
Abstract
Peatlands are a major source of methylmercury that contaminates downstream aquatic food webs. The large store of mercury (Hg) in peatlands could be a source of Hg for over a century even if deposition is dramatically reduced. However, the reliability of Hg mass balances can be questioned due to missing long-term land-atmosphere flux measurements. We used a novel micrometeorological system for continuous measurement of Hg peatland-atmosphere exchange to derive the first annual Hg budget for a peatland. The evasion of Hg (9.4 µg m−2 yr−1) over the course of a year was seven times greater than stream Hg export, and over two times greater than wet bulk deposition to the boreal peatland. Measurements of dissolved gaseous Hg in the peat pore water also indicate Hg evasion. The net efflux may result from recent declines in atmospheric Hg concentrations that have turned the peatland from a net sink into a source of atmospheric Hg. This net Hg loss suggests that open boreal peatlands and downstream ecosystems can recover more rapidly from past atmospheric Hg deposition than previously assumed. This has important implications for future levels of methylmercury in boreal freshwater fish and the estimation of historical Hg accumulation rates from peat profiles.
Similar content being viewed by others

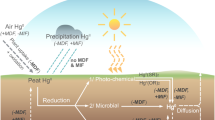
Introduction
Global anthropogenic mercury (Hg) emissions have increased the amount of Hg cycling between vegetation, surface soil, ocean, and the atmosphere by about a factor of three compared to pre-industrial levels1. Gaseous elemental mercury (GEM) has an atmospheric lifetime of 0.8–1.7 years after emission, resulting in long range transport before deposition on the earth surface2. This poses a threat to humans and wildlife even in remote areas3. Current global models assume that Hg deposition to non-contaminated terrestrial surfaces and subsequent re-emission are similar in magnitude. However, large uncertainty remains mostly due to a strong spatial and temporal bias in direct flux measurements towards Hg-enriched sites, and/or short-term, daytime and summertime measurements4,5.
Peatlands play an important role in Hg cycling because they constitute a major source of methylmercury to adjacent streams and lakes6. Methylmercury is the Hg compound most prone to bioaccumulation in aquatic food webs3. The large amount of Hg in peatlands relative to wet bulk deposition and export in surface water runoff indicates that they have accumulated Hg from past atmospheric deposition7. In high latitude regions almost all freshwater fish have Hg concentrations exceeding European limits for good ecological status (0.02 mg Hg kg−1 fish muscle8,9). To predict future Hg levels in fish of these regions, the annual net ecosystem flux of Hg from peatlands needs to be quantified.
Since the store of Hg in peat is so large compared to runoff fluxes, it is generally assumed that it will take several decades, if not centuries, before a reduction in atmospheric deposition could lead to any reduction in the Hg pools of peatlands, thus putting the prospect of substantial recovery far into the future7,10. Such calculations, however, neglect GEM emission to the atmosphere which preliminary measurements indicated are substantial11,12,13.
Earlier research has found that below a certain threshold of atmospheric GEM concentration, terrestrial surfaces switch from being net sinks to net sources of Hg5,14,15,16,17,18,19,20,21. The exact value of this threshold will vary, though, depending on the nature of the surfaces involved, ambient air characteristics and other physical and chemical conditions22,23,24 or atmospheric turbulence25,26. Specific studies have shown that at ambient atmospheric GEM concentrations (~1.5 ng m−3), vegetated terrestrial surfaces are net sinks of atmospheric GEM which is incorporated into leaf tissue in forests18, grasslands27, peatlands28, or the arctic tundra29. However, Hg land-atmosphere exchange at a peatland scale not only represents uptake of Hg by vegetation but also re-emission of GEM induced by photochemical processes at the peat surface and non-photochemical abiotic reduction by natural organic matter30,31,32,33. Agnan et al.5 reported that over barren, litter-covered and vegetated soils, the GEM flux, on average, switches from net deposition to net emission below an atmospheric GEM concentration of 2.75 ng m−3.
We hypothesize that recent declines in atmospheric GEM concentrations have fallen below the threshold at which some peatlands switch from being a net sink of atmospheric Hg to being a source of Hg to the atmosphere due to increasing emission of GEM from the peatland surface. If this proves to be the case, the loading of Hg from such peatlands to aquatic food webs would be reduced substantially faster than previously anticipated. To test this hypothesis, we established the first full year, Hg mass balance for a boreal peatland. The key to achieving this was continuous measurement of land-atmosphere GEM exchange. Together with data on export of total mercury (THg) in stream runoff, wet bulk deposition, and peat Hg storage, these measurements allowed us to estimate the major components of the annual Hg mass balance for a boreal peatland.
Results and Discussion
The most challenging component of a catchment scale Hg mass balance is the long-term continuous measurement of land-atmosphere exchange of GEM. We used a novel, dual-inlet, single detector relaxed eddy accumulation (REA) system to quantify GEM fluxes13. REA is a direct measurement approach that overcomes uncertainties associated with other micrometeorological techniques (aerodynamic gradient and modified Bowen-ratio methods) that rely on concentration measurements at different heights. Compared to other GEM-REA systems, the advanced REA design used in this study is fully automated and simultaneously collects GEM emission from, and deposition to, the ground surface13.
The system was deployed in the center of an open area of the nutrient poor, minerogenic Degerö Stormyr peatland [64°11′N, 19°33′E] located near Vindeln in the county of Västerbotten, Sweden (see Supplementary Fig. S1). The study area is representative of one of the dominant mire ecosystems in the boreal region. Our study was conducted within the mire complex that is drained by the Vargstugbäcken stream. The catchment covers an area of 2.7 km2 of which 70% is peatland34. The REA sampling inlets were mounted 3.5 m above the peatland surface, with a ca. 2.5 ha footprint (see Supplementary Fig. S2). Continuous measurements integrating exchange over such a large area overcome much of the measurement uncertainty that would otherwise be introduced with small-scale dynamic flux chambers. Chambers cover only small fractions of the peatland surface and are normally only deployed during short time periods, thus introducing large spatial and temporal uncertainties5,35.
We found that the annual Hg mass balance at the Degerö Stormyr peatland was clearly dominated by net GEM evasion due to substantial net emission between May and October (Fig. 1a). The annual GEM emission was 9.4 µg m−2 and the wet bulk deposition was 3.9 µg m−2. GEM is the predominant atmospheric form of Hg and can contribute substantially to net Hg deposition over vegetated surfaces28,29. Multi-year gaseous oxidized Hg (GOM) and particulate bound Hg (PBM) concentrations measured at northern European sites in southern Scotland36 and the western coast of Sweden37 revealed low average levels for GOM < 1 pg m−3 and PBM < 4 pg m−3. We thus expect that dry deposition of GOM and PBM has only a marginal effect on the Hg mass balance for the Degerö Stormyr peatland. This assumption is confirmed by Enrico et al.28 who found that dry deposition of GOM and PBM contributed less than 1% to the total annual deposition flux to the Pinet peat bog in the French Pyrenees. This was examined using stable Hg isotope signatures. This clearly indicates that GEM dry deposition and Hg wet deposition are the dominant deposition pathways. These conclusions are valid even when considering the findings by Gustin et al.38 that demonstrated that GOM concentrations monitored with the instruments used in the studies above (refs36,37) are generally underestimated by a factor of 1.6 to 12 depending on the composition of GOM. Even if being ~10 times higher the contribution from GOM will be minor.

Year-round measurement of gaseous elemental Hg (GEM) concentrations in ambient air, GEM peat-atmosphere flux, water table levels, snow cover, dissolved gaseous Hg (DGM) concentrations in porewater and its supersaturation with respect to atmospheric GEM concentrations at the Degerö Stormyr peatland. (a) Monthly average of the net gaseous elemental Hg (GEM) flux, atmospheric GEM concentration, snow depth and water table level (WTL). Negative GEM fluxes represent GEM dry deposition to the peatland, while positive fluxes represent GEM volatilization. Empty symbols represent GEM flux measurements over snowpack measured in March 2014. The figures at the top of the panel represent total numbers of 60-min averages of both GEM flux and atmospheric GEM concentration per month. (b) Monthly averages and standard errors of the dissolved gaseous Hg (DGM) concentration measured on a weekly basis between May and October. For comparison, the degree of DGM supersaturation of the peat pore water and the GEM flux is indicated. Number of observations for bulk DGM is displayed.
Annual discharge export of Hg from the catchment area amounted to 1.3 µg m−2 between July 2013 and June 2014 (Fig. 2). The average annual THg export in runoff during the four previous years 2009–2012 was 1.7 µg m−2 yr−1, ranging between a low of 1.3 µg m−2 yr−1 in 2010 to a high of 2.3 µg m−2 yr−1 in 2012. The THg export in discharge has commonly been thought of as the dominant output pathway for most boreal catchments10,39. At the study site, however, the peatland-atmosphere evasion was seven times larger than stream discharge of Hg. The stream discharge originates from within the topographically-defined catchment because it is the local high point with no regional groundwater sources contributing to the catchment water balance34. This is supported by a previous water balance assessment indicating a low potential for other hydrological sources coming from outside the delineated catchment area40. The potential contribution of THg from the mineral soils (30% of total catchment area) to the calculated fluxes is also considered low since dissolved organic carbon (DOC), which is strongly associated with dissolved THg, originates almost entirely from the peatland area of the catchment41. This suggestion is supported by: 1) δ13C values of DOC in the peat profile near the origin of the outlet stream that are extremely enriched (−5%) compared to measurements in the mineral soils41 (−25%) and 2) measurements of the DOC in the peat profile near the stream initiation point for Vargstugbäcken (40.8 ± 12 mg C L−1)41 compared to the low DOC (~2 mg C L−1) in the groundwater from forested podzol soils entering boreal peatlands similar to Degerö Stormyr42.

Annual Hg mass balance at the Degerö Stormyr peatland (June 18, 2013 – June 17, 2014). Net emission of gaseous elemental Hg (GEM) and total Hg (THg) in discharge are the major Hg output pathways. Wet bulk deposition is considered as the only Hg input pathway. The units for the flux represented by each arrow are in µg m−2 yr−1. Uncertainties for GEM flux, total Hg inputs in wet bulk deposition and outputs in stream discharge are indicated (see section “Methods” for details).
Observed GEM emission during the growing season was 11 µg m−2 and substantially exceeded Hg wet bulk deposition during the same time period (2.5 µg m−2). During the growing season the peatland emitted GEM to the atmosphere at an average rate ± standard error of the mean (SE) of 3 ± 0.5 ng m−2 h−1 (Fig. 3a). Outside the growing season (October, November and April) the net GEM flux was close to zero (−0.1 µg m−2) with emission dominating from 9 AM to 8 PM and deposition during the night (Fig. 3b). Measurements during March revealed net GEM deposition of 0.4 µg m−2 month−1 from air to the snow covered surface. Winter time GEM exchange also showed a diurnal pattern indicating photochemical GEM production at the snow surface and subsequent emission of previously deposited GEM in the afternoon (Fig. 3c).

Diel patterns of gaseous elemental Hg (GEM) exchange between the peatland surface and the atmosphere. Flux data were smoothed with a 6-point moving average. The red bars (average ± SE) indicate emission and the green bars dry deposition of gaseous elemental Hg (GEM) during (a) the growing season (May-September) (b) non-growing season (October, November, and April) and (c) snow covered winter time period (represented by data from March). The black dots show the average atmospheric GEM concentrations. Vertical orange dashed lines separate between day (Rg ≥ 5 Wm−2) and night (Rg < 5 Wm−2). Rg is the hourly average incoming solar radiation. The number of observations of GEM flux and atmospheric GEM concentration are given.
Our data clearly demonstrate the importance of continuous, long-term measurements to derive complete, unbiased annual budgets of the land-atmosphere GEM exchange. There are several factors which corroborate the overall rates of evasion and temporal variation. The largest evasion rates during the summer correspond to peak pore water concentrations of dissolved gaseous mercury (DGM) (Fig. 1b). Field studies over water surfaces have identified photochemically induced Hg2+ reduction43,44 and dark abiotic reduction32,45 as main processes controlling the concentration of DGM in the superficial peat porewater. This suggests the importance of peat porewater DGM concentrations as a driving factor for the net annual peatland-atmosphere exchange of GEM. The near-surface mire porewater was supersaturated in DGM relative to atmospheric GEM concentrations on 86% of the measurement occasions between May and October, peaking in July and August (Fig. 1b).
These findings indicate that the overall mass transfer of GEM from the water-saturated peat layers to the surface is less limited by diffusion than anticipated23,46,47. Analysis of GEM concentrations in dry, mineral soils, in contrast, revealed consistent GEM uptake and downward redistribution into the soil48 which is indicated by lower soil GEM levels compared to atmospheric GEM concentrations.
Summertime evasion also corresponds to the highest levels of incident radiation that can drive photoreduction of Hg2+ to Hg0 at the peat surface. Surface chamber measurements over boreal peatlands have demonstrated diel patterns with daytime GEM evasion and nighttime GEM deposition, indicating photochemical reduction at the surface and subsequent GEM evasion49. The gross ecosystem production of vascular plants at the measurement site peaks during late June throughout July50. The concurrent maximum plant uptake of GEM in July is likely reflected by about 30% lower GEM net emission rates as compared to June and August.
The unexpectedly high annual net evasion may also be consistent with the pattern of superficial peat Hg concentration-depth profiles. The average ± SE concentration of Hg within the top 34 cm was 57 ± 8 ng g−1 dry matter (DM). That is within the range of 10–115 ng g−1 DM representing typical soil THg concentrations from boreal forest catchments51. Peat THg concentrations increased with depth to a peak of about 110 ng g−1 dated to ca 1950 (Fig. 4). This pattern is similar to that of other Hg concentration profiles from open peatlands in Canada52, Scotland53 and Sweden54. This might indicate that the decrease in THg concentration towards the surface (24 ng THg g−1 in the top 10 cm) not only represents the degree of peat decomposition and lower Hg deposition rates but also increasing evasion of Hg to the atmosphere after ca 1985. This assumes that the halving of atmospheric GEM concentrations in Europe between ca 1980 and 20005,55 increased evasion of Hg from the peatland surface enough to switch the peatland from being a net sink for Hg to a net source to the surrounding environment, primarily by evasion to the atmosphere.

Average depth profile of total Hg (THg) at Degerö Stormyr. THg concentrations from peat cores with 95% confidence interval (n = 8). The zero level denotes the surface of living Sphagnum moss. The red area indicates the amount of legacy Hg stored in the uppermost peat when allowing for a “background” concentration of 20 ng g−1.
Since 1990 atmospheric GEM concentration in Western Europe has been decreasing at a rate of 2.2 ± 0.15% yr−1 reflecting emission reductions in these regions56. The novel Hg flux database presented in Agnan et al.5 clearly showed that the air concentration of GEM was the only variable that correlated to land-atmosphere exchange at sites with background soil Hg concentrations (r = −0.312, p < 0.001, n = 263). However, even if significant, the negative correlation to air GEM concentrations only explained 11% of the variation in GEM fluxes in this set of data from around the globe. This indicates the importance of local conditions (e.g. different landscape, surface properties and micrometeorology) for the strength and even direction of the GEM flux.
Recent advances in the interpretation of Hg isotope signatures have also revealed high mobility of Hg in peat soils. Jiskra et al.33 deduced substantial re-emission upon non-photochemical reduction by natural organic matter and estimated that 30% of the historically deposited Hg on their study site has already been re-emitted to the atmosphere. They calculated a peat Hg re-emission flux of 5 µg m−2 yr−1 that is about 50% of the net GEM flux measured with REA in this study.
The vertical distribution of Hg in surface peat might however also result from decay of organic matter and cannot be interpreted only as a result of changes in Hg deposition and GEM evasion57. The surface peat decay rate constant at Degerö Stormyr is 0.0158 yr−1 58 resulting in a vertical THg profile similar to the ones observed. However, our results indicate that besides changes in peat mass production and decay processes, GEM evasion has the potential to alter the signal in the vertical peat profile from atmospheric Hg deposition. Potential gaseous losses of Hg and other elements (selenium, sulphur and arsenic) must be carefully considered when interpreting historical and present accumulation rates59. As stated by Biester et al.60 and Jiskra et al.33, GEM emission provides an alternative explanation for discrepancies between peat and lake sediment archives in the ratio of Hg accumulation rates from preindustrial to modern times61.
Accumulation rates of Hg in peat during the industrial period until ca. 1990 are estimated to have been up to 15 times greater than those during the pre-industrial era for northern Europe62. Between 1990 and 2011, Hg emissions from European sources decreased by ~60% due to both abatement strategies and the closing of many coal-fired power plants, chlor-alkali plants, etc.63. This was associated with roughly a halving of global atmospheric GEM concentrations55. In Sweden atmospheric GEM went down from more than 3 ng m−3 in the 1980s to northern hemispheric background concentrations of 1.3–1.9 ng m−3 after 199564,65.
We hypothesize that this reduction in deposition of atmospheric Hg to terrestrial ecosystems has impacted the bi-directional land-atmosphere exchange of GEM in remote areas. More specifically at Degerö Stormyr, where the current levels of wet bulk Hg deposition are low relative to our observed evasion, the emission of legacy Hg suggests that the peat soil pool is the dominant source for Hg loss from open boreal peatlands. If this situation with high re-emission of GEM is sustained, it will lead to a much faster than anticipated reduction in the pool of Hg from the Degerö Stormyr peatland, and eventually a reduction in the supply of Hg to downstream aquatic food webs.
To make an initial estimate of the time it would take to reduce the supply of Hg to downstream freshwaters, we assumed that the observed levels of evasion will remain constant while no considerable change occurs in either the wet bulk deposition, discharge or the growing season water balance66. With a further assumption that catchment runoff is dominated by Hg from the uppermost peat34,41, it would take around 90 years to deplete the entire pool of legacy Hg in the uppermost 34 cm to a background concentration level of 20 ng THg g−1 (DM) in the peat. Exchange patterns of GEM might vary from year-to-year at the same location and between nearby sites due to changes in meteorological conditions, atmospheric Hg deposition, or plant composition and growth. However, based on our knowledge of carbon cycling, the inter-annual variability of CO2 and CH4 exchange is limited. The 12-year average ± standard deviation (SD) net ecosystem exchange at Degerö Stormyr was −58 ± 21 g C m−2 yr−1 67 and the average ± SD (2003–2014) total stream carbon export for a 12-year period was 12.2 ± 3.4 g C m−2 yr−1 34. Most of the biogeochemical and biogeophysical processes that drive the transformation and transport of the various carbon species are suggested to constitute the major drivers of Hg transformations and transport39.We conclude that even with inter-annual and between mire variations, the GEM emission to the atmosphere may constitute a major loss term in the Hg balance of boreal peatlands. This will lead to decreased Hg contamination of downstream aquatic ecosystems in the coming decades, provided that atmospheric Hg levels do not increase. Evasion from peatlands associated with lowered atmospheric GEM levels would thereby attenuate some of the negative effects of past Hg deposition on ecosystems and human health.
The proven reliability of the novel REA system for long-term measurements of land atmosphere GEM exchange13 creates possibilities for similar long-term measurements at other peatlands as well as over other ecosystems, including forests and oceans. If the reversal of the net flux of Hg from the atmosphere into peatlands is occurring at a global scale, and possibly even for other ecosystems, then it will increase the societal value of the ambitious goals set by regulatory frameworks such as the UN Minamata Convention for reducing anthropogenic Hg emissions to the atmosphere68.
Methods
Research site
The annual Hg mass balance was estimated for the period June 18, 2013 – June17, 2014, for the nutrient-poor, boreal mire, Degerö Stormyr (64°11′N, 19°33′E, altitude 270 m above sea level), Västerbotten, Sweden (see Supplementary Fig. S1). The study focused on an open peatland area of 1.9 km2 with an average peat depth of 3–4 m (max. 8 m). The GEM flux source area is characterized by uniform microtopography and vegetation, dominated by lawn and carpet plant communities (see Supplementary Fig. S2). The climate is cold and humid with persistent snow cover during ~6 months. The 30-year (1981–2010) mean annual temperature is 1.8 °C and the mean precipitation is 614 mm, of which about 35% falls as snow69.
Peat sampling and Hg analysis
In autumn 2015 two replicate peat profiles were taken in every cardinal direction from the REA tower (N = 8, see Supplementary Fig. S2). The 34 cm long profiles (100 cm2 cross section) were cut in 2 cm increments, freeze-dried for 5 days and then weighed for the determination of their dry mass and bulk density (see Supplementary Tab. S1 and Fig. S3). The THg in peat samples was analyzed using a SMS100 (Perkin Elmer, Waltham, USA) through thermal decomposition atomic absorption spectrometry according to EPA method 7473. Certified reference lake sediment (IAEA SL1 [130 ng g−1]) and pine needle material (PINE1575a [40 ng g−1]) were used for calibration. Replicate samples and the reference material were analyzed regularly (10% of the sampling sequence). The precision was ≤5% relative to the standard deviation.
Hg in wet bulk deposition
Atmospheric wet bulk deposition was sampled continuously at the EMEP station Bredkälen (63°51′N, 15°20′E; 210 km west of Degerö Stormyr) using IVL wet bulk samplers. Cumulative wet bulk deposition was derived from these THg concentrations and the precipitation at the reference climate station Kulbäcksliden, located 1 km east of Degerö Stormyr (see Supplementary Table S2).
Peatland-atmosphere exchange of gaseous elemental Hg
The dual-inlet, single detector REA system was set up in the center of the peatland (15 m south of a fully equipped EC tower70). Sampling inlets were mounted 3.5 m above the ground. For a more detailed description see Supplementary Fig. S4 and in Osterwalder et al.13). The REA sampling interval was 30 min and the data coverage during the nine months of measurements was 33%. Between December 2013 and February 2014 no measurements were performed. Monthly averages of GEM flux were computed based on an annual total of 4,075 60-min values. The numbers of hourly GEM flux observations were well balanced over 24 hours (see Fig. 3). To determine the net annual GEM flux we calculated averages for each of the 24 hours during the day for each month. This yielded the diurnal pattern for that month. The sum of this diurnal curve is then multiplied by the number of days for the integrating time period. The annual sum of GEM fluxes is then derived by cumulated monthly averages. The GEM flux source area covered 2.5 ha of homogenous wet lawns and carpet plant communities during snow-free conditions. The Hg detector and the two gold-cartridge pairs were calibrated every second month by injection of different volumes of Hg saturated air from a temperature controlled Hg vapor calibration unit (Tekran Model 2505, Toronto, Canada). GEM recovery was monitored automatically using the GEM reference gas and Hg zero-air generator. The system was regularly set into a reference mode (2s-simulated wind signal) to check for sampling line bias and to investigate the precision of concentration difference measurements. The detection limits were derived from the absolute standard deviation of the residuals from orthogonal linear regression fitting71. Residuals (N = 960) did not show any trend with time or GEM concentration. The detection limit (1σ) was 0.05 and 0.04 ng m−3 for gold cartridge Pair 1 and Pair 2, respectively. In total 53% and 52% of the measured GEM differences were above these limits. Please note that the average flux values reported in the main text include the measured data below the detection limit (average exchange rates would otherwise be overestimated).
Hg in catchment discharge
Continuous discharge was measured in a heated flume at the catchment outlet averaged to hourly values. Stream water was sampled twelve times (monthly) for THg during the measurement period. The THg analyses were performed at the Stockholm University Department of Applied Environmental Science following the US EPA standard method 1631 (US EPA, 2002) using a 10.035 Millennium Merlin 1631 CV-AFS (PSA, Orpington, UK). Annual THg export in discharge was calculated by multiplying the interpolated daily THg concentration with daily discharge quantity (see Supplementary Table S3 and Fig. S5).
Dissolved gaseous Hg and other environmental parameters
Dissolved gaseous elemental Hg concentration in the pore water was measured weekly during June – October 2013 and May–June 2014 using a Tekran Automated Purging System developed and described in Lindberg et al.72. Instruments to determine meteorological parameters were mounted at 2 m height above the mire surface on the same tower as the eddy covariance system. Air temperature and humidity were determined by an MP100 temperature and moisture sensor (Rotronic AG, Bassersdorf, Switzerland) equipped with a ventilated shield. Global radiation was measured using a Li200sz sensor (LI-COR, Lincoln, USA). Water table levels were measured in a lawn plant community using a float and counterweight system attached to a potentiometer73. The snow depth was measured by a Sr-50 ultrasonic sensor (Campbell Scientific Logan, USA) mounted on the REA tower.
Uncertainty estimates for the peatland Hg mass balance
The uncertainty of the annual land-atmosphere GEM flux was calculated by error propagation of the hourly averages for each day of every month. The monthly errors were then propagated to get an estimated uncertainty of the annual GEM flux which was ±6%. The uncertainty associated with the precipitation inputs was ±20%. This is a conservative estimate since annual deposition values are composed of 26 biweekly measurements where the uncertainty of each Hg analysis is ±7% for concentrations >0.25 ng/L according to the analytical laboratory performing these analyses (IVL Swedish Environmental Research Institute). The uncertainty of the Hg output via catchment discharge was ±12%. That value was estimated using a propagation of error calculation that includes the error in discharge measurements and the error in the THg concentration measurements in runoff (±7%). The nine year inter-annual variation in discharge and total carbon export (sum of DOC, DIC, CO2 and CH4) are very similar. For discharge the standard errors were ±10% and for total C export ±9%, using the same method applied in this paper to estimate the uncertainty in the export of Hg in discharge34. The uncertainty of the peatland Hg budget is ±24%.
References
Driscoll, C. T., Mason, R. P., Chan, H. M., Jacob, D. J. & Pirrone, N. Mercury as a global pollutant: Sources, pathways, and effects. Environ. Sci. Technol. 47, 4967–4983 (2013).
Ariya, P. A. et al. Mercury physicochemical and biogeochemical transformation in the atmosphere and at atmospheric interfaces: A review and future directions. Chem. Rev. 115, 3760–3802 (2015).
United Nations Environment Programme. Global Mercury Assessment 2013: Sources, emissions, releases and environmental transport, UNEP Chemicals Branch, Geneva, Switzerland, 42 pp. (2013).
Amos, H. M., Jacob, D. J., Streets, D. G. & Sunderland, E. M. Legacy impacts of all-time anthropogenic emissions on the global mercury cycle. Glob. Biogeochem. Cycle 27, 410–421 (2013).
Agnan, Y., Le Dantec, T., Moore, C. W., Edwards, G. C. & Obrist, D. New constraints on terrestrial surface atmosphere fluxes of gaseous elemental mercury using a global database. Environ. Sci. Technol. 50, 507–524 (2016).
St. Louis, V. L. et al. Importance of wetlands as sources of methylmercury to boreal forest ecosystems. Can. J. Fish Aquat. Sci. 51, 1065–1076 (1994).
Grigal, D. F. Mercury sequestration in forests and peatlands: A review. J. Environ. Qual. 32, 393–405 (2003).
Chalmers, A. T. et al. Mercury trends in fish from rivers and lakes in the United States, 1969–2005. Environ. Monit. Assess. 175, 175–191 (2011).
Åkerblom, S., Bignert, A., Meili, M., Sonesten, L. & Sundbom, M. Half a century of changing mercury levels in Swedish freshwater fish. Ambio 43, 91–103 (2014).
Meili, M. et al. Critical levels of atmospheric pollution: criteria and concepts for operational modelling of mercury in forest and lake ecosystems. Sci Total Environ. 304, 83–106 (2003).
Selvendiran, P., Driscoll, C. T., Montesdeoca, M. R. & Bushey, J. T. Inputs, storage, and transport of total and methyl mercury in two temperate forest wetlands. J. Geophys. Res. 113, G00C01, https://doi.org/10.1029/2008JG000739 (2008).
Converse, A. D., Riscassi, A. L. & Scanlon, T. M. Seasonal variability in gaseous mercury fluxes measured in a high-elevation meadow. Atmos. Environ. 44, 2176–2185 (2010).
Osterwalder, S. et al. A dual-inlet, single detector relaxed eddy accumulation system for long-term measurement of mercury flux. Atmos. Meas. Tech. 9, 509–524 (2016).
Hanson, P. J., Lindberg, S. E., Tabberer, T. A., Owens, J. G. & Kim, K.-H. Foliar exchange of mercury vapor: evidence for a compensation point. Water, Air, Soil Pollut. 80, 373–382 (1995).
Ericksen, J. A. & Gustin, M. S. Foliar exchange of mercury as a function of soil and air mercury concentrations. Sci Total Environ. 324, 271–279 (2004).
Graydon, J. A. et al. Investigation of mercury exchange between forest canopy vegetation and the atmosphere using a new dynamic chamber. Environ. Sci.Technol. 40, 4680–4688 (2006).
Xin, M. & Gustin, M. S. Gaseous elemental mercury exchange with low mercury containing soils: investigation of controlling factors. Appl. Geochem. 22, 1451–1466 (2007).
Fu, X. et al. Depletion of atmospheric gaseous elemental mercury by plant uptake at Mt. Changbai, Northeast China. Atmos. Chem. Phys. 16, 12861–12873 (2016).
Poissant, L., Pilote, M., Yumvihoze, E. & Lean, D. Mercury concentrations and foliage/atmosphere fluxes in a maple forest ecosystem in Québec. Canada. J. Geophys. Res. 113, D10307, https://doi.org/10.1029/2007JD009510 (2008).
Bash, J. O. & Miller, D. R. Growing season total gaseous mercury (TGM) flux measurements over an Acer rubrum L. stand. Atmos. Environ. 43, 5953–5961 (2009).
Edwards, G. C. & Howard, D. A. Air-surface exchange measurements of gaseous elemental mercury over naturally enriched and background terrestrial landscapes in Australia. Atmos. Chem. Phys. 13, 5325–5336 (2013).
Carpi, A. & Lindberg, S. E. Application of a Teflon (TM) dynamic flux chamber for quantifying soil mercury flux: Tests and results over background soil. Atmos. Environ. 32, 873–882 (1998).
Poissant, L. & Casimir, A. Water-air and soil-air exchange rate of total gaseous mercury measured at background sites. Atmos. Environ. 32, 883–893 (1998).
Gustin, M. S., Biester, H. & Kim, C. S. Investigation of the light-enhanced emission of mercury from naturally enriched substrates. Atmos. Environ. 36, 3241–3254 (2002).
Wallschläger, D. et al. Factors affecting the measurement of mercury emissions from soils with flux chambers. J. Geophys. Res.-Atmos. 104, 21859–21871 (1999).
Lindberg, S. E. et al. Increases in mercury emissions from desert soils in response to rainfall and irrigation. J. Geophys. Res.-Atmos. 104, 21879–21888 (1999).
Fritsche, J. et al. Elemental mercury fluxes over a sub-alpine grassland determined with two micrometeorological methods. Atmos. Environ. 42, 2922–2933 (2008).
Enrico, M. et al. Atmospheric Mercury Transfer to Peat Bogs Dominated by Gaseous Elemental Mercury Dry Deposition. Environ. Sci. Technol. 50, 2405–2412 (2016).
Obrist, D. et al. Tundra uptake of atmospheric elemental mercury drives Arctic mercury pollution. Nature 547, 201–204 (2017).
Alberts, J. J., Schindler, J. E., Miller, R. W. & Nutter, D. E. Elemental mercury evolution mediated by humic acid. Science 184, 895–897 (1974).
Bergquist, B. A. & Blum, J. D. Mass-dependent and -independent fractionation of Hg isotopes by photoreduction in aquatic systems. Science 318, 417–420 (2007).
Gu, B. et al. Mercury reduction and complexation by natural organic matter in anoxic environments. Proc. Natl. Acad. Sci. USA 108, 1479–1483 (2011).
Jiskra, M. et al. Mercury deposition and re-emission pathways in boreal forest soils investigated with Hg isotope signatures. Environ. Sci. Technol. 49, 7188–7196 (2015).
Leach, J. A., Larsson, A., Wallin, M. B., Nilsson, M. B. & Laudon, H. Twelve year interannual and seasonal variability of stream carbon export from a boreal peatland catchment. J. Geophys. Res. Biogeosci. 121, 2016JG003357 (2016).
Gustin, M. S. et al. Nevada STORMS project: Measurement of mercury emissions from naturally enriched surfaces. J. Geophys. Res.-Atmos. 104, 21831–21844 (1999).
Kentisbeer, J. et al. Patterns and source analysis for atmospheric mercury at Auchencorth Moss, Scotland. Environ. Sci.: Processes Impacts 16, 1112–1123 (2014).
Wängberg, I., Nerentorp Mastromonaco, M. G., Munthe, J. & Gårdfeldt, K. Airborne mercury species at the Råö background monitoring site in Sweden: distribution of mercury as an effect of long-range transport. Atmos. Chem. Phys. 16, 13379–13387 (2016).
Gustin, M. S., Amos, H. M., Huang, J., Miller, M. B. & Heidecorn, K. Measuring and modeling mercury in the atmosphere: a critical review. Atmos. Chem. Phys. 15, 5697–5713 (2015).
Shanley, J. B. & Bishop, K. Mercury Cycling in terrestrial Watersheds. In: Mercury in the Environment: Pattern and Process, edited by: Bank, M. S. University of California Press, Berkeley, California. 1st edition. pp. 119–141 (2012).
Peichl, M. et al. Energy exchange and water budget partitioning in a boreal minerogenic mire. J. Geophys. Res.: Biogeosciences 118, 1–13 (2013).
Campeau, A. et al. Aquatic export of young dissolved and gaseous carbon from a pristine boreal fen: implications for peat carbon stock stability. Glob Change Biol. Accepted Author Manuscript. https://doi.org/10.1111/gcb.13815.
Lidman, F., Köhler, S. J., Mörth, C.-M. & Laudon, H. Metal Transport in the Boreal Landscape-The Role of Wetlands and the Affinity for Organic Matter. Environ. Sci. Technol. 48, 3783–3790 (2014).
Amyot, M., McQueen, D. J., Mierle, G. & Lean, D. R. S. Sunlight-Induced Formation of Dissolved Gaseous Mercury in Lake Waters. Environ. Sci. Technol. 28, 2366–2371 (1994).
Feng, X. B. et al. Seasonal variation of gaseous mercury exchange rate between air and water surface over Baihua reservoir, Guizhou, China. Atmos. Environ. 38, 4721–4732 (2004).
Zheng, W., Liang, L. Y. & Gu, B. H. Mercury reduction and oxidation by reduced natural organic matter in anoxic environments. Environ. Sci. Technol. 46, 292–299 (2012).
Kim, J. & Fitzgerald, W. Sea-Air Partitioning of Mercury in the Equatorial Pacific-Ocean. Science 231, 1131–1133 (1986).
Kuss, J., Holzmann, J. & Ludwig, R. An Elemental Mercury Diffusion Coefficient for Natural Waters Determined by Molecular Dynamics Simulation. Environ. Sci. Technol. 43, 3183–3186 (2009).
Obrist, D., Pokharel, A. K. & Moore, C. Vertical Profile Measurements of Soil Air Suggest Immobilization of Gaseous Elemental Mercury in Mineral Soil. Environ. Sci. Technol. 48, 2242–2252 (2014).
Kyllönen, K., Hakola, H., Hellen, H., Korhonen, M. & Verta, M. Atmospheric Mercury Fluxes in a Southern Boreal Forest and Wetland. Water Air Soil Pollut. 223, 1171–1182 (2012).
Peichl, M., Sonnentag, O. & Nilsson, M. B. Bringing color into the picture: Using digital repeat photography to investigate phenology controls of the carbon dioxide exchange in a boreal mire. Ecosystems 18, 115–131 (2015).
Skyllberg, U., Qian, J., Frech, W., Xia, K. & Bleam, W. F. Distribution of mercury, methyl mercury and organic sulphur species in soil, soil solution and stream of a boreal forest catchment. Biogeochemistry 64, 53–76 (2003).
Givelet, N., Roos-Barraclough, F. & Shotyk, W. Predominant anthropogenic sources and rates of atmospheric mercury accumulation in southern Ontario recorded by peat cores from three bogs: comparison with natural “background” values (past 8000 years). J. Environ. Monit. 5, 935–949 (2003).
Farmer, J. G. et al. Historical accumulation rates of mercury in four Scottish ombrotrophic peat bogs over the past 2000 years. Sci. Total Environ. 407, 5578–5588 (2009).
Rydberg, J. et al. Importance of vegetation type for mercury sequestration in the northern Swedish mire, Rodmossamyran. Geochim. Cosmochim. Acta 74, 7116–7126 (2010).
Slemr, F. et al. Worldwide trend of atmospheric mercury since 1977. Geophys. Res. Lett. 30, 1516 (2003).
Zhang, Y. et al. Observed decrease in atmospheric mercury explained by global decline in anthropogenic emissions. Proc. Natl. Acad. Sci. USA 113, 526–531 (2016).
Biester, H., Martinez-Cortizas, A., Birkenstock, S. & Kilian, R. Effect of Peat Decomposition and Mass Loss on Historic Mercury Records in Peat Bogs from Patagonia. Environ. Sci. Technol. 37, 32–39 (2003).
Olid, C., Nilsson, M. B., Eriksson, T. & Klaminder, J. The effects of temperature and nitrogen and sulfur additions on carbon accumulation in a nutrient-poor boreal mire: Decadal effects assessed using 210Pb peat chronologies. J. Geophys. Res.-Biogeosci. 119, 392–402 (2014).
Vriens, B., Lenz, M., Charlet, L., Berg, M. & Winkel, L. H. E. Natural wetland emissions of methylated trace elements. Nat. Commun. 5, 3035 (2014).
Biester, H., Bindler, R., Martinez-Cortizas, A. & Engstrom, D. R. Modeling the past atmospheric deposition of mercury using natural archives. Environ. Sci. Technol. 41, 4851–4860 (2007).
Amos, H. M. et al. Observational and modeling constraints on global anthropogenic enrichment of mercury. Environ. Sci. Technol. 49, 4036–4047 (2015).
Steinnes, E. & Sjøbakk, T. E. Order-of-magnitude increase of Hg in Norwegian peat profiles since the outset of industrial activity in Europe. Environ. Pollut. 137, 365–370 (2004).
UNECE. Hemispheric transport of air pollution 2010. Part B: Mercury, edited by: Pirrone, N. and Keating, T. UNPublication, Geneva, Switzerland (2012).
Iverfeldt, Å., Munthe, J., Brosset, C. & Pacyna, P. Long-term changes in concentration and deposition of atmospheric mercury over Scandinavia. Water Air Soil Pollut. 80, 227–233 (1995).
Wängberg, I. et al. Trends in air concentration and deposition of mercury in the coastal environment of the North Sea Area. Atmospheric Environ. 41, 2612–2619 (2007).
Teutschbein, C., Grabs, T., Karlsen, R. H., Laudon, H. & Bishop, K. Hydrological response to changing climate conditions: Spatial streamflow variability in the boreal region. Water Resour. Res. 51, 9425–9446 (2015).
Peichl, M. et al. A 12-year record reveals pre-growing season temperature and water table level threshold effects on the net carbon dioxide exchange in a boreal fen. Environ. Res. Lett., 9, https://doi.org/10.1088/1748-9326/9/5/055006 (2014).
Selin, N. E. Global change and mercury cycling: challenges for implementing a global treaty. Environ. Toxicol. Chem. 33, 1202–1210 (2014).
Laudon, H. et al. The Krycklan Catchment Study - A flagship infrastructure for hydrology, biogeochemistry, and climate research in the boreal landscape. Water Resour. Res. 49, 7154–7158 (2013).
Sagerfors, J. et al. Annual CO2 exchange between a nutrient-poor, minerotrophic, boreal mire and the atmosphere. J. Geophys. Res.-Biogeosci. 113, G01001 (2008).
Zhu, W., Sommar, J., Lin, C.-J. & Feng, X. Mercury vapor air-surface exchange measured by collocated micrometeorological and enclosure methods - Part II: Bias and uncertainty analysis. Atmos. Chem. Phys. 15, 5359–5376 (2015).
Lindberg, S. E., Vette, A. F., Miles, C. & Schaedlich, F. Mercury speciation in natural waters: Measurement of dissolved gaseous mercury with a field analyzer. Biogeochemistry 48, 237–259 (2000).
Roulet, N., Hardill, S. & Comer, N. Continuous measurement of the depth of water-table (inundation) in wetlands with fluctuating surfaces. Hydrol. Processes 5, 399–403 (1991).
Acknowledgements
We thank Markus Meili at Stockholm University for the analyses of THg in discharge, Lisa Okon and Pianpian Wu from SLU, Uppsala for the measurements of THg in the peat cores and Ingvar Wängberg from IVL for providing wet bulk deposition data. The crew of the Field-based Forest Research Unit in Vindeln, Sweden is acknowledged for technical and maintenance support. The study was supported by the Swedish research infrastructures, ICOS and SITES, both financed by the Swedish Research Council and partner universities. The research was financed by the Swiss National Foundation (Doc.Mobility fellowship, #P1BSP2_148458), the Swedish Research Council (Grant 2009-15586-68819-37) and SMaReF (Grant 639-2013-6978).
Author information
Authors and Affiliations
Contributions
S.O., C.A., K.B. and M.B.N. designed the study and drafted the manuscript. S.O. analyzed the data and conducted most of the field work. J.H., H.L. and S.Å. contributed both to the interpretation of data and to the development of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Osterwalder, S., Bishop, K., Alewell, C. et al. Mercury evasion from a boreal peatland shortens the timeline for recovery from legacy pollution. Sci Rep 7, 16022 (2017). https://doi.org/10.1038/s41598-017-16141-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16141-7
This article is cited by
-
Plant demethylation in global mercury cycling
Nature Food (2024)
-
Quantifying soil accumulation of atmospheric mercury using fallout radionuclide chronometry
Nature Communications (2024)
-
Comparing ecosystem gaseous elemental mercury fluxes over a deciduous and coniferous forest
Nature Communications (2023)
-
Mercury deposition and redox transformation processes in peatland constrained by mercury stable isotopes
Nature Communications (2023)
-
Vegetation uptake of mercury and impacts on global cycling
Nature Reviews Earth & Environment (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
