
Abstract
Arboviruses that replicate in mosquitoes activate innate immune response within mosquitoes. Regulatory non-coding microRNAs (miRNA) are known to be modulated in mosquitoes during chikungunya infection. However, information about targets of these miRNAs is scant. The present study was aimed to identify and analyze targets of miRNAs that are regulated during chikungunya virus (CHIKV) replication in Aedes aegypti cells and in the mosquito. Employing next-generation sequencing technologies, we identified a total of 126 miRNAs from the Ae. aegypti cell line Aag2. Of these, 13 miRNAs were found to be regulated during CHIKV infection. Putative targets of three of the most significantly regulated miRNAs- miR-100, miR-2b and miR-989 were also analyzed using quantitative PCRs, in cell lines and in mosquitoes, to validate whether they were the targets of the miRNAs. Our study expanded the list of miRNAs known in Ae. aegypti and predicted targets for the significantly regulated miRNAs. Further analysis of some of these targets revealed that ubiquitin-related modifier is a target of miRNA miR-2b and plays a significant role in chikungunya replication.
Similar content being viewed by others


Introduction
Innate immunity in insects has been described as the germ line-encoded anti-infection response of the host1. The response is executed via several mechanisms such as phagocytosis2, antimicrobial peptides3, melanotic encapsulation4, and nitrogen intermediates5. In addition to these mechanisms, insects employ other pathways such as RNA interference (RNAi), immune deficiency (IMD), and toll and JAK/STAT signaling pathways to provide defense against pathogens, including viruses6,7. Amongst viruses, some are transmitted by arthropods and are called arboviruses, many of which are of importance due to the impact they have on human health8. Mostly transmitted by the Ae. aegypti and Culex tritaeniorhynchus mosquitoes, these viruses are characterized by their requirement to alternate between a vertebrate host and the mosquito.
Chikungunya virus (CHIKV) is an arbovirus belonging to the genus Alphavirus, family Togaviridae. Its genome is a single-stranded positive-sense RNA that transcribes mRNA from two open reading frames (ORFs): one that expresses nonstructural proteins and another that expresses structural proteins. In addition to these two ORFs, the genome contains a 5′-terminal cap and a 3′-terminal poly(A) tract along with 5′ and 3′ untranslated regions (UTRs) that contain signals important for replication of the RNA9. Whereas much information is available with respect to the innate and adaptive immune response of CHIKV in the mammalian hosts10,11, little is known about the innate immune responses in the vector during CHIKV replication12. Amongst all innate immune responses, RNAi assumes the first level of defense against RNA viruses in mosquitoes.
RNAi is a conserved, sequence-specific, gene-silencing phenomenon that is induced by double-stranded RNA. In the case of a viral infection in insects, viral replication intermediates in the form of double-stranded RNA (dsRNA) trigger RNAi that acts as a defense mechanism13. Whereas exogenous RNAi is triggered by the presence of siRNA generated through the processing of dsRNA and virus-derived siRNAs (vsiRNAs), another class of small RNAs that are regulatory are the microRNAs (miRNA)14. A class of small, non-coding RNAs of 19–24 nt in length, miRNAs regulate gene expression post-transcriptionally by binding to complementary regions in the 3′ UTR of target mRNAs15. Reports have revealed the role of miRNAs in several cellular processes16, including regulating virus replication17. However, studies on the cellular targets through which these miRNAs may act are scant, thereby not providing enough information on the mode of action of miRNAs in insect immunity.
A recent study by our group revealed that CHIKV replication in Aedes albopictus cells regulated vector miRNAs18. The present study was initiated to study the impact of the cellular targets of some of the miRNAs that are regulated in Ae. aegypti. Upon CHIKV infection in an Ae. aegypti cell line, namely Aag2, significantly regulated miRNAs were identified through next-generation sequencing, targets of the significantly regulated miRNAs were predicted through computational approaches, and pathway analysis of these targets were performed. Furthermore, through loss-of-function assays, targets of the selected miRNAs were validated using quantitative PCR, both in Ae. aegypti cells and in Ae. aegypti mosquitoes. The present study also showed that miR-2b targets the 3′UTR of ubiquitin-related modifier (URM) to control CHIKV replication.
Results
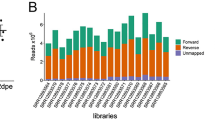
Global miRNA profiling of Ae. aegypti was performed using high-throughput small RNA sequencing. Small RNA libraries obtained from a CHIKV-infected Ae. aegypti cell line (Aag2) at 12 and 24 hours post-infection (h.p.i.) were compared with Ae. aegypti miRNAs from an uninfected Aag2 cell line. These time points were chosen in order to evaluate the vector miRNAs during early-phase CHIKV infection. Post-sequencing, small RNA reads from all the three libraries were separately mapped on the Ae. aegypti genome. The mapping showed 76.12% reads of uninfected library, 82.34% reads of 12 h.p.i. library, and 51.63% reads of 24 h.p.i. library. The mapped reads were further used for the identification of known miRNAs, including up to one mismatch using data analysis pipeline established in-house18. Unmapped reads of 12 and 24 h.p.i. libraries were further mapped on to the CHIKV genome. Details of small RNA analysis of the datasets are shown in Supplementary Tables S2 and S3.
Ae. aegypti miRNAs are regulated upon CHIKV infection
To analyze the relative abundance and expression profiling of miRNAs, tags per million (TPM) of total RNA reads for each miRNA in all libraries were calculated. The TPM values were compared between libraries and a heat map of all predicted miRNAs was generated and visualized. We normalized the row values to have mean of 0 and a variance of 1 (Fig. 1A ). Log-fold change and p-value of the predicted miRNAs were calculated using edgeR packages (trimmed mean of M-values; TMM) described elsewhere18. miRNAs with p-value ≤ 0.05 were considered significant. The analysis revealed a total of thirteen miRNAs to be significant in all analyses (Fig. 1B ). Three miRNAs (miR-2b, miR-2951–5p, and miR-10–5p) were found to be significantly regulated (p ≤ 0.05) in the 12 h.p.i. library when compared with the uninfected library: two miRNAs (miR-10-5p and miR-2b) were upregulated, and one miRNA (miR-2951-5p) was found to be downregulated. Similarly, eleven miRNAs were significantly regulated in the 24 h.p.i. library, with five miRNAs (miR-34-3p, miR-317-5p, miR-278-5p, miR-2b, and miR-71-5p) showing overexpression and six miRNAs (miR-998-3p, miR-989, miR-285, miR-2779, miR-2951-3p, and miR-100) being underexpressed.

Heatmap of regulated (Ae. aegypti cell line) Aag2 miRNAs upon different time points of CHIKV infection. (A) Heatmap of top 50 regulated miRNAs. (B) Heatmap of significantly regulated miRNAs.
Validation of miRNA expression using quantitative real-time PCR
To validate the small RNA sequencing results, we carried out quantitative real-time PCR (qRT-PCR) analysis for randomly picked six miRNAs. Four out of the six miRNAs showed expression profiles similar to those observed in small RNA sequencing analysis, whereas miR-100 and miR-71-5p showed different patterns. Quantitative RT-PCR showed that at 24 h.p.i., miR-989 and miR-71-5p were downregulated and miR-2b, miR-184-3p, and miR-278-5p along with miR-2b were upregulated. This scenario changed at 48 h.p.i. in the case of miR-2b, miR-184-3p, and miR-71-5p: these miRNAs showed significant upregulation in their expression at 48 h.p.i. miR-2b showed a six-fold increase (p < 0.001), miR-184-3p showed a 7.5-fold increase (p < 0.0001), miR-71-5p showed a 7.7-fold increase (p < 0.0001), whereas changes in the expression profiles of miR-278-5p, miR-989, and miR-100 remained statistically insignificant (Fig. 2). With respect to the two miRNAs that showed a different expression pattern from small RNA sequencing, namely, miR-71-5p and miR-100, we sought to understand the discrepancy in the results between the two techniques. In the case of miR-71-5p, sequencing data analysis showed TPM values to be 15.63, 23.93, and 58.85 for the uninfected, 12 h.p.i. and 24 h.p.i. libraries, respectively. It should be noted that even though the regulation was statistically significant in small RNA data analysis, expression of this miRNA might be too low, which could be a reason for the insignificant qRT-PCR expression profiling even at 24 h. With respect to miR-100, the TPM in the library were higher: 172.42, 288.68, and 9.81 in the uninfected, 12 h.p.i., and 24 h.p.i. library, respectively. However, this pattern was reversed in qPCR analysis, which warrants further studies. Earlier studies reported that such discrepancies are possible due to the inherent probing methods of the technologies19,20,21. All RT-PCR-amplified products were cloned and sequenced to validate the amplification of correct miRNAs. BLAST results revealed specific miRNAs amplified by RT-PCR, thereby further validating small RNA sequencing analysis.

qRT-PCR analysis of selected six miRNAs showing differential regulation upon CHIKV infection at 12, 24, and 48 h, validating deep sequencing results. Data were expressed as mean ± SEM; ****p < 0.0001 vs. control group.
Target prediction of significant miRNAs and functional analysis
To understand the function of miRNAs, it is important to study their targets. miRNAs are known to function in clusters to regulate various biological processes22,23, which prompted us to predict the targets of all the differentially regulated miRNAs that were regulated under different experimental conditions. Targets with the minimum free energy not exceeding −20 kcal/mol and p ≤ 0.05 were considered significant and were further subjected to pathway enrichment analysis (Supplementary Figure S1 ).
Expression profiling of targets upon miRNA inhibition in Ae. aegypti cell lines and mosquitoes
In order to confirm the targets that were predicted, we selected three miRNAs, namely, miR-2b, miR-100, and miR-989 for target prediction, that were among the most significantly differentially expressed in small RNA sequencing, further downstream validations and biological relevance in other systems. A total of 785, 1024, and 294 targets were predicted for miR-2b, miR-989, and miR-100, respectively. Further analysis was performed on the basis of the involvement of targets in immune and signaling pathways, and their binding energies and two targets for each of the three miRNAs were validated further. For miR-2b, two genes, namely, URM (AAEL008680) and ubiquitin (AAEL006511), were selected on the basis of statistical significance and their relevance in immune-related and signaling pathways. In the case of miR-100, cdc42 (AAEL011500) and sumo-ligase (AAEL015099) were selected. Similarly, for miR-989, sh2/sh3 adaptor (AAEL013539) and vacuolar ATP synthase (AAEL012819) were selected for validation.
To understand the role of miRNAs in the expression of the putative targets, miRNAs were knocked down by either transfecting miRNA-specific antagomirs in Ae. aegypti cells or by injecting in the mosquitoes, and the time points that exhibit maximum silencing were ascertained (Supplementary Figure S2). Upon maximum knockdown of the miRNAs, the expression levels of these miRNA targets were elucidated through qRT-PCR at 24 and 48 h after miRNA silencing. Expression of URM was found to be significantly regulated by miR-2b inhibition as compared to ubiquitin at 24 h, but at 48 h, ubiquitin showed a marked increase in its expression. In the case of miR-100 and miR-989, their targets did not show significant difference in their expression levels after antagomir transfection at both 24 and 48 h (Fig. 3A). To further confirm our findings in mosquitoes, we performed loss-of-function assays of miRNAs in Ae. aegypti mosquitoes. The miRNA-specific antagomirs were injected into the thorax of female mosquitoes and time points similar to those for the experiment performed in the Aag2 cell line—24 and 48 h after nanoinjection—were chosen for the validation of targets identified in mosquitoes. Expression of URM was found to be significantly regulated when miR-2b was inhibited as compared to ubiquitin, whereas miR-100 and miR-989 targets did not show much difference in their expression levels (Fig. 3B ). On the basis of these findings, we hypothesized that URM is a target of miR-2b. To confirm this, we performed a luciferase assay in HEK 293-T cells to evaluate the binding of miR-2b to the 3′UTR of URM. Empty pmR-mCherry vector and pmirGLO vector containing 3′UTR of URM served as controls; sequence-specific binding of miR-2b to the 3′UTR of URM was evaluated by mutating the miRNA-binding site at the 3′UTR and then the assay was performed. The assay revealed a >80% reduction in the luciferase activity in those constructs with wild-type 3′UTR of URM. However, constructs with a mutated binding site showed loss of binding, and the luciferase activity reverted to control values. Taken together, our results establish that URM is indeed a target of miR-2b (Fig. 3C).

(A) Aag2 cell line transfected with antagomir for miR-2b, miR-989, and miR-100, 24 and 48 h after transfection, showing expression levels of ubiquitin, URM, sh2/sh3 adaptor, vacuolar ATP synthase, cdc42, and sumoligase. (B) Mosquito injected with antagomir for miR-2b, miR-989, and miR-100, 24 and 48 h after transfection, showing expression levels of ubiquitin, URM, sh2/sh3 adaptor, vacuolar ATP synthase, cdc42, and sumoligase. (C) Luciferase assay showing relative percentage of luciferase/renilla luminescence for miR-2b binding to 3′UTR of URM and to mutated 3′UTR of URM. Data were expressed as mean ± SEM except in 3 C, which is expressed in SD; ****p < 0.0001 vs. control group.
URM affects CHIKV replication
To understand the role of URM during CHIKV infection better, we dissected the expression pattern of this transcript during CHIKV infection in the presence and absence of miR-2b. As a first step, we confirmed whether silencing miR-2b regulated CHIKV infection. For this purpose, we inhibited miR-2b using antagomir in the Aag2 cell line and infected it with CHIKV virus. After 24 hours of infection, we checked the CHIKV viral genomic RNA and found that inhibiting miR-2b increased CHIKV replication (Fig. 4A ). We observed an increase in viral replication when miR-2b is silenced, thereby proving that miR-2b may be playing a role in regulating CHIKV infection in Ae. aegypti. Next, we sought to understand the role of URM during CHIKV infection, as it is a target of miR-2b. We first checked the expression of URM in Ae. aegypti mosquitoes 24 and 48 h after CHIKV infection. We observed more than a 2.5-fold reduction in URM expression upon CHIKV infection, which further reduced in a day-dependent manner (Fig. 4B). This finding prompted us to hypothesize that URM may have an impact on CHIKV replication. To test this, we evaluated CHIKV replication upon silencing URM. Following dsRNA-induced silencing of URM and subsequent infection of CHIKV in Ae. aegypti mosquitoes, we evaluated the CHIKV viral genomic RNA 24 and 48 h.p.i. We observed a 50% reduction (p < 0.001) in CHIKV viral genomic RNA in 24 h and a more drastic 87% (p < 0.0001) at 48 h.p.i., whereas in GFP dsRNA-transfected cells that served as the control, the CHIKV viral genomic RNA remained similar to control infection (Fig. 4C ). This finding was surprising, as we hypothesized that a reduction in URM would increase CHIKV viral genomic RNA, as the presence of virus seems to reduce the expression of URM, as can be observed in Fig. 4B. We suspected that miR-2b may be playing a role in this phenomenon and could be involved in regulating URM during CHIKV replication. To test our hypothesis, we silenced miR-2b in the mosquitoes and then tested the expression of URM upon CHIKV challenge in these mosquitoes. We observed that although there was a reduction in URM expression upon CHIKV infection, this effect was totally reversed when miR-2b was silenced; expression of URM that was downregulated upon miR-2b silencing increased by 3.5-fold (p = 0.005) when the miRNA-silenced mosquitoes were infected with CHIKV (Fig. 4D ).

(A) Relative CHIKV viral genomic RNA during miR-2b inhibition (B) Relative expression of URM 24 and 48 h after CHIKV infection. (C) Relative CHIKV viral genomic RNA 24 and 48 h after infection in Ae. aegypti with dsRNA transfections. (D) Effect of miR-2b inhibitor on URM expression in CHIKV-infected and uninfected state. Data were expressed as mean ± SEM; ****p < 0.0001 vs. control group.
Discussion
Ae. aegypti spreads many viral diseases such as dengue and chikungunya and has more recently created global havoc with Zika transmission, causing much harm to healthcare and public health management. The most effective way of controlling these infections is through vector control. Central to vector control is the understanding of mechanisms that the vector employs to tackle the viruses it carries. Viral infection creates a competition between the vector and the virus in taking over the cellular machinery to run each of their own regime and successfully establish each of their hold. During this process, the host activates genes of several pathways to control the virus and the virus attempts to either activate the factors that help in membrane budding, which in turn will help its replication, or suppress the pathways inhibiting its replication and expansion24,25. The purpose of our present study was to identify the cellular factors that may play a role in viral replication. One approach is to identify the vector factors that may be regulated upon infection using several methodologies identifying host factors that regulate viral infection26. Another approach is to identify those regulators that regulate these factors within the cell, such as miRNAs, and then identify their targets22. These targets may further be validated for their functions to understand their mode of action during specific conditions. The second approach provides information at a more biological level, offering insights both into cellular transcripts and into their modulators such as miRNAs that could be regulating them in a more centralistic manner. Therefore, we utilized the second method in the present study.
An earlier report on Ae. aegypti had identified 86 distinct miRNAs27; our data expanded the list of known miRNAs to 124. Whereas the earlier study used Roche 454 platform for generating high-throughput data, our study utilized Ilumina sequencing, generating much more data, which could be a plausible reason for the identification of more number of miRNAs. The new miRNAs have already been described in earlier studies on Ae. albopictus 18,28, thereby proving that these are bonafide miRNAs.
Regulation of miRNAs during arboviral replication in Ae. albopictus and Ae. aegypti has been previously studied29,30. miRNAs regulated in these studies were found to be regulated in the present study as well. For instance, miR-285 was reported to be among the highest expressing miRNAs in the saliva of CHIKV-infected Ae. aegypti 30. Similarly, miR-317 and miR-2951-3p were found to be upregulated during dengue virus infection in Ae. albopictus 29,31 and miR-34-3p was shown to be involved in Wolbachia infection32. Whereas our earlier study on Ae. albopictus miRNAs highlighted miR-2b and miR-100 to be regulated upon CHIKV infection18, miR-100 has been shown to attenuate human cytomegalovirus replication via the mTOR pathway33 and have also been implicated in pathways related to cell cycle progression with targets such as plk1, while also playing a role in apoptosis34. Similarly, miR-989 has been predicted to be an important miRNA targeting insect immunity in other mosquito species as well35,36. The time points of our study coincided with the early replication events of CHIKV in the vector. Studies have shown that replication kinetics of the virus differ in the host and the vector, with the virus replicating at a much slower pace in the vector than in the host, probably allowing for persistent infection in the vector to enable efficient virus transmission throughout the life of the vector9. In addition to the several mechanisms that may be employed by the virus, host-derived factors may also play a role. Due to the significance of miRNAs in mosquito biology, we hypothesized that vector miRNAs and their targets could also be involved in regulating viral replication.
Using RNAhybrid, multiple targets for the selected miRNA were predicted, taking the seed binding region into consideration. The targets were analyzed by Gene Ontology (GO) terms and selected for further validation. Bioinformatics has been useful in identifying targets of miRNAs in the recent past37. Prediction tools identified a total of 8928 targets for all the regulated miRNAs taken together; on the basis of binding energies and relevant pathways, a total of six targets were chosen for the three miRNAs, to study whether the expression of these transcripts was affected by silencing the respective miRNAs that supposedly target them. Loss-of-function and gain-of-function analyses have been earlier employed to study the role of miRNAs in mosquitoes36,38. In the present study, out of the six transcripts, only one target, URM, showed promising results, clearly emphasizing the importance of validating miRNA targets by wet lab validations to confirm that the candidate is indeed a target of miRNA. Whereas luciferase assays provide information on binding of the miRNA to the 3′UTR of the target gene, the expression profile of the putative targets provides indirect information of the action of these miRNAs on the targets39.
In our study, we tested the effect of silencing of miRNAs on their putative targets in both in vitro conditions and mosquitoes. In the case of miR-100, of the two targets chosen (sumoligase and cdc42), cdc42 was found to be regulated in the cell line; however, we did not observe any significant regulation in the mosquito. Sumoligase expression was, however, almost negligibly regulated by the knockdown of miR-100. Cdc42 is a highly conserved small GTPase of the Rho family, acting as molecular switch in a wide range of signaling pathways such as vesicle trafficking and cell polarity40; sumoligase has been shown to play a role in immunity to virus infection41. Neuraminidase to the host cell surface is also regulated by cdc42 affecting influenza virus replication42. The miR-989-predicted target sh2/sh343 and vacuolar ATP synthase44 have been found to play a role in viral infections. But sh2/sh3 adaptor and vacuolar ATP synthase were not found to be regulated when the cells or mosquitoes were transfected or injected with the respective antagomir, negating the possibility of them being a target of miR-989. Targets of miR-2b showed the most promising results. URM and ubiquitin were selected, as both of these targets have been previously reported to be significant in viral replication and infection45,46. URMs such as ISG15 and FAT-10 have been the shown to play a role in innate immunity and viral infection47,48. In our study, we found URM to be differentially regulated upon its antagomir treatment in Ae. aegypti cell line and Ae. aegypti mosquito. The expression level of URM increased 24 h after transfection with antagomir both in the cell line and in the mosquitoes. In the case of mosquitoes, there was more than 20-fold increase in its expression after 48 h of miRNA silencing, clearly emphasizing the effect miR-2b has on the target. To further validate the target of miR-2b, we performed a luciferase reporter assay that shows significant binding to the 3′UTR of URM. Most significant in our findings was the pattern of URM expression during CHIKV infection and the manner in which it was altered when miR-2b was silenced in mosquitoes.
Based on all of our findings, we propose a model that may explain the mechanism by which CHIKV may be regulated in Ae. aegypti by miR-2b through its regulation of a host factor, namely, URM (Fig. 5). Our data reveals that miR-2b expression is upregulated during CHIKV infection in Ae. aegypti. Our data also reveals that miR-2b binds to URM and negatively regulates it. Several reports have implicated URM in tRNA thiolation49,50, which has been shown to be essential for virus replication in other systems51. When there is a decrease tRNA thiolation, there is a negative impact on viral replication and this is the probable mechanism by which Ae. aegypti may be regulating CHIKV replication. Upon CHIKV replication, Ae. aegypti increases the expression of miR-2b, which in turn decreases the expression of its target, URM, thereby reducing the overall tRNA thiolation in the cell. This, in turn, results in controlling CHIKV replication in the vector. The present study throws light on the regulation of 126 miRNAs in Ae. aegypti upon CHIKV infection. Further analysis of these miRNAs and their targets revealed that the URM transcript is an important target regulated by miR-2b controlling CHIKV replication. These findings were validated using cell lines and mosquitoes, making these results significant in studying insect immunity. The present study also provides insights into the miRNAs that could be taken up for further studies for their possible role in CHIKV replication. This study can be taken forward to investigate the possible role of URM in tRNA thiolation and its effect in CHIKV replication.

Proposed model for miR-2b affecting CHIKV replication through URM inhibition.
Methods
Cell culture and virus infection
Ae. aegypti cell line (Aag2) was maintained at 28 °C and 5% CO2 in Schneider media (Invitrogen, CA) supplemented with 10% FBS and antibiotics. The cells were infected with MOI 10 of a CHIKV isolate (accession no. JF950631.1) that was characterized during an outbreak in 201052. The cells were infected in triplicates and harvested 12 and 24 h.p.i and processed further for small RNA sequencing.
Rearing of Ae. aegypti mosquito and CHIKV infection
Ae. aegypti eggs were allowed to hatch into first instar larvae which were fed on fish food until fourth instar. Upon development, the pupae were transferred to water-filled plastic containers inside cloth cages and allowed to emerge. Upon emergence, the adult mosquitoes were reared under controlled conditions at 28 ± 2 °C, 70–75% humidity, and fed with 2% sterile glucose solution. Ae. aegypti mosquitoes were infected by blood-feeding using 500 μL of polyethylene glycol purified CHIKV mixed in 2 mL of rabbit blood. Previously starved mosquitoes were fed for 20 min at room temperature through membrane feeding. After blood feeding, fully fed mosquitoes were separated and taken for further processing for time point studies. All experiments related to mosquitoes and virus were performed in bio-containment facilities ACL-2 and BSL-3, respectively.
Sample preparation for small RNA sequencing
CHIKV-infected Aag2 cells were harvested at 12 and 24 h.p.i. and stored in Trizol (Invitrogen, CA) until RNA extraction. Small RNA population from the total RNA was enriched and extracted using miRNeasy kit (Qiagen, Germany) as per manufacturer protocol. Agilent 2100 Bioanalyzer RNA Nano 6000 kit was used to check the quantity and quality of isolated RNA. Uninfected and infected samples were outsourced for small RNA sequencing by Illumina Genome Analyzer II (Agilent, CA).
Data analysis
Data analysis was carried out using an in-house PERL-based pipeline developed for analysing small RNA data as described elsewhere18. The pipeline makes use of tools like Bowtie, RNAfold, and RNAplot for analysing the data53,54,55. Briefly, Ae. aegypti genome and coding region sequences were downloaded from VectorBase using the BioMart tool56. Pre-miRNA and mature miRNA sequences of arthropods were downloaded from the miRBase database V.1957. Other non-coding RNAs (ncRNA) sequences were fetched from the ncRNA database58. The downloaded sequences were indexed separately. After a quality check and adaptor trimming of small RNA reads, reads with length ≥18 nt were matched to mature miRNA sequences to identify known miRNAs. Tags per million (TPM) values were calculated for matched miRNAs. UTR sequences of Ae. aegypti genes were downloaded from VectorBase and targets of significant miRNAs were predicted using RNAhybrid tool59. The targets were filtered on the basis of complementarities of the miRNAs with the targets and energy of the miRNA:target, i.e, not exceeding −20 kcal/mol. Functional enrichment of the predicted targets was performed by WEGO webtool60 using GO term accession of each target gene fetched from VectorBase using the BioMart tool. The dataset analysed during the current study are available in ArrayExpress (accession number E-MTAB-5222).
Transfection in Aag2 cell lines and CHIKV infection
Antagomirs for miR-989, miR-100, and miR-2b complementary to mature miRNA sequence were synthesized from Ambion (Life Technologies, CA). In addition, scrambled miRNA provided by the manufacturer was used as the negative control in all experiments. Antagomir transfections were performed with Attractene transfection reagent (Qiagen, Germany) in Aag2 cell line as per the manufacturer instructions. Briefly, 0.3 × 106 Aag2 cells were seeded into 6-well plates to reach 75% confluency before proceeding to transfection. A volume of 100 pmol of antagomir for each miRNA, miR-2b, miR-100, and miR-989, along with the negative control was transfected and the cells were incubated at 28 °C for 4 h before changing with fresh media. The cells were collected 24, 48, and 72 h after transfection and processed for RNA isolation. Aag2 cells that were transfected with miR-2b antagomir were infected with CHIKV after 24 h and cells were collected 24 h.p.i for RNA isolation.
Antagomir injections in mosquitoes
Female mosquitoes (4–5 days old) were divided into four batches of 100 mosquitoes each. The first batch was injected with 69 nL of negative control (scrambled miRNA). The second, third, and fourth batches of mosquitoes were injected, with 69 nL of 100 µM miR-2b, miR-100, and miR-989 antagomirs, respectively. Mosquitoes were collected at two time points, 24 and 48 h, and were stored in Trizol (Invitrogen, CA) at −80 °C until RNA extraction. Time-wise knockdown of miRNA expression following injection was checked by miRNA qRT-PCR. In the case of subsequent blood feeding studies, nanoinjected mosquitoes were allowed to revive for 24 h prior to membrane feeding. Every experiment was repeated at least three times.
miRNAs, site-directed mutagenesis, and 3′UTR cloning
All the mature miRNAs were mapped against Ae. aegypti genome using Bowtie, and 250 bp flanking region from each side were extracted using in-house PERL script. miRNA miR-2b binding site 3′UTR of URM were amplified with primers containing the desired mutation for binding sites of miRNA miR-2b and cloned into the pmirGLO vector (Promega, WI) for lucifersae assay. Pre-miRNAs were PCR amplified and cloned into pmR-mCherry vector (Clontech Lab, CA) along with the 3′UTR of CHIKV which was cloned into pmirGLO vector (Promega, WI).
Luciferase reporter assay
Luciferase activities were measured with a luminometer according to manufacturer recommendation (Glomax20/20 Luminometer, Promega, WI) 24 h after transfection using the Dual-Glo luciferase reporter assay system (Promega, WI). Renilla luciferase activity was normalized using firefly luciferase activity for each sample.
dsRNA preparation for URM and mosquito injection
For dsRNA preparation, URM and green fluorescent protein (GFP, as control) were cloned into pGEMT-easy vector (Promega, WI) and were in vitro transcribed with T7 and SP6 polymerase using Promega’s Riboprobe combination kit (Promega, WI). Further dsRNAs were purified using the Trizol (Invitrogen, CA) method described earlier and stored until further use. For dsRNA-mediated silencing, 800 ng of total dsRNA was injected into a mosquito. In the case of subsequent blood-feeding studies, nanoinjected mosquitoes were allowed to recover for 24 h prior to membrane feeding. For time-series experiments, female mosquitoes were collected 24 and 48 h after feeding and stored in Trizol (Invitrogen, CA) at −80 °C until RNA extraction.
Sample collection and RNA isolation
Antagomir-injected mosquitoes were collected 24 and 48 h after injection and Aag2 cell line 24, 48, and 72 h after transfection. RNA was isolated by the Trizol (Invitrogen, CA) method and kept at −80 °C until further use. The isolated RNA was used for qPCR reaction for the miRNA and host factor-expression profiling.
Quantitative RT-PCR
Expression profiling was carried out by quantitative RT-PCR. For miRNAs, 1 µg of RNA was polyadenylated, reverse transcribed, and quantified by qRT-PCR using NCode miRNA First Strand cDNA Synthesis and qRT-PCR kit (Thermo Fisher Scientific, MA). qRT-PCR reactions were set up using 1:10 diluted cDNA as template following manufacturer instructions. For other transcripts, cDNA was used directly for quantitative RT-PCR using QuantiTect SYBR Green RT-PCR Kit (Qiagen, Germany). Experiments were conducted a minimum of two times, with each experiment set up in triplicates. For miRNA and transcript expression profiling, 5.8 s rRNA was used as an endogenous control. Expression levels were then calculated using the 2−ΔΔCT method. Details of the qRT-PCR primers used in the study are provided in Supplementary Table S1.
Statistical analysis
Differentially expressed significant miRNAs were predicted using edgeR package in R. The p-value cut-off was determined from the data, with the significance threshold selected as 0.05. Statistical analysis of experimental data was conducted using GraphPad Prism (version 5) using Student’s t-test when comparing two conditions. Data from different treatments were subjected to analysis of variance (ANOVA). For multiple comparisons, Dunnett’s test was performed. Values of p < 0.05 considered significant have been represented with an asterisk in the figures.
Data availability
Small RNA sequencing data were deposited in ArrayExpress (accession no. E-MTAB-5222).
References
Boman, H. G. Gene-encoded peptide antibiotics and the concept of innate immunity: an update review. Scandinavian journal of immunology 48, 15–25 (1998).
Hillyer, Jn. F., Schmidt, S. L. & Christensen, B. M. Rapid phagocytosis and melanization of bacteria and Plasmodium sporozoites by hemocytes of the mosquito Aedes aegypti. Journal of Parasitology 89, 62–69 (2003).
Lowenberger, C. Innate immune response of Aedes aegypti. Insect biochemistry and molecular biology 31, 219–229 (2001).
Christensen, B. M. & Forton, K. F. Hemocyte-mediated melanization of microfilariae in Aedes aegypti. The Journal of parasitology, 220–225 (1986).
Luckhart, S., Vodovotz, Y., Cui, L. & Rosenberg, R. The mosquito Anopheles stephensi limits malaria parasite development with inducible synthesis of nitric oxide. Proceedings of the National Academy of Sciences 95, 5700–5705 (1998).
Donald, C. L., Kohl, A. & Schnettler, E. New insights into control of arbovirus replication and spread by insect RNA interference pathways. Insects 3, 511–531 (2012).
Fragkoudis, R., Attarzadeh-Yazdi, G., Nash, A. A., Fazakerley, J. K. & Kohl, A. Advances in dissecting mosquito innate immune responses to arbovirus infection. Journal of General Virology 90, 2061–2072 (2009).
Weaver, S. C. & Reisen, W. K. Present and future arboviral threats. Antiviral research 85, 328–345 (2010).
Strauss, J. H. & Strauss, E. G. The alphaviruses: gene expression, replication, and evolution. Microbiological reviews 58, 491–562 (1994).
Pingen, M. et al. Host Inflammatory Response to Mosquito Bites Enhances the Severity of Arbovirus Infection. Immunity 44, 1455–1469 (2016).
Ryman, K. D. & Klimstra, W. B. Host responses to alphavirus infection. Immunological reviews 225, 27–45 (2008).
McFarlane, M. et al. Characterization of Aedes aegypti innate-immune pathways that limit Chikungunya virus replication. PLoS Negl Trop Dis 8, e2994 (2014).
Blair, C. D. & Olson, K. E. The role of RNA interference (RNAi) in arbovirus-vector interactions. Viruses 7, 820–843 (2015).
Campbell, C. L., Black, W. C., Hess, A. M. & Foy, B. D. Comparative genomics of small RNA regulatory pathway components in vector mosquitoes. Bmc Genomics 9, 1 (2008).
Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. cell 116, 281–297 (2004).
Chawla, G. & Sokol, N. S. MicroRNAs in Drosophila development. Int Rev Cell Mol Biol 286, 1–65 (2011).
Pfeffer, S. & Baumert, T. F. Impact of microRNAs for pathogenesis and treatment of hepatitis C virus infection. Gastroentérologie clinique et biologique 34, 431–435 (2010).
Shrinet, J., Jain, S., Jain, J., Bhatnagar, R. K. & Sunil, S. Next generation sequencing reveals regulation of distinct Aedes microRNAs during chikungunya virus development. PLoS Negl Trop Dis 8, e2616 (2014).
Qian, K. et al. Identification and validation of human papillomavirus encoded microRNAs. PLoS One 8, e70202 (2013).
Saldaña, M. A. et al. Zika virus alters the microRNA expression profile and elicits an RNAi response in Aedes aegypti mosquitoes. PLoS neglected tropical diseases 11, e0005760 (2017).
Su, Y. et al. Small RNA sequencing reveals a role for sugarcane miRNAs and their targets in response to Sporisorium scitamineum infection. BMC genomics 18, 325 (2017).
Hausser, J. & Zavolan, M. Identification and consequences of miRNA-target interactions [mdash] beyond repression of gene expression. Nature Reviews Genetics 15, 599–612 (2014).
Wang, Y., Luo, J., Zhang, H. & Lu, J. microRNAs in the same clusters evolve to coordinately regulate functionally related genes. Molecular biology and evolution 33, 2232–2247 (2016).
Martin-Serrano, J. & Neil, S. J. D. Host factors involved in retroviral budding and release. Nature Reviews Microbiology 9, 519–531 (2011).
Sundquist, W. I. & Kräusslich, H.-G. HIV-1 assembly, budding, and maturation. Cold Spring Harbor perspectives in medicine 2, a006924 (2012).
Lenarcic, E. M., Landry, D. M., Greco, T. M., Cristea, I. M. & Thompson, S. R. Thiouracil cross-linking mass spectrometry: a cell-based method to identify host factors involved in viral amplification. Journal of Virology 87, 8697–8712 (2013).
Li, S., Mead, E. A., Liang, S. & Tu, Z. Direct sequencing and expression analysis of a large number of miRNAs in Aedes aegypti and a multi-species survey of novel mosquito miRNAs. Bmc Genomics 10, 1 (2009).
Skalsky, R. L., Vanlandingham, D. L., Scholle, F., Higgs, S. & Cullen, B. R. Identification of microRNAs expressed in two mosquito vectors, Aedes albopictus and Culex quinquefasciatus. Bmc Genomics 11, 1 (2010).
Liu, Y. et al. The expression profile of Aedes albopictus miRNAs is altered by dengue virus serotype-2 infection. Cell & bioscience 5, 16 (2015).
Maharaj, P. D., Widen, S. G., Huang, J., Wood, T. G. & Thangamani, S. Discovery of mosquito saliva microRNAs during CHIKV infection. PLoS neglected tropical diseases 9, e0003386 (2015).
Su, J. et al. Identification of microRNAs expressed in the midgut of Aedes albopictus during dengue infection. Parasites & vectors 10, 63 (2017).
Mayoral, J. G., Etebari, K., Hussain, M., Khromykh, A. A. & Asgari, S. Wolbachia infection modifies the profile, shuttling and structure of microRNAs in a mosquito cell line. PLoS One 9, e96107 (2014).
Wang, F.-Z. et al. Human cytomegalovirus infection alters the expression of cellular microRNA species that affect its replication. Journal of Virology 82, 9065–9074 (2008).
Studach, L. L. et al. Polo−like kinase 1 inhibition suppresses hepatitis B virus X protein-induced transformation in an in vitro model of liver cancer progression. Hepatology 50, 414–423 (2009).
Hussain, M., Frentiu, F. D., Moreira, L. A., O’Neill, S. L. & Asgari, S. Wolbachia uses host microRNAs to manipulate host gene expression and facilitate colonization of the dengue vector Aedes aegypti. Proceedings of the National Academy of Sciences 108, 9250–9255 (2011).
Jain, S., Rana, V., Tridibes, A., Sunil, S. & Bhatnagar, R. K. Dynamic expression of miRNAs across immature and adult stages of the malaria mosquito Anopheles stephensi. Parasites & vectors 8, 1 (2015).
Akhtar, M. M., Micolucci, L., Islam, M. S., Olivieri, F. & Procopio, A. D. Bioinformatic tools for microRNA dissection. Nucleic acids research 44, 24–44 (2016).
Puthiyakunnon, S. et al. Functional characterization of three MicroRNAs of the Asian tiger mosquito, Aedes albopictus. Parasites & vectors 6, 1 (2013).
Maudet, C. et al. Functional high-throughput screening identifies the miR-15 microRNA family as cellular restriction factors for Salmonella infection. Nature communications 5 (2014).
Harris, K. P. & Tepass, U. Cdc42 and vesicle trafficking in polarized cells. Traffic 11, 1272–1279 (2010).
Brown, J. R. et al. SUMO Ligase Protein Inhibitor of Activated STAT1 (PIAS1) Is a Constituent Promyelocytic Leukemia Nuclear Body Protein That Contributes to the Intrinsic Antiviral Immune Response to Herpes Simplex Virus 1. Journal of Virology 90, 5939–5952 (2016).
Wang, S. et al. Transport of influenza virus neuraminidase (NA) to host cell surface is regulated by ARHGAP21 and Cdc42 proteins. Journal of Biological Chemistry 287, 9804–9816 (2012).
Tan, S.-L. et al. NS5A, a nonstructural protein of hepatitis Cvirus, binds growth factor receptor-bound protein 2 adaptor protein in a Src homology 3 domain/ligand-dependent manner and perturbs mitogenic signaling. Proceedings of the National Academy of Sciences 96, 5533–5538 (1999).
Fongsaran, C. et al. Involvement of ATP synthase β subunit in chikungunya virus entry into insect cells. Archives of virology 159, 3353–3364 (2014).
Rajsbaum, R. & Garcia-Sastre, A. Unanchored ubiquitin in virus uncoating. Science 346, 427–428 (2014).
Randow, F. & Lehner, P. J. Viral avoidance and exploitation of the ubiquitin system. Nature cell biology 11, 527–534 (2009).
Morales, D. J. et al. Novel mode of ISG15-mediated protection against influenza A virus and Sendai virus in mice. Journal of Virology 89, 337–349 (2015).
Nguyen, N. T. H., Kim, W.-J., Kim, N. & Yoo, J.-Y. Ubiquitin-like modifier FAT10 attenuates RIG-I mediated antiviral signaling by segregating activated RIG-I from its signaling platform. Scientific reports 6 (2016).
Maynard, N. D., Macklin, D. N., Kirkegaard, K. & Covert, M. W. Competing pathways control host resistance to virus via tRNA modification and programmed ribosomal frameshifting. Molecular systems biology 8, 567 (2012).
Isaacson, M. K. & Ploegh, H. L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell host & microbe 5, 559–570 (2009).
Albers, S. & Czech, A. Exploiting tRNAs to Boost Virulence. Life 6, 4 (2016).
Shrinet, J. et al. Genetic characterization of Chikungunya virus from New Delhi reveal emergence of a new molecular signature in Indian isolates. Virology journal 9, 1 (2012).
Hofacker, I. L. et al. Fast folding and comparison of RNA secondary structures. Monatshefte für Chemie/Chemical Monthly 125, 167–188 (1994).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10, 1 (2009).
Lorenz, R. et al. ViennaRNA Package 2.0. Algorithms for Molecular Biology 6, 1 (2011).
Giraldo-Calderon, G. I. et al. VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic acids research, gku1117 (2015).
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic acids research 42, D68–D73 (2014).
He, S. et al. NONCODEv2. 0: decoding the non-coding. Nucleic acids research 36, D170–D172 (2008).
Rehmsmeier, M., Steffen, P., Höchsmann, M. & Giegerich, R. Fast and effective prediction of microRNA/target duplexes. Rna 10, 1507–1517 (2004).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucleic acids research 34, W293–W297 (2006).
Acknowledgements
Small RNA libraries were sequenced at NxGenBio Life Sciences, New Delhi. We acknowledge Mr. Dinesh Singh for his assistance with mosquito experiments.
Author information
Authors and Affiliations
Contributions
S.S. designed the research. S.K.D., J.S. and J.J. performed the research and analyzed the data. S.K.D., J.S. and S.S. wrote the paper. The research work was supervised by S.A. and S.S. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dubey, S.K., Shrinet, J., Jain, J. et al. Aedes aegypti microRNA miR-2b regulates ubiquitin-related modifier to control chikungunya virus replication. Sci Rep 7, 17666 (2017). https://doi.org/10.1038/s41598-017-18043-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18043-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
