
Abstract
Lepus oiostolus (L. oiostolus) is a species endemic to the Qinghai-Tibet Plateau. However, the absence of a reference genome limits genetic studies. Here, we reported a high-quality L. oiostolus genome assembly, with scaffolds anchored to 24 chromosomes and a total assembled length of 2.80 Gb (contig N50 = 64.25 Mb). Genomic annotation uncovered 22,295 protein-coding genes and identified 49.84% of the sequences as transposable elements. Long interspersed nuclear elements (LINEs) constitute a high proportion of the genome. Our study is at the first time to report the chromosome-scale genome for the species of the L. oiostolus. It provides a valuable genomic resource for future research on the evolution of the Leporidae.
Similar content being viewed by others

Background & Summary
The emergence and rapid development of new sequencing technologies, such as single-molecule real-time sequencing (SMRT) by Pacific Biosciences (PacBio)1, nanopore sequencing by Oxford Nanopore Technologies2 10x genomics3, optical and chromosome-contact maps from BioNano Genomics4, and Hi-C sequencing5 have facilitated the construction of high-quality genome assemblies6. Integrative application of these techniques elevate human genome assembly to a Telomere-to-Telomere (T2T) gapless level. Subsequent mining of the human T2T genome unveils genetic variations of mitoplasts, centromeres and other previously unassembled regions, which significantly enriched the understanding of human genome diversity, evolution and disease occurrence7. Other studies on important livestock also showed the important values of high quality genome assemblies. Qiu et al. presented the draft genome sequence of a female domestic yak generated using Illumina-based technology and related to sensory perception and energy metabolism, as well as an enrichment of protein domains involved in sensing the extracellular environment and hypoxic stress8. Zhang et al. used long-read resequencing data for 6 wild and 23 domestic yaks and identified genes that are predominantly related to the nervous system, behaviour, immunity, and reproduction and may have been targeted by artificial selection during yak domestication9. Liu et al. constructed a graph-genome for 47 genomes of 7 cross-fertile bovine species and validated a significant association of the selected stratified SVs with gene expression10. These aforementioned research greatly deepened the understanding of the genetic basis of high-altitude adaptation of bovine species. Decoding of high-quality genome assemblies is increasingly becoming a powerful approach serving clarification of the genetic mechanisms of adaptive evolution.
L. oiostolus is a species endemic to the Qinghai-Tibet Plateau (QTP) and is also commonly known as the gray-tailed hare. It belongs to the genus Lepus11. It is distributed at an elevation of 2,100–4,000 m in the alpine zone of the Qinghai-Tibet Plateau in China, making it the highest-altitude rabbit species in the world. In contrast, the typical habitat of rabbits rarely exceeds 600 m at altitude12. For instance, Oryctolagus cuniculus, a closely related species of L. oiostolus, has been used as a model organism for biomedical research. Previous physiological studies demonstrated that L. oiostolus has developed unique tolerance to plateau environment, making this species an excellent model for investigation of genetic mechanisms contributing to high-altitude adaptation13. Although deciphering of genome assemblies of plateau animals have significantly broaden our knowledge horizon concerning plateau adaptation and evolution, the genome information of L. oiostolus is still lacking8,14,15.
In this study, we assembled a chromosome-scale genome of L. oiostolus by combining Illumina and PacBio data with Hi-C technology. The final genome size was 2.85 Gb with N50 sizes of 64.25 Mb and a complete Benchmarking Universal Single-Copy Orthologs (BUSCO) score of 96.2%. A total of 2.80 Gb (98.1%) genome sequences were further clustered and ordered into 24 chromosomes. Genome annotation predicted 22,295 protein-coding genes. The availability of a complete and detailed genome assembly is essential to basic biological research. This paper provides a valuable genomic resource for research into the molecular mechanisms and evolution of L. oiostolus.
Methods
Sample collection and sequencing
L. oiostolus samples for genome sequencing were collected from Haiyan County, Haibei Prefecture, Qinghai Province, China (100°98’E, 36°90’N). Genomic DNA was extracted from muscle tissue and blood using the TIANamp Genomic DNA kit. Agarose gel electrophoresis (0.7% agarose gel) was used to assess overall DNA quality. DNA purity was assessed using a NanoDrop One Spectrophotometer (Thermo Fisher Scientific), and DNA concentration was determined using a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA).
We implemented a hybrid strategy combining Illumina short-read sequencing, PacBio long-read sequencing, and Hi-C sequencing technologies to obtain better sequencing data. For Illumina short reads, a genomic library with insert sizes of 150 bp was constructed using the NextEra DNA Flex X Library Prep Kit (Illumina, San Diego, CA, USA). Qubit 2.0, Agilent 2100, and qPCR were used to ensure the quality of the library, and then the library was subsequently sequenced on the Illumina NovaSeq. 6000 platform (Illumina, San Diego, CA, USA). For PacBio long-read sequencing, PCR-free SMRT bell libraries were constructed and sequenced on the PacBio Sequel II sequencing platform. To generate a chromosomal-level assembly of the L. oiostolus genome, a Hi-C library was generated by the DpnII restriction enzyme following in situ ligation protocol14. The formaldehyde cross-linked DNAs were digested with DpnII and treated with biotin, which were ligated to the ends of the fragmented DNA sequences. Cyclization of DNA after end-repair was used to identify the location of intersecting DNA. After reversing the crosslinking, ligated DNA fragments were fragmented into 300–700 bp sizes, followed by a biotin-streptavidin purification for library construction. Finally, the Hi-C libraries were quantified and sequenced with PE150 sequencing read lengths on the Illumina platform. Finally, 79.03 Gb of PacBio reads (Table S1) and 177.15 Gb of filtered Illumina short-read sequencing data (Table S2) were obtained from the L. oiostolus genome.
Genome survey and assembly
Using the kmer-freq subroutine in the software GCE v1.0.0, the genome size, heterozygosity, and duplication ratio were estimated based on the k-mer distribution of 19-mers extracted from Illumina short reads. A total of 223.91 Gb raw PacBio subreads were filtered and corrected with the CCS pipeline v6.0.0 (parameters: -min-passes 3 –min-snr 2.5 –top-passes 60)15. The resulting CCS reads were subjected to hifiasm v0.14.216 for de novo assembly. We corrected the primary contigs with the pilon v1.2317 using 89.12 Gb (70.35×) of Illumina paired-end reads. Using the samtools v1.918 and bwa v0.7.1714, after which the processed reads from the Illumina sequencing platform were mapped to the reference genome. The mapping rate and coverage were counted to confirm the consistency and integrity of the assembled genome. The genome size is 2.83 Gb with 0.72% heterozygosity and 36.55% duplicate repeatability, as estimated by K-mer-based methods (Table S3; Figure S1). The resulting assembly was 2.86 Gb with a contig N50 of 64.25 Mb and the longest contig being 147.07 Mb in length (Table 1; Fig. 1). 99.99% of Illumina short reads were successfully aligned to our assembly (Table S4).

Distribution of various elements on the chromosomes of L. oiostolus. From the outer circle to the inner circle: (A) Chromosomes karyotype. (B) Gene density. (C) Distribution of GC content in the genome. (D) Repetitive elements density. (E) Schematic presentation of major inter-chromosomal relationships in the L. oiostolus genome.
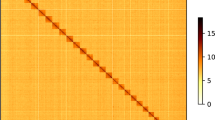
Hi-C analysis was used to generate the contig assembly at the chromosome level. The raw data were filtered using a perl script as implemented in the software LACHESIS v1.5715. To obtain uniquely mapped read pairs, the cleaned data were aligned to the initial genome assembly using BWA-MEM. Approximately 1.18 Gb of clean data were generated (Table S5). The hicup v0.8.016 was used to evaluate the validity of the Hi-C data based on uniquely mapped read pairs. We only used valid read pairs for the draft genome re-correction and chromosome-level genome assembly. We further applied ALLHIC pipeline v0.9.1217 to link the contigs into 24 pseudo chromosomes. Finally, our team manually curated the Hi-C scaffolding based on the chromatin contact matrix in the Juicebox v1.11.0818. The scaffold assembly was obtained using the ALLHIC pipeline v0.9.1217 with 748.17 million uniquely mapped reads from cleaned Hi-C data (Table S6). A total of 2.80 Gb of sequences were anchored onto 24 pseudo chromosomes, accounting for 98.08% of the initial assembly (Figure S2, Table 2). In addition, Hi-C data were mapped against the Hi-C scaffold assembly, showing 74.31% valid sequences (Table S6). Genome-wide analysis of chromatin interactions showed a well-organized pattern of Hi-C signals along the diagonals, indicating a high-quality chromosomal genome assembly for L. oiostolus (Figure S2).
Genome structure prediction and annotation
Both homology-based and de novo prediction methods were used to identify repetitive DNA elements. For the de novo prediction, RepeatModeler v1.0.1119 was used to construct an ab initio database of predicted repetitive elements, and RepeatMasker v4.0.920 was used to annotate the repetitive elements in the database. RepeatMasker and RepeatProteinMask were then used to search the genome sequence for known repetitive elements, with the genome sequences used as queries against the RepBase database v27.06 (http://www.girinst.org/repbase). Tandem repeats were also identified with the TRF method. Repetitive elements play an essential role in genome evolution. In the repeat annotation, a total of 1,423, 017,427 bp of transposon elements (TEs) comprise 49.84% of the L. oiostolus genome. Among all the classifications of TEs, LINE constituted the largest portion (Table 3, Figure S3).
Genome structure analysis was conducted using homology-based prediction, de novo prediction, and RNA-seq-based prediction. For homology-based prediction, the amino acid sequences from Aotus nancymaae, Callithrix jacchus, Carlito syrichta, Ochotona princeps, and Ochotona curzoniae were aligned to the L. oiostolus assembly by using Exonerate v2.4.021. For de novo gene prediction, Augustus v3.3.222,23, Genscan v1.024, and GlimmerHMM v3.0.425 were used to predict coding regions in the genome with internal gene models. We used Stringtie v2.1.126 to align assembled transcripts to the L. oiostolus genomic sequence and then TransDecoder v5.1.024 from the Trinity package to identify likely open reading frames within the transcripts. Finally, the gene sets obtained from the predictions of various methods were integrated using Maker v2.31.1025. After the characterization of repeat sequences, we used Maker v2.31.1025 to predict a consistent set of genes with 24,410 total genes annotated. Overall, 251 complete BUSCOs (98.4%), including 239 single-copy (93.7%) and 12 duplicate BUSCOs (4.7%), and one fragmented BUSCO (0.4%) were identified in the gene annotations (Table 4). Three genes (1.2%) were recognized as missing BUSCOs in our genome (Table 4).
Functional annotation of the predicted genes in the L. oiostolus genome was performed by alignment to the UniProt, Pfam, GO, KEGG, KEGG pathway, Interproscan, and NR databases using Diamond BLASTP v2.11.0 (parameter -evalue 1e-5)27 and KOBAS v3.028. Motifs, conserved sequences, and domains were annotated using InterProScan v5.33-72.029 and hmmscan v3.1 (parameter e-value 0.01)30. A total of 22,295 protein-encoding genes were functionally annotated in the L. oiostolus genome (Table S7).
Identification of non-coding RNA genes
Noncoding RNAs, including small nuclear RNAs (snRNA) and microRNAs (miRNA), were identified using INFERNAL v1.1.231 based on the Pfam database32. Predictions of tRNAs were generated using tRNAscan-SE v1.2333. The rRNAs and their subunits were predicted using RNAmmer v1.234. 463 miRNAs, 2,293 snRNAs, 1,053 tRNAs, and 541 rRNAs were predicted in the L. oiostolus genome (Table S8).
Data Records
The assembled genome has been deposited at DDBJ/ENA/GenBank under the accession JAWMBE00000000035. The raw reads were also deposited at Sequence Read Archive (SRA) at NCBI, respectively36, under Bioproject PRJNA1026309. Data of the gene functional annotations, and repeat annotation had been deposited at figshare37.
Technical Validation
Code availability
All software used in this work is in the public domain, with parameters being clearly described in Methods. If no detail parameters were mentioned for a software, default parameters were used as suggested by developer.
References
Eid, J. et al. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138 (2009).
Jain, M. et al. Improved data analysis for the MinION nanopore sequencer. Nature Methods 12, 351–356 (2015).
Zheng, G. X. Y. et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nature Biotechnology 34, 303–311 (2016).
Lam, E. T. et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nature Biotechnology 30, 771–776 (2012).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Bickhart, D. M. et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nature Genetics 49, 643–650 (2017).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Qiu, Q. et al. The yak genome and adaptation to life at high altitude. Nature Genetics 44, 946–949 (2012).
Zhang, S. et al. Structural variants selected during Yak domestication inferred from long-read whole-genome sequencing. Mol Biol Evol 38, 3676–3680 (2021).
Liu, X. et al. Evolutionary origin of genomic structural variations in domestic yaks. Nature Communications 14, 5617 (2023).
Kao, Y. & Feng, T. On the subspecies of the Chinese greytailed hare, Lepus oiostolus Hodgson. Acta Zootaxonomica Sinica 1, 19–30 (1964).
Wang, Y., Luo, Z. & Feng, Z. Taxonomic revision of Yunnan Hare, Lepus comus G.Allen with description of two new subspecies. Zoological research 6, 101 (1985).
Liu, K., Yang, J. & Yuan, H. Recent progress in research on the gut microbiota and highland adaptation on the Qinghai-Tibet Plateau. J Evol Biol 34, 1514–1530 (2021).
Belton, J. M. et al. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Xu, T., Yang, Z., Jiang, L., Xing, X. & Zuo, X. A connectome computation system for discovery science of brain. Science Bulletin 60, 86–95 (2015).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE 9, e112963 (2014).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Tempel, S. in Mobile Genetic Elements: Protocols and Genomic Applications (ed Yves Bigot) 29-51 (Humana Press, 2012).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Stanke, M. et al. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–W439 (2006).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8, 1494–1512 (2013).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome research 18, 188–196 (2008).
Shumate, A., Wong, B., Pertea, G. & Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLOS Computational Biology 18, e1009730 (2022).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinformatics 10, 421 (2009).
Bu, D. et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Research 49, W317–W325 (2021).
Jones, P. et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Finn, R. D. et al. HMMER web server: 2015 update. Nucleic Acids Research 43, W30–W38 (2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Kalvari, I. et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Research 49, D192–D200 (2020).
Chan, P. P. & Lowe, T. M. in Gene Prediction: Methods and Protocols (ed Martin Kollmar) 1-14 (Springer New York, 2019).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research 35, 3100–3108 (2007).
Feng, S. Chromosome-scale genome assembly of Lepus oiostolus (Lepus, Leporidae). GenBank https://identifiers.org/ncbi/insdc:JAWMBE000000000 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP470953 (2023).
Feng, S. Chromosome-scale genome assembly of Lepus oiostolus (Lepus, Leporidae). figshare https://doi.org/10.6084/m9.figshare.24303109.v1 (2023).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of Eukaryotic, Prokaryotic, and Viral genomes. Mol Biol Evol 38, 4647–4654 (2021).
Acknowledgements
We thank Dr. Daniel Petticord at the University of Cornell for his assistance with English language and grammatical editing of the manuscript. This work was supported by the Project of Qinghai Science & Technology Department (2024-SF-102).
Author information
Authors and Affiliations
Contributions
S.F. and Y.Y.Z conceived and designed the experiments; S.F., Y.Y.Z., Z.T.H., E.N.X. and D.F.R. conducted the experiments; Y.Y.Z., Z.T.H., E.N.X., D.F.R., J.L. and Y.Z.Y. performed the data; S.F. and Y.Y.Z. wrote the paper; and all authors read, edited and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Feng, S., Zhang, Y., He, Z. et al. Chromosome-scale genome assembly of Lepus oiostolus (Lepus, Leporidae). Sci Data 11, 183 (2024). https://doi.org/10.1038/s41597-024-03024-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03024-6
