
Abstract
Sesame is prized for its oil. Genetic improvement of sesame can be enhanced through marker-assisted breeding. However, few simple sequence repeat (SSR) markers and SSR-based genetic maps were available in sesame. In this study, 7,357 SSR markers were developed from the sesame genome and transcriptomes, and a genetic map was constructed by generating 424 novel polymorphic markers and using a cross population with 548 recombinant inbred lines (RIL). The genetic map had 13 linkage groups, equalling the number of sesame chromosomes. The linkage groups ranged in size from 113.6 to 179.9 centimorgans (cM), with a mean value of 143.8 cM over a total length of 1869.8 cM. Fourteen quantitative trait loci (QTL) for sesame charcoal rot disease resistance were detected, with contribution rates of 3–14.16% in four field environments; ~60% of the QTL were located within 5 cM at 95% confidence interval. The QTL with the highest phenotype contribution rate (qCRR12.2) and those detected in different environments (qCRR8.2 and qCRR8.3) were used to predict candidate disease response genes. The new SSR-based genetic map and 14 novel QTLs for charcoal rot disease resistance will facilitate the mapping of agronomic traits and marker-assisted selection breeding in sesame.
Similar content being viewed by others

Introduction
Sesame (Sesamum indicum L.), a member of the family Pedaliaceae 1, is grown widely in tropical and subtropical areas2. It is considered one of the first oil seed crops known to humanity, and its domestication has been dated to 3,050−3,500 BC based on charred sesame remains recovered from archaeological excavations3, 4. Sesame is prized for its high oil content (~55% in the seeds), which exceeds that from other oilseed crops such as rapeseed, peanut, soybean, and sunflower. It is popular among consumers of east and south Asian countries for its high-quality oil and the broad utility of its seeds. Sesame seeds contain special anti-oxidative furofuran lignans such as sesamin and sesamolin5, 6, which demonstrate pharmacological properties thought to decrease blood lipids7 and lower cholesterol levels8. Despite these elite characteristics, studies on sesame remain insufficient compared with other crops, contributing to its low and unstable yield capacity9. According to the Food and Agriculture Organisation (FAO), the average yield of sesame was only 576 kg/ha in 2014 (http://www.fao.org), ranking second from lowest among 22 oil crops worldwide. Solid genetic studies are necessary to improve its resistance to biotic stress from fungal and bacterial diseases such as stem charcoal rot, Fusarium wilt, and powdery mildew.
Charcoal rot is one of the most damaging diseases, typically inducing 30% decrease or even complete loss of sesame crops. The disease is caused by Macrophomina phaseolina (Tassi) Goid. M., a seed- and soil-borne fungal pathogen that can infect nearly 500 plant species in more than 100 families10,11,12,13, including important crops such as peanut, cabbage, pepper, chickpea, soybean, sunflower, sweet potato, alfalfa, sesame, potato, sorghum, wheat, and corn11, 14,15,16. In sesame, charcoal rot occurs mainly at the end stage of flowering to maturity, with black spots initiating from the root or stem during hot and dry weather or under unfavourable environmental stresses17,18,19. Controlling the disease in a safe and efficient way is a current pivotal problem facing plant pathologist, geneticist, and breeders. Although breeding cultivars with integrated resistant genes is expected to be fundamentally the best choice, progress in genetic improvement efforts has been slow due to the lack of information regarding the gene–for–gene relationship between sesame and M. phaseolina fungus20.
Genetic mapping provides the foundation for genetic study, especially for discovering and manipulating the loci or genes underlying simple and complex traits in crop plants21, 22. The construction of genetic linkage maps, QTL mapping, and evolutionary analyses performed in standard molecular biology laboratories have provided SSR markers the primary choice for marker-assisted selection (MAS), based largely on the properties of co-dominance, reproducibility, and relative abundance in complete genomes23,24,25. However, molecular genetic research in sesame had lagged for decades, and some SSR markers were developed only recently26,27,28,29. The first map for sesame was constructed by Wei et al. based on amplified fragment length polymorphisms30 and was last updated in 2013.
Since few SSRs have been validated, genetic maps and gene mapping have been hampered in sesame. Several SNP-based maps were constructed by restriction-site-associated DNA sequencing (RAD-seq) and specific length amplified fragment sequencing (SLAF-seq) technologies using next generation sequencing platforms in recent years31,32,33. However, such genetic maps and SNP tags are not easy to be used by most sesame researchers in different molecular laboratories, because of the lack of available sequence information and special instruments.
The present study was designed to develop a greater number of SSR markers based on sesame genome and transcriptome sequences34, 35, and construct a genetic map with these co-dominant markers. Furthermore, the loci associated with sesame charcoal rot resistance were screened. The SSR-based genetic map will provide an essential and effective tool for QTL mapping of genes to identify various traits in sesame, facilitating future gene exploration and discovery of elite sesame cultivars for more productive breeding.
Results and Discussion
Polymorphic genomic-SSR development
A total of 110,495 genomic-SSR loci were detected in the sesame genome using the microsatellite identification tool (MISA) software. Of these, 39.1% were mono-nucleotides, 34.3% were di-nucleotides, 17.7% were tri-nucleotides, and 27.2% were compound SSRs. Relying on a previously published sesame transcriptome, 7,702 cDNA-SSR loci were investigated28. Here, 5,587 genomic-SSRs and 1,770 cDNA-SSRs were selected for designing primer pairs to synthesise markers. PCR analysis showed that 498 of the markers were polymorphic between the parents, ZZM2748 and Zhongzhi No. 13, accounting for 6.8% of the total synthesised markers. As these markers were developed based on the sesame genome, such differences might represent general polymorphisms of sesame SSR loci. However, SSRs with varying repeat units differed in their polymorphisms. The highest levels of polymorphisms were found with dinucleotide repeat units (11.9%), followed by compound (7.8%) and mononucleotide (7.4%) repeat units. Tetra- and pentanucleotide repeat unit SSRs showed lower polymorphic rates, <2%.
SSR-based genetic map construction
All 498 polymorphic SSR markers were used to genotype the 548 RILs generated from the cross between ZZM2748 and Zhongzhi No. 13. After filtering out markers that lacked polymorphic alleles and those with significantly distorted segregation ratios (P < 0.01)36, 37, the remaining 462 markers were used to construct a genetic map using Joinmap 4.0. Finally, 424 SSR markers were mapped to the genetic map and distributed into 13 linkage groups (LG). All the 424 mapped markers were newly developed and published (Table S1). The LGs were numbered from LG1 to LG13 (Table 1, Fig. 1) and corresponded to the 13 assembled pseudomolecule chromosomes of sesame38.

The simple sequence repeat (SSR)-based genetic map of the sesame genome and the mapped quantitative trait loci (QTL). Positions of the QTL for sesame charcoal rot response are indicated with red rectangles centred at the peak of each location.
The lengths of the 13 LGs ranged from 113.6 to 179.9 cM, with a mean value of 143.8 cM, and the resulting map length was 1869.8 cM in total. The number of markers in each LG ranged from 17 (LG5) to 53 (LG8). The interval distances between adjacent markers varied from 0.1 to 36.2 cM, with a mean interval distance of 5.1 cM across different LGs; 81.4% of markers showed interval distances less than 10 cM relative to adjacent markers. The highest marker density was observed in LG7 (an average of 2.2 cM between adjacent markers), followed by LG8 (3.2 cM) (Fig. 1); LG5 showed the lowest density average (8.1 cM). Obviously, the total marker number in the map was less than those presented in previously published genetic maps that were constructed using next-generation sequencing technology, such as RAD-seq (1,230 and 1,522)31, 38 and SLAF-seq (1,233)32. As few SSR-based genetic maps were available previously and a large number of markers need to be designed and screened for polymorphisms with proper segregation of the population for genotyping, the construction of such a SSR-based genetic map was very time consuming, which lasted for 5 years. However, considering the small and diploid genome of sesame (357 Mb), the current map is competent for genetic analysis including genetic or QTL mapping.
Mapping the QTL associated with charcoal rot resistance
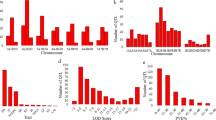
The charcoal rot response of the 548 RILs and the two parents was evaluated in four growing environments including Luohe (2015), Jinxian (2015), and Yangluo (2014 and 2015). Jinxian and Yangluo are in southern China along the Yangtze River, and they typically suffered from serious charcoal rot disease during the sesame planting seasons. Luohe, in northern China, also experienced serious charcoal rot disease during the sesame planting season. In 2014, the disease index (DI) at the Yangluo site ranged from 0.01 to 0.81 and averaged 0.31. In 2015, the ranges of DI in the three sites were 0.14–0.96 (Luohe, average 0.60), 0.0–0.87 (Jinxian, average 0.42), and 0.05–0.89 (Yangluo, average 0.42) respectively (Fig. 2). The DIs of Zhongzhi No. 13 were generally <0.15, whereas ZZM2748 displayed DIs >0.50 in different environments. ANOVA revealed significant differences (P = 0.01) in DIs among RILs, environments, and line × environment interactions, but not among environmental replicates (Table S2). The DIs from the four environments showed that Yangluo and Jinxian were highly correlated, but they all showed low correlations with Luohe. Those findings reveal the diversities among M. phaseolinas pathogenic races in southern and northern China, as well as among environmental conditions (Fig. 3).

Disease index (DI) variations across different environments in the RIL population. DI, the charcoal rot disease index of the recombinant inbred lines (RIL). YL 2014, Yangluo environment in 2014; YL 2015, Yangluo environment in 2015; JX 2015, Jinxian environment in 2015; LH 2015, Luohe environment in 2015.

Scatter plots of the co-relationships between the DIs from the four locations. YL 2014, Yangluo environment in 2014; YL 2015, Yangluo environment in 2015; JX 2015, Jinxian environment in 2015; LH 2015, Luohe environment in 2015.
Using the composite interval mapping method implemented in Windows QTL Cartographer 2.5 software (Microsoft, Inc., Redmond, WA, USA)39, 14 QTLs were found to be significantly associated with sesame charcoal rot disease resistance, with contribution rates of 3–14.16% (mean, 6.95%; Table 2, Fig. 1). A previous study showed enhanced infection of M. phaseolina when soil water was deficient, and several overlapped or pleiotropic QTLs for drought tolerance and charcoal rot resistance were reported in other crops40, 41. Sesame is generally drought tolerant, as it originated from the tropical regions of Africa or India42, and several of these loci may function in drought tolerance. However, such cases require further investigation, as no genes or QTLs for sesame drought-tolerance have yet been identified.
The 95% confidence intervals of the 14 mapped QTLs ranged from 0.8 to 25 cM; 60% were located within 5 cM (Table S3). Of these QTL, 2 loci (qCRR8.2 and qCRR8.3) were detected in all four environments, and both were located in LG8. QTL qCRR3.1, qCRR9.1, qCRR13.1, qCRR5.1, and qCRR12.2 were repetitively detected in three environments; the first three were found in Yangluo (2014 and 2015) and Jinxian (2015), and the last two were detected in Yangluo (2014 and 2015) and Luohe (2015) (Table 2). Five QTLs (qCRR3.2, qCRR3.3, qCRR8.1, qCRR12.1, and qCRR12.2) were detected in two environments. Locus qCRR8.1 was associated with the DI from Yangluo and Jinxian in 2015, while the other four loci were found in Yangluo in 2014 and 2015. The loci.qCRR3.4 and qCRR12.3 were detected only in Jinxian, while qCRR13.2 was detected only in Luohe.
Sesame charcoal rot resistance-related QTLs were distributed differently among the LGs. LG3 contained the greatest number of mapped loci, with four QTL, followed by LG8 and LG13, with three QTLs each (Table 2). In LG12, the loci qCRR12.2 had the highest phenotype contribution rate (14.16%). After mapping the flanking markers to the sesame genome34, the locus qCRR12.2 fell into a region that included 15 genes. Annotation of these genes predicted a cluster of plant receptor-like serine threonine kinase (RLK) genes (SIN_1001382, SIN_1001381, SIN_1001380, SIN_1001379, SIN_1001377, SIN_1001376, SIN_1001373, SIN_1001372). It has been hypothesised that the RLK gene family expansion allowed accelerated evolution among domains implicated in signal reception, playing a central role in signalling during pathogen recognition43. We also focused on qCRR8.2 and qCRR8.3 for their common function in different environments. The physical mapping regions of the two loci on the sesame genome showed they coincidently contained several homologous plant disease resistance genes encoding nucleotide-binding sites (NBSs) (Fig. 4) 44. Genes encoding NBSs are the largest class of disease resistance genes in plants44. Thus, these mapped loci may represent an important locus for sesame resistance to charcoal rot disease.

The predicted nucleotide-binding site (NBS)-encoding resistance genes in the qCRR8.2 and qCRR8.3 regions. Distributions of NBS-encoding resistance gene models (R-genes) along the sesame genome are denoted with short coloured lines. The 14 R-genes fell into the QTL listed in the box.
Conclusions
This study provided a novel genetic map for sesame and generated 424 polymorphic SSR markers. The mean interval between adjacent markers was 5.1 cM. Based on the constructed genetic map, 14 QTLs for sesame charcoal rot disease resistance were detected, with contribution rates of 3–14.16%; ~60% of these were located within 5 cM (95% confidence interval). The QTL with the highest phenotype contribution rate was qCRR12.2. Two loci (qCRR8.2 and qCRR8.3) were detected in plants grown in all the four trial environments. QTL mapping revealed several candidate genes that were predicted to confer disease resistance. Thus, the genetic map of sesame is competent for mapping genes or QTLs. Moreover, the genetic map consisted of SSR markers, which will be more easily to be supplemented in a common molecular laboratory rather than SNP-based maps. Thus, this study will provide a useful reference for gene mapping and genetic studies in sesame.
Methods
Plant material
This study employed a RIL population (F8) with 548 lines generated from the cross between ZZM2748 (P1, male parent) and Zhongzhi No. 13 (P2). ZZM2748 is susceptible to sesame charcoal rot. Zhongzhi No. 13 showed high resistance to the disease, and it was de novo sequenced in 201434. Both materials were preserved at the National Medium-term Sesame GenBank of China (Wuhan, China).
Evaluation for charcoal rot response
As charcoal rot is a common disease in most sesame traditional planting areas, the natural responses of the RIL populations (ZZM2748 × Zhongzhi No. 13, F8:9) were evaluated in three typical sites that had suffered from M. phaseolina, including Luohe (2015), Jinxian (2015), and Yangluo (2014 and 2015), during the normal sesame planting season (June–September). In each field trial, the 548 lines were grown in a randomised complete block design with three replicate plots, each comprising three 2-m rows spaced 40 cm apart with a plant spacing of 10–20 cm. The highly susceptible material ZZM2748 was planted in every tenth plot as a check.
When over 50% of plants in each growing area exhibited apparent charcoal rot symptoms at the end stage of flowering, an investigation was performed on all plants having disease progressions of differing degrees (I) as follows: 0 = normal plant without disease spots; 1 = less than 1/3 of the plant and less than 1/4 of the capsules exhibited charcoal rot; 3 = 1/3 to 2/3 of the plant and 1/4 to 1/2 of the capsules exhibited charcoal rot; 5 = over 2/3 of the plant and 1/2 to 3/4 of the capsules exhibited charcoal rot; 7 = the entire plant died. The disease index (DI) was calculated based on the following formula45:
(where Xi represents the plant number with disease degree I).
SSR marker development
A Perl script MISA tool (http://pgrc.ipk-gatersleben.de/misa) was used to search microsatellite sites in the sesame genome (http://ocri-genomics.org/Sinbase) and transcriptome sequences28, 34, 35, 38. The SSRs with mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide repeat units and compound units comprising two or more repeat motifs but interrupted by ≤100 bases were identified, and the minimum repeat numbers were defined as ten for mono-, six for di-, and five for tri-, tetra-, penta- and hexa-nucleotide repeat SSRs. Following the steps, the two Perl scripts p3_in.pl and p3_out.pl were used to handle the data generated from MISA and to format input data for primer design.
Based on the SSR flanking sequences, PRIMER346 software was employed to design the primer pairs. The major parameters were adjusted as follows: primer length of 18–23 bases (optimal, 20 bases), GC content of 40–70% (optimal, 50%), annealing temperatures of 50–60 °C (optimal, 55 °C), and PCR product size of 100–400 bp (optimal, 200 bp). The markers developed based on sesame genome sequence were named with the prefix “ZMM”, “D”, or “ID” (genomic SSR), and those from transcriptomes were named with the prefix “ZM” (cDNA-SSR). All primer pairs were synthesised by GenScript Co., Ltd. (Nanjing, China).
DNA extraction and PCR
At the early flowering stage, healthy young leaves of ZZM2748 and Zhongzhi No. 13, and the 548 RILs (F8) were selected and used for total genomic DNA extraction employing the cetyltrimethylammonium bromide (CTAB) method32, 47. DNA concentration qualities were estimated using an ND-1000 spectrophotometer (NanoDrop, Wilmington, DE, USA) at 260 nm, and the quality was confirmed by 0.8% agarose gel electrophoresis using a lambda DNA standard.
A 10-μl reaction mixture used for PCR contained 2.5 μl of template DNA (20 ng/ml), 0.2 μl of 5 mM/μl dNTP, 0.2 μl of 5 U/ml Taq polymerase (Bicolor, Shanghai, China), 0.45 μl of 18 pmol/μl of each primer, 1 μl of 10 × buffer, and 5.2 μl of ddH2O. PCR was performed on a thermocycle instrument (S1000, Bio-Rad, Hercules, CA, USA) using the following conditions: 10 min at 94 °C, then 30 cycles of 45 s at 94 °C, 45 s at 50−60 °C, and 1 min at 72 °C, followed by a final extension at 72 °C for 10 min. The PCR products were separated with 6% denaturing polyacrylamide gels and visualised after silver staining.
Linkage map construction
The polymorphic SSR markers were used to genotype 548 lines. Using the software JoinMap 448, the segregation ratios of these markers were evaluated using the chi-squared test, and significantly distorted (P < 0.01) markers were removed. With a logarithm of minimum odds (LOD) score of 4.0, the remaining markers were grouped and ordered according to their pair-wise recombination frequencies. The Kosambi mapping function was chosen to translate the recombination frequencies into map distances in cM. The goodness of fit of the calculated regression map for each tested position was checked with default parameters.
QTL analysis
The frequency distributions of the mean phenotypic data for all RILs in each trial were analysed using R package software (https://www.r-project.org/). The QTLs related to charcoal rot resistance in sesame were detected with Windows QTL Cartographer 2.5 software (Microsoft, Inc., Redmond, WA, USA) using the composite interval mapping method. An associated peak with LOD score over 2.5 was judged as the presence of a QTL, and the statistical significance of the QTL effect was determined based on 1,000 permutations. The detected QTLs were named according to trait and LG location, referring to the rules of wheat gene nomenclature (http://wheat.pw.usda.gov/ggpages/wgc/98/Intro.htm).
References
The Angiosperm Phylogeny, G. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 161, 105–121, doi:10.1111/j.1095-8339.2009.00996.x (2009).
Ashri, A. In Plant Breed. Rev. Vol. 16 (ed J. Janick) Ch. 5, (John Wiley & Sons, Inc., 2010).
Ram, R., Catlin, D., Romero, J. & Cowley, C. In Advances in new crops (ed J. Janick and J.E. Simon) 225–228 (Timber Press, 1990).
Dorothea Bedigian, J. R. H. Evidence for cultivation of sesame in the ancient world. Econ. Bot. 40, 18, doi:10.1007/BF02859136 (1986).
Chang, L. W., Yen, W. J., Huang, S. C. & Duh, P. D. Antioxidant activity of sesame coat. Food Chem. 78, 347–354 (2002).
Fukuda, Y., Nagata, M., Osawa, T. & Namiki, M. Contribution of lignan analogues to antioxidative activity of refined unroasted sesame seed oil. J. Am. Oil Chem. Soc. 63, 1027–1031 (1986).
Hirata, F. et al. Hypocholesterolemic effect of sesame lignan in humans. Atherosclerosis 122, 135–136 (1996).
Chen, P. R. et al. Dietary sesame reduces serum cholesterol and enhances antioxidant capacity in hypercholesterolemia. Nutr Res 25, 559–567 (2005).
Cagirgan, M. I. & Uzun, B. Comparison of determinate and indeterminate lines of sesame for agronomic traits. Field Crops Res. 96, 13–18 (2006).
Dossa, K. et al. Insight into the AP2/ERF transcription factor superfamily in sesame and expression profiling of DREB subfamily under drought stress. BMC Plant Biol. 16, 171, doi:10.1186/s12870-016-0859-4 (2016).
Gomez-Cadena, A. et al. Immune-system-dependent anti-tumor activity of a plant-derived polyphenol rich fraction in a melanoma mouse model. Cell Death Dis. 7, e2243, doi:10.1038/cddis.2016.134 (2016).
Garcia, A. V. & Hirt, H. Salmonella enterica induces and subverts the plant immune system. Frontiers in microbiology 5, 141, doi:10.3389/fmicb.2014.00141 (2014).
Koen, E. et al. Nitric oxide is a major player in plant immune system. Medecine sciences: M/S 29, 309–316, doi:10.1051/medsci/2013293018 (2013).
McDowell, J. M. & Meyers, B. C. A transposable element is domesticated for service in the plant immune system. Proc. Natl. Acad. Sci. USA 110, 14821–14822, doi:10.1073/pnas.1314089110 (2013).
Claverie, J. et al. Fighting bacterial infections: the plant immune system is also very efficient!. Medecine sciences: M/S 32, 335–339, doi:10.1051/medsci/20163204008 (2016).
Asai, S. & Shirasu, K. Plant cells under siege: plant immune system versus pathogen effectors. Curr. Opin. Plant Biol. 28, 1–8, doi:10.1016/j.pbi.2015.08.008 (2015).
Abdel-Razik, A. A., Ahmed, H. A. M., Hassan, M. H. A. & Khaled, S. A. Management of Charcoal Rot of Sesame by Seed Soaking in Medicinal Plant Extracts and Hot Water. Plant Pathology Journal 26, 372–379 (2010).
Ahuja, D. B. et al. Development of integrated pest management technology for sesame (Sesamum indicum) and its evaluation in farmer participatory mode. Indian J. Agric. Sci. 79, 808–812 (2009).
Wang, L. H. et al. Variations in the isolates of Macrophomina phaseolina from sesame in China based on amplified fragment length polymorphism (AFLP) and pathogenicity. African Journal of Microbiology Research 5, doi:10.5897/ajmr11.306 (2011).
Jones, J. D. & Dangl, J. L. The plant immune system. Nature 444, 323–329, doi:10.1038/nature05286 (2006).
Tanksley, S. D. et al. High density molecular linkage maps of the tomato and potato genomes. Genetics 132, 1141–1160 (1992).
Varshney, R. K. et al. The first SSR-based genetic linkage map for cultivated groundnut (Arachis hypogaea L.). Theor. Appl. Genet. 118, 729–739 (2009).
Wu, J. et al. High-density genetic linkage map construction and identification of fruit-related QTLs in pear using SNP and SSR markers. J. Exp. Bot. 65, 5771–5781, doi:10.1093/jxb/eru311 (2014).
Ma, J. Q. et al. Construction of a SSR-based genetic map and identification of QTLs for catechins content in tea plant (Camellia sinensis). PLoS One 9, e93131, doi:10.1371/journal.pone.0093131 (2014).
Andargie, M., Pasquet, R. S., Gowda, B. S., Muluvi, G. M. & Timko, M. P. Construction of a SSR-based genetic map and identification of QTL for domestication traits using recombinant inbred lines from a cross between wild and cultivated cowpea (V. unguiculata (L.) Walp.). Mol. Breed. 28, 413–420, doi:10.1007/s11032-011-9598-2 (2011).
Zhang, H., Wei, L., Miao, H., Zhang, T. & Wang, C. Development and validation of genic-SSR markers in sesame by RNA-seq. BMC Genomics 13, 316, doi:10.1186/1471-2164-13-316 (2012).
Wang, L., Zhang, Y., Qi, X., Gao, Y. & Zhang, X. Development and characterization of 59 polymorphic cDNA-SSR markers for the edible oil crop Sesamum indicum (Pedaliaceae). Am. J. Bot. 99, e394–398, doi:10.3732/ajb.1200081 (2012).
Wei, W. et al. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 12, 451, doi:10.1186/1471-2164-12-451 (2011).
Wei, X. et al. Development of simple sequence repeat (SSR) markers of sesame (Sesamum indicum) from a genome survey. Molecules 19, 5150–5162, doi:10.3390/molecules19045150 (2014).
Zhang, H. et al. Genetic analysis and QTL mapping of seed coat color in sesame (Sesamum indicum L.). PLoS One 8, e63898, doi:10.1371/journal.pone.0063898 (2013).
Wu, K. et al. High-density genetic map construction and QTLs analysis of grain yield-related traits in Sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Biol. 14, 274, doi:10.1186/s12870-014-0274-7 (2014).
Zhang, Y. et al. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Plant Biol. 13, 141, doi:10.1186/1471-2229-13-141 (2013).
Sedivy, E. J., Wu, F. & Hanzawa, Y. Soybean domestication: the origin, genetic architecture and molecular bases. New Phytol., doi:10.1111/nph.14418 (2017).
Wang, L. et al. Genome sequencing of the high oil crop sesame provides insight into oil biosynthesis. Genome Biol. 15, R39, doi:10.1186/gb-2014-15-2-r39 (2014).
Wang, L., Yu, J., Li, D. & Zhang, X. Sinbase: An Integrated Database to Study Genomics, Genetics and Comparative Genomics in Sesamum indicum. Plant Cell Physiol., doi:10.1093/pcp/pcu175 (2014).
Suzuki, C. K. & Bielas, J. H. Editorial overview: Molecular and genetic bases of disease. Curr. Opin. Genet. Dev. 38, iv–vi, doi:10.1016/j.gde.2016.09.001 (2016).
Arking, D. & Rommens, J. Editorial overview: Molecular and genetic bases of disease: Enter the post-GWAS era. Curr. Opin. Genet. Dev. 33, 77–79, doi:10.1016/j.gde.2015.10.001 (2015).
Wang, L. et al. Updated sesame genome assembly and fine mapping of plant height and seed coat color QTLs using a new high-density genetic map. BMC Genomics 17, 31, doi:10.1186/s12864-015-2316-4 (2016).
Silva Lda, C., Wang, S. & Zeng, Z. B. Composite interval mapping and multiple interval mapping: procedures and guidelines for using Windows QTL Cartographer. Methods Mol. Biol. 871, 75–119, doi:10.1007/978-1-61779-785-9_6 (2012).
Finkina, E. I., Melnikova, D. N., Bogdanov, I. V. & Ovchinnikova, T. V. Lipid Transfer Proteins As Components of the Plant Innate Immune System: Structure, Functions, and Applications. Acta naturae 8, 47–61 (2016).
Tenkouano, A., Miller, F. R., Frederiksen, R. A. & Rosenow, D. T. Genetics of nonsenescence and charcoal rot resistance in sorghum. Theor. Appl. Genet. 85, 644–648, doi:10.1007/BF00220925 (1993).
Wang, L. et al. Deep resequencing reveals allelic variation in Sesamum indicum. BMC Plant Biol. 14, 225, doi:10.1186/s12870-014-0225-3 (2014).
Afzal, A. J., Wood, A. J. & Lightfoot, D. A. Plant receptor-like serine threonine kinases: roles in signaling and plant defense. Mol. Plant-Microbe Interact. 21, 507–517, doi:10.1094/MPMI-21-5-0507 (2008).
Meyers, B. C. et al. Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J. 20, 317–332 (1999).
Zhang, X. & Feng, X. Descriptors and Data Standard for Sesame (Sesamum indicum L.) (China Agriculture Press, 2006).
Rozen, S. & Skaletsky, H. In Methods Mol. Biol. Vol. 132, 365–386 (Humana Press, 2000).
Doyle, J. J. & Doyle, J. L. Isolation of plant DNA from fresh tissue. Focus, 13–15 (1990).
Stam, P. Construction of integrated genetic linkage maps by means of a new computer package: Join Map. The Plant Journal 3, 739–744, doi:10.1111/j.1365-313X.1993.00739.x (1993).
Acknowledgements
The studies conducted by the Oil Crops Research Institute of the Chinese Academy of Agricultural Sciences, were supported by the Agricultural Science and Technology Innovation Project of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2013-OCRI), the Fundamental Research Funds for Central Non-profit Scientific Institution and China Agriculture Research System (no. CARS–15).
Author information
Authors and Affiliations
Contributions
X.R.Z. and L.H.W. contributed to the design of the research. L.H.W. and Y.X.Z. constructed the RIL population. Y.X.Z., X.M.Z, G.B.X. and D.H.L. performed the field trial and evaluated the phenotypes. L.H.W. and X.W. developed the markers. X.F.Z., X.D.Z. and Y.G. performed PCR analysis and genotyping. L.H.W. and X.R.Z. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
The Additional file 1 with the information of the 424 SSR markers and QTL regions is available with the online version of the paper.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Zhang, Y., Zhu, X. et al. Development of an SSR-based genetic map in sesame and identification of quantitative trait loci associated with charcoal rot resistance. Sci Rep 7, 8349 (2017). https://doi.org/10.1038/s41598-017-08858-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-08858-2
This article is cited by
-
The status of in vitro regeneration and genetic transformation in the recalcitrant oil seed crop Sesamum indicum L
In Vitro Cellular & Developmental Biology - Plant (2023)
-
Omics technologies towards sesame improvement: a review
Molecular Biology Reports (2023)
-
Development of molecular resources for the genetic improvement of noug (Guizotia abyssinica (L.f) Cass): a mini review
CABI Agriculture and Bioscience (2022)
-
The mechanism of sesame resistance against Macrophomina phaseolina was revealed via a comparison of transcriptomes of resistant and susceptible sesame genotypes
BMC Plant Biology (2021)
-
QTL mapping of yield-related traits in sesame
Molecular Breeding (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
