doi.org/10.1021/jp026576n (2003).
Alebic-Juretic, A., Cvitas, T. & Klasinc, L. Heterogeneous polycyclic aromatic hydrocarbon degradation with ozone on silica gel carrier. Environ. Sci. Technol. 24, 62–66 (1990).
Ray, D., Kurková, R., Hovorková, I. & Klán, P. Determination of the Specific Surface Area of Snow Using Ozonation of 1,1-Diphenylethylene. Environ. Sci. Technol. 45, 10061–10067 (2011).
Hasegawa, T., Kajiyama, M. & Yamazaki, Y. Surface photochemistry of alkyl aryl ketones: energy transfer and the effect of a silica-gel surface on electronic states of excited molecules. J. Phys. Org. Chem. 13, 437–442 (2000).
Brunauer, S., Emmett, P. H. & Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 60, 309–319 (1938).
Kuznetsov, B. V., Rakhmanova, T. A., Popovicheva, O. B. & Shonija, N. K. Water adsorption and energetic properties of spark discharge soot: Specific features of hydrophilicity. J. Aerosol Sci. 34, 1465–1479 (2003).
Rockne, K. J., Taghon, G. L. & Kosson, D. S. Pore structure of soot deposits from several combustion sources. Chemosphere 41, 1125–1135 (2000).
Lu, P. et al. Research on soot of black smoke from ceramic furnace flue gas: characterization of soot. J. Hazard. Mater. 199–200, 272–281 (2012).
Ferry, D., Suzanne, J., Nitsche, S., Popovitcheva, O. B. & Shonija, N. K. Water adsorption and dynamics on kerosene soot under atmospheric conditions. J. Geophys. Res. Atmos. 107, AAC 22-21–AAC 22-10 (2002).
Masel, R. I. Principles of Adsorption and Reaction on Solid Surfaces. (John Wiley & Sons, 1996).
Ammann, M., Poschl, U. & Rudich, Y. Effects of reversible adsorption and Langmuir-Hinshelwood surface reactions on gas uptake by atmospheric particles. Phys. Chem. Chem. Phys. 5, 351–356 (2003).
Shiraiwa, M. et al. The role of long-lived reactive oxygen intermediates in the reaction of ozone with aerosol particles. Nat. Chem. 3, 291–295 (2011).
Zhang, D. & Zhang, R. Laboratory Investigation of Heterogeneous Interaction of Sulfuric Acid with Soot. Environ. Sci. Technol. 39, 5722–5728 (2005).
Henning, S. et al. Hygroscopic growth and droplet activation of soot particles: uncoated, succinic or sulfuric acid coated. Atmos. Chem. Phys. 12, 4525–4537 (2012).
Ferrari, A. C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 143, 47–57 (2007).
Beams, R., Gustavo Cancado, L. & Novotny, L. Raman characterization of defects and dopants in graphene. J. Phys. Condens. Matter 27, 083002 (2015).
Romanías, M. N. et al. Investigation of the Photochemical Reactivity of Soot Particles Derived from Biofuels Toward NO2. A Kinetic and Product Study. J. Phys. Chem. A 119, 2006–2015 (2015).
Zhang, S., Zeng, X. T., Xie, H. & Hing, P. A phenomenological approach for the Id/Ig ratio and sp3 fraction of magnetron sputtered a-C films. Surf. Coat. Technol. 123, 256–260 (2000).
Zuberi, B. et al. Hydrophilic properties of aged soot. Geophys. Res. Lett. 32, L01807 (2005).
Lammel, G. et al. Long-range Atmospheric Transport of Polycyclic Aromatic Hydrocarbons is Worldwide Problem - Results from Measurements at Remote Sites and Modelling. Acta chimica Slovenica 62, 729–735 (2015).
Tomikawa, K. & Kanno, H. Raman Study of Sulfuric Acid at Low Temperatures. J. Phys. Chem. A 102, 6082–6088 (1998).
Chiang, H. P. et al. FT-Raman, FT-IR and normal-mode analysis of carcinogenic polycyclic aromatic hydrocarbons. Part I-a density functional theory study of benzo(a)pyrene (BaP) and benzo(e)pyrene (BeP). J. Raman Spectroscopy 32, 45–51 (2001).
Acknowledgements
Authors would like to thank Science and Engineering Research Board, Department of Science and Technology, Government of India for supporting the study under IRHPA (Intensification of Research in High Priority Areas) scheme. The authors are thankful to Tapan K. Dutta for providing the Benzo(a)pyrene, Soumedra Singh for providing the DBD reactor; Swaroop Biswas for HPLC measurements; Barun Mahata, Sibjyoti Debnath, Abhisek Sarkar and Avisek Banerjee for technical helps.
Author information
Authors and Affiliations
Contributions
D.R. conceived the idea of the work, designed and constructed the set-up, performed the experiments, analyzed the data and wrote the original manuscript. A.S. and T.B. took the Raman spectra of the samples, analyzed the data and wrote the Raman spectroscopy portion of the manuscript. S.R., A.C. and S.K.G. made critical comments based on which the final manuscript was prepared.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ray, D., Bhattacharya, T., Chatterjee, A. et al. Hygroscopic Coating of Sulfuric Acid Shields Oxidant Attack on the Atmospheric Pollutant Benzo(a)pyrene Bound to Model Soot Particles. Sci Rep 8, 129 (2018). https://doi.org/10.1038/s41598-017-18292-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18292-z
This article is cited by
-
Process-oriented analysis of aircraft soot-cirrus interactions constrains the climate impact of aviation
Communications Earth & Environment (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
