
Abstract
Hippophae gyantsensis, which is a native tree species in China, is ideal for windbreak and sand-fixing forests. It is an economically and ecologically valuable tree species distributed exclusively in the Qinghai-Tibet Plateau in China. In our study, we assembled a chromosome-level genome of H. gyantsensis using Illumina sequencing, Nanopore sequencing and chromosome structure capture technique. The genome was 716.32 Mb in size with scaffold N50 length of 64.84 Mb. A total of 716.25 Mb genome data was anchored and orientated onto 12 chromosomes with a mounting rate of up to 99.99%. Additionally, the genome was found to comprise approximately 56.84% repeat sequences, of which long terminal repeats(LTRs) that accounted for 33.19% of the entire genome. Meanwhile, a total of 32,316 protein-coding genes were predicted, and 91.07% of these genes were functionally annotated. We also completed a series of comparative genomic analyses to provide researchers with useful reference material for future studies on seabuckthorn.
Similar content being viewed by others

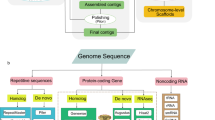
Background & Summary
Hippophae gyantsensis, which is a small tree of the family Elaeagnaceae, is an endemic species in the Qinghai-Tibet Plateau in China1. It is mainly distributed in the western part of the Qinghai-Tibet Plateau at an altitude range of 3,500–5,000 m2. Seabuckthorn plants have good drought resistance, barren resistance, saline-alkali resistance and cold resistance3,4. In addition, seabuckthorn has fast growth rate, strong reproduction and nitrogen fixation ability5. It is an excellent native tree species for windbreak, sand fixation and afforestation in Tibet and other places6. All seabuckthorn are rich in active substances, such as seabuckthorn flavonoids, vitamins, etc., they have great nutritional value7,8,9. Moreover, due to its unique geographical distribution and likely hybrid origin, H. gyantsensis is also an excellent species for studying the systematic geography of the Qinghai-Tibet Plateau10.
So far, the research on H. gyantsensis that have been conducted to date have mainly focused on its drought resistance, morphological characteristics, ecological distribution, and origin. Although the H. gyantsensis chloroplast genome has been published11 and phylogenetic trees have been constructed using partial nuclear sequences and chloroplast sequences12,13,14,15,16, because of a lack of whole-genome sequences, the genetic and evolutionary relationships of H. gyantsensis are still unclear17,18,19,20,21. For example, there is some controversy regarding whether H. gyantsensis is a hybrid of Hippophae rhamnoides Linn. subsp. yunnanensis Rousi and Hippophae neurocarpa or an independent species2. So a chromosome-level H. gyantsensis genome sequence will be a useful resource for research on the inheritance of H. gyantsensis and the genetic relationships of seabuckthorn. In this study, we assembled the H. gyantsensis whole-genome sequence and compared it with the genomes of 13 other representative plants.
Methods
Sample collection and genomic DNA sequencing
The H. gyantsensis plant used for the de novo genome assembly was collected in Jiangzi, Xizang province, China. High-molecular weight genomic DNA was extracted from the leaves according to the cetyltrimethylammonium bromide (CTAB) method22. Libraries were constructed by MGIEasy Universal DNA Library Prep Kit V1.0 (CAT#1000005250, MGI) following the standard protocol. The qualified libraries were sequenced on DNBSEQ-T7RS platform in GrandOmics Biosciences Co., Ltd. (Wuhan, China). Fastp v0.23.223 was used to filter the raw Illumina sequencing data. ONT regular DNA were extracted using the Grandomics Genomic DNA Kit following the manufacturer’s guidelines. The quality of the extracted DNA was assessed and then long DNA fragments were recovered from the high-quality samples using the Blue Pippin system (Sage Science, Beverly, MA) by gel cutting. Then the Pippin HT system (Sage Science, USA) was employed to extract the size-selected long DNA fragments. After completing the damage repair and end repair steps, the 3’ ends of the recovered long DNA fragments were modified via the addition of A. The DNA was purified using magnetic beads prior to the ligation of a sequencing adapter using an SQK-LSK110 kit. Finally, the constructed DNA library was accurately quantified using a Qubit ® 3.0 fluorometer and added to the sequencing buffer. The solution was thoroughly mixed and then added to the Flow cell, which was transferred to the primed Nanopore PromethION sequencer for sequencing. A total of 55.86 Gb clean Illumina short-read data and 102.9 Gb Nanopore read data were generated.
Hi-C sequencing
For chromosome conformation capture (Hi-C) sequencing, genomic DNA was extracted for the Hi-C library from H. gyantsensis, we constructed the Hi-C library and obtained sequencing data via the DNBSEQ-T7RS platform. Firstly, Cells were treated with 2% final concentration fresh formaldehyde to induce the formation of crosslinks (DNA–protein and protein–protein), after which the cells were lysed and samples were extracted and assessed in terms of quality. The high-quality samples were used for the ‘Hi-C fragment’ preparation process. Specifically, a restriction endonuclease was used to digest chromatin. The effect of this digestion was determined. After the biotin labeling, blunt end ligation, and DNA purification steps, the Hi-C samples were prepared. The DNA quality was evaluated and the high-quality DNA was retained. A standard library construction procedure was completed, which was followed by the optimization of PCR conditions and amplification. The amplified products were sampled for the ‘Hi-C fragment connection point quality control test’ before the construction of the sequencing library was completed. The library that passed the quality control step was sequenced using the DNBSEQ-T7RS platform. Finally, 183 Gb Hi-C data was generated.
Transcriptome sequencing
After extracting the total RNA from samples in CTAB-LiCI method22, the eukaryotic mRNA was enriched from total RNA using Dynabeads mRNA Purification Kit (Cat#61006, Invitrogen). The mRNA was fragmented using fragmentation reagent in MGIEasy RNA Library Prep Kit V3.1 (Cat# 1000005276, MGI). The first cDNA strand was synthesized using random hexanucleotide primers and mRNA as the template, after which the second cDNA strand was synthesized by adding buffer, dNTPs, RNase H, and DNA polymerase I. Following an end repair step, a poly(A) tail and a sequencing adapter were added. The cDNA fragments were amplified by PCR and purified with MGIEasy DNA Clean beads (CAT#1000005279, MGI). The MGIEasy Circularization module (CAT # 1000005260, MGI) was used to perform thermal denaturation and circularization of the double stranded PCR products. The single stranded circle DNA (ssCirDNA) was formed as the final library for sequencing on the DNBSEQ-T7RS platform. A total of 34.23 Gb data was generated.
Chromosome-level genome assembly
We performed a k-mer analysis of the H. gyantsensis genome to select the appropriate genome assembly procedure. Jellyfish v2.3.024 was used to analyze the clean Illumina data and determine the k-mer frequency distribution. Finally, GENOMESCOPE v2.025 was used to estimate the genome size and heterozygosity according to the frequency distribution structure. The estimated genome size was 718.61 Mb, with a heterozygosity of 1.28% (Fig. 1a).

Chromosome-level genome assembly of the H. gyantsensis (a) K-mer analysis of H. gyantsensis. (b) Hi-C interaction heatmap for the H. gyantsensis genome. (c) Distribution of LTR -RTs insertion times for H. gyantsensis.
We combined the clean Illumina and Nanopore reads for the de novo genome assembly. Specifically, NextDenovo v2.4.0 (https://github.com/Nextomics/NextDenovo) was used to initially assemble the Nanopore data and then NextPolish v1.4.126 and the filtered Illumina data were used to correct the assembled structure. PurgeDup v1.2.527 was used to eliminate redundancy and generate the final haploid assembly. Then we used the ragtag28 script to rearrange the chromosomes of the previous assembly results with the H. rhamnoides genome as a reference. Finally, Juicer was used to align the Hi-C reads to the assembled draft genome and then the default parameters of 3D-DNA29 were used to map the contigs to the chromosome-level scaffolds (Fig. 1b). Chromosome identification numbers and orientations were refined according to the previously published H. rhamnoides genome30 and named chr1 to chr12. We assembled a chromosome-level H. gyantsensis genome with a total scaffold length of 716 Mb. Additionally, its 12 chromosomes accounted for 99.99% of the total length. The contig N50 of the genome assembly was 23 Mb and the scaffold N50 was 64 Mb. We evaluated the genome quality using Benchmarking Universal Single-Copy Orthologs (BUSCO v5.4.5)31. The results indicated the genome was 98.8% complete. Furthermore, 1,614 expected embryophyta genes were identified (Table 1).
Repeat and gene annotation
Repeats were annotated using Extensive de novo TE Annotator (EDTA v2.1.2)32, which is a comprehensive tool that integrates multiple prediction tools. After obtaining the TE library annotated by EDTA, TEsorter v1.333 was used to reclassify ‘LTR-unknown’ and then deepTE was used to classify test.tesorter.unk. Finally, we combined the three obtained TE databases and determined that 56.84% of the H. gyantsensis genome consisted of repetitive elements, which were primarily long terminal repeats (LTRs) (33.19%) and terminal inverted repeats (TIRs) (15.71%) (Table 2). We also analyzed the timing of the LTR insertions in the H. gyantsensis genome. A total of 2,481 full-length Long terminal repeat-retrotransposons (LTR -RTs) were inserted over a period of approximately 1 million years (Fig. 1c).
Gene annotation
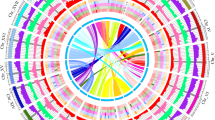
To annotate genes, we first used RepeatMasker v4.1.2-pl34 and the TE library to mask the whole-genome sequence. Gene annotations were performed by integrating evidence from homology-, De novo- and transcriptome-based information. Braker v3.0335 were employed for the gene structure annotation.Then the annotation results as the input of Maker236 for an additional annotation to obtain a higher quality model. A total of 32,316 genes were predicted, with an average gene length of 3,541 bp. TBtools37 was used to visualize the gene density, GC content, Gypsy density, Copia density, and chromosomal synteny of 12 chromosomes (Fig. 2).

Circos plot of the genomic landscape of H. gyantsensis. (a) Gene density. (b) repeat sequences density. (c) Gypsy density. (d) Copia density. (e) GC content. (f) Interspecies collinearity.
Genes were functionally annotated in two ways. First, DIAMOND v2.0.1538 was used to align protein sequences with the sequences in the NCBI non-redundant protein (NR) and Swiss-Prot databases39 Second, eggNOG-mapper40 was used to annotate protein sequences according to the Kyoto Encyclopedia of Genes and Genomes (KEGG)41 Gene Ontology (GO)42 and Pfam databases. Thus, 91.07% of the predicted genes were annotated on the basis of at least one of these databases (Table 3).
Gene family evolution analysis
Gene families were analyzed using protein sequences from H. gyantsensis and 13 other plant species (H. rhamnoides, Amborella trichopoda, Arabidopsis thaliana, Cannabis sativa, Hippophae tibetana, Morus notabilis, Oryza sativa, Populus trichocarpa, Fragaria daltoniana, Rhamnella rubrinervis, Vitis vinifera, Ziziphus jujuba, and Prunus persica). The default parameters of OrthoFinder v2.5.443 were used to identify orthogroups in the 14 species. A total of 416,260 genes from the 14 species were classified into 27,387 gene families (Fig. 3a). There were no significant differences in the number of gene families among the three Hippophae species. However, these three species had more gene families than the other analyzed species, with the exception of V. vinifera. The results of the analysis of the gene families in the three seabuckthorn species were visualized using a Venn diagram44 Fig. 3b). These three species shared 12,527 homologous gene families, but 517 gene families were unique to H. gyantsensis.

Gene family evolution analysis (a). Clusters of gene families from H. gyantsensis and other species. (b) Venn diagram of OGs shared by the three seabuckthorn species. (c) Divergence time tree of Hippophae gyantsensis and 13 other species. The number of expanded gene families (red) and the number of contracted gene families (blue) are indicated to the right of each species branch The yellow/red/blue circles and the corresponding numbers reflect the expansion (remain or gain) or contraction (loss) of gene families in specific species.
The 596 single-copy genes that were identified in the 14 species using OrthoFinder were used to construct phylogenetic trees. The single-copy homologous gene coding sequences were extracted using the seqkit tool45 and aligned with MUSCLE v3.8.3146 Next, the seqkit tool was used to connect the sequences to form supergenes, after which trimAL v1.4.rev1547-gt 0.6 -cons 60) was used to trim the supergene sequences. The trimmed sequences were used to construct the phylogenetic tree according to the maximum likelihood method using RAxML48
The MCMCtree program in the PAML v4.10.6 package49 as used to estimate the divergence time for each node in the phylogenetic tree. Divergence times in the TimeTree database50(http://www.timetree.org) were used as calibration time-points, including the divergence times for V. vinifera and P. persica [109.8–122.4 million years ago (mya)], F. daltoniana and P. persica (49.1–77.1 mya), and C. sativa and M. notabilis (48.9–70.9 mya). The phylogenetic tree with divergence times and the sorted gene family results were used as the input to construct the phylogenetic tree with gene family expansion and contraction information using the CAFÉ V5 program51 According to the constructed phylogenetic tree (Fig. 3c), H. gyantsensis (family: Elaeagnaceae) diverged from Z. jujuba (family: Rhamnaceae) and M. notabilis (family: Moraceae) approximately 78.01 mya. Notably, the phylogenetic tree indicated that H. gyantsensis diverged first among the three Hippophae species. Moreover, H. tibetana and H. rhamnoides are more closely related to each other than to H. gyantsensis. Extensive gene family expansions and contractions occurred after the divergence of the genus Hippophae.
Identification of WGD events and analysis of genome collinearity
We used the WGDI toolkit (v0.6.4)52 to detect WGD events. DIAMOND v2.0.1538 was used to identify homologous genes (e-values no higher than 1e-5). The WGDI toolkit was also used to identify collinear genes (parameter: ‘-icl’). The ‘ks’ parameter in WGDI was modified to calculate the KS values, whereas the ‘bi’ and ‘c’ parameters were modified to screen the collinearity results (ks-col = ks_YN00). Finally the ‘kp’ parameter was modified to calculate the KS peak. The results were visualized using the ‘kf’ parameter (Fig. 4a). Three collinear block peaks (0.315 ± 0.001, 0.483 ± 0.002, and 2.029 ± 0.002) were detected for H. gyantsensis. Three collinear block peaks were also detected for H. rhamnoides and H. tibetana, which was consistent with the results of previous studies that suggested two lineage-specific polyploidization events occurred in the genus Hippophae within a relatively narrow timeframe 30,53.

WGD event and collinearity analysis of H. gyantsensis. (a) Distribution of the number of synonymous substitutions per synonymous site (Ks) confirming the occurrence of a whole-genome duplication (WGD) event in H. gyantsensis. (b) Whole-genome synteny between H. gyantsensis and H. rhamnoides (c) Whole-genome synteny between H. gyantsensis and H. tibetana.
In this study, MCscan (Python version)54 was used for the genome-wide collinearity analysis involving H. gyantsensis, H. tibetana, and H. rhamnoides. In addition, JCVI55 and TBtools37 were used to draw collinearity figures according to the gene collinearity between species. Chromosome 2 of H. tibetana differed significantly from the corresponding chromosomes in H. gyantsensis and H. rhamnoides (Fig. 4b,c).
Data Records
The genomic Illumina sequencing data were deposited in the Sequence Read Archive at NCBI SRR2538250554.
The genomic Nanopore sequencing data were deposited in the Sequence Read Archive at NCBI SRR2538249956 and SRR2538249857.
The transcriptome Illumina sequencing data were deposited in the Sequence Read Archive at NCBI SRR25382500-SRR2538250358,59,60,61. The Hi-C sequencing data were deposited in the Sequence Read Archive at NCBI SRR2538250462. The final chromosome assembly were deposited in the GenBank at NCBI JAUQSU00000000063.
The final gene structure annotation, repeat annotation, and gene functional prediction were deposited in the Figshare database64.
Technical Validation
DNA quantifcation and qualification
For all sequencing samples, whether DNA samples or RNA samples, we performed sample quality testing, the detailed steps have been mentioned in the Method section
Assessment of genome assemblies
In addition to using Benchmarking Universal Single-Copy to evaluate genome quality(mentioned in method section), we used BWA-MEM2 v2.2.165 to align Illumina short reads to the H. gyantsensis genome to evaluate the accuracy of the final genome assembly. Analysis showed that 97.84% of the short reads were successfully mapped to the H. gyantsensis genome. We further assessed the base quality of genome assembly by estimating the quality value score (QVS) using Inspector version 1.0.166, which showed a high QVS of 33.18. These fndings indicate that the quality of our assembled genome is high. In addition, the LAI package inside the LTR_retriever v.2.9.067 is used to evaluate the LTR Assembly Index, and the result file of EDTA is used as input. The results showed that the LAI value of the genome was 11.7. In summary, the genome can provide a good reference for subsequent work.
Code availability
nextDenovo: input_type = raw, read_type = ont, read_cutoff = 1k, seed_cutoff = 34747, sort_options = -m 20 g -t 14, minimap2_options_raw = -t 14, pa_correction = 8, correction_options = -p 14, minimap2_options_cns = -t 14, minimap2_options_map = -t 14, nextgraph_options = -a 1
NextPolish: sgs_options = -max_depth 100 -bwa, lgs_options = -min_read_len 1k -max_depth 100, lgs_minimap2_options = -x map-ont
TEsorter: -db rexdb-plant
Repeatmasker: -pa 14 -s -xsmall
Blastp: E-value ≤ 1e-5
Swiss-Prot: E-value ≤ 1e−5
Nr: E-value ≤ 1e−5
Orthofinder: -S diamond -M msa -T fasttree
trimAl: -gt 0.6 -cons 60
RAxML: raxmlHPC-PTHREADS -m PROTGAMMAJTT -f a -p 123 -x 123 -# 100
Wgdi: pvalue = 0.05
Other commands and pipelines used in data processing were executed using their corresponding default parameters.
References
Lian, Y. S. New discoveries of the genus Hippophae L. (Elaeagnaceae). J. Syst. Evol. 26, 235–237 (1988).
Jia, D. R. et al. Diploid hybrid origin of Hippophaë gyantsensis (Elaeagnaceae) in the western Qinghai–Tibet Plateau. Biol. J. Linn. Soc. 117, 658–671 (2016).
He, C. Y., Zhang, G. Y., Zhang, J. G., Duan, A. G. & Luo, H. M. Physiological, biochemical, and proteome profiling reveals key pathways underlying the drought stress responses of Hippophae rhamnoides. Proteomics 16, 2688–2697, https://doi.org/10.1002/pmic.201600160 (2016).
Stobdan, T., Angchuk, D. & Singh, S. B. Seabuckthorn: an emerging storehouse for researchers in India. Curr. Sci. India 94, 1236–1237 (2008).
Zhou, X., Tian, L., Zhang, J. F., Ma, L. & Li, X. J. Rhizospheric fungi and their link with the nitrogen-fixing Frankia harbored in host plant Hippophae rhamnoides L. J. Basic Microbiol. 57, 1055–1064, https://doi.org/10.1002/jobm.201700312 (2017).
Nan, J. B., Yang, G. H., Wu, T. Y. & Lin, L. Comparative study on drought resistance of three species of Hippophae in Tibet. J. Northwest A&F Univ. (in Chinese) 49, 37–47 (2021).
Suryakumar, G. & Gupta, A. Medicinal and therapeutic potential of sea buckthorn (Hippophae rhamnoides L.). J. Ethnopharmacol. 138, 268–278, https://doi.org/10.1016/j.jep.2011.09.024 (2011).
Dhyani, D., Maikhuri, R. K. & Dhyani, S. Seabuckthorn: an underutilized resource for the nutritional security and livelihood improvement of rural communities in Uttarakhand Himalaya. Ecol. Food Nut.r 50, 168–180, https://doi.org/10.1080/03670244.2011.552375 (2011).
Qian, X. S. & Jin, J. H. Medical research and development of sea-buckthorn. Chin. Wild Plant Res. 34, 68–72 (2015).
Xu, T. et al. Climate heterogeneity shapes phylogeographic pattern of Hippophae gyantsensis (Elaeagnaceae) in the east Himalaya-Hengduan Mountains. Ecol. Evol. 13, e10182–e10182, https://doi.org/10.1002/ece3.10182 (2023).
Wang, L., Wang, J., He, C., Zhang, J. & Zeng, Y. Characterization and comparison of chloroplast genomes from two sympatric Hippophae species (Elaeagnaceae). J. Forestry Res. 32, 307–318, https://doi.org/10.1007/s11676-019-01079-5 (2021).
Wang, L. Y. et al. Genomic SSR and EST-SSR markers for phylogenetic and pedigree reconstructions—A comparison in sea buckthorn. Plant Breed. 140, 167–183, https://doi.org/10.1111/pbr.12889 (2021).
Jia, D. R. & Bartish, I. V. Climatic changes and orogeneses in the late miocene of Eurasia: the main triggers of an expansion at a continental scale? Front. Plant Sci. 9, 1400, https://doi.org/10.3389/fpls.2018.01400 (2018).
Sun, K. et al. Molecular phylogenetics of Hippophae L. (Elaeagnaceae) based on the internal transcribed spacer (ITS) sequences of nrDNA. Plant Syst. Evol. 235, 121–134, https://doi.org/10.1007/s00606-002-0206-0 (2002).
Bartish, I. V., Jeppsson, N., Nybom, H. & Swenson, U. Phylogeny of Hippophae (Elaeagnaceae) inferred from parsimony analysis of chloroplast DNA and morphology. Syst. Bot. 27, 41–54, https://doi.org/10.1043/0363-6445-27.1.41 (2002).
Sheng, H. M. et al. Analysis of the genetic diversity and relationships among and within species of Hippophae (Elaeagnaceae) based on RAPD markers. Plant Syst. Evol. 260, 25–37, http://www.jstor.org/stable/23655536 (2006).
Swenson, U. & Bartish, I. V. Taxonomic synopsis of Hippophae (Elaeagnaceae). Nord. J. Bot. 22, 369–374, https://doi.org/10.1111/j.1756-1051.2002.tb01386.x (2002).
Rousi, A. The genus Hippophaë L. A taxonomic study. Ann. Bot. Fenn. 8, 177–227 (1971).
Lian, Y. S., Chen, X. L., Sun, K. & Ma, R. J. Clarification of the systematic position of Hippophae goniocarpa (Elaeagnaceae). Bot. J. Linnean Soc. 142, 425–430, https://doi.org/10.1046/j.1095-8339.2003.00184.x (2003).
Hyvönen, J. On phylogeny of Hippophae (Elaeagnaceae). Nord. J. Bot. 16, 51–62, https://doi.org/10.1111/j.1756-1051.1996.tb00214.x (1996).
Bartish, I. V. & Thakur, R. In The seabuckthorn genome (ed P C. Sharma) 23-66 (Springer International Publishing, 2022).
Pahlich, E. & Gerlitz, C. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemistry 19, 11–13 (1980).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. Fastp: an ultra-fast all-in-one fastq preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Hu, J., Fan, J. P., Sun, Z. Y. & Liu, S. L. Nextpolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2020).
Guan, D. F. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Alonge, M. et al. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 20, 224, https://doi.org/10.1186/s13059-019-1829-6 (2019).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://www.science.org/doi/10.1126/science.aal3327 (2017).
Wu, Z. F. et al. The genome of Hippophae rhamnoides provides insights into a conserved molecular mechanism in actinorhizal and rhizobial symbiosis. New Phytol. 235, 276–291, https://doi.org/10.1111/nph.18017 (2022).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245, https://doi.org/10.1007/978-1-4939-9173-0_14 (2019).
Ou, S. J. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275, https://doi.org/10.1186/s13059-019-1905-y (2019).
Zhang, R. Z. et al. TEsorter: an accurate and fast method to classify LTR-retrotransposons in plant genomes. Hortic. Res. 9, uhac017, https://doi.org/10.1093/hr/uhac017 (2022).
Tempel, S. Using and understanding RepeatMasker. Methods Mol. Biol. 859, 29–51, https://doi.org/10.1007/978-1-61779-603-6_2 (2012).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. Whole-genome annotation with Braker. Methods Mol. Biol. 1962, 65–95, https://doi.org/10.1007/978-1-4939-9173-0_5 (2019).
Holt, C. & Yandell, M. Maker2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491, https://doi.org/10.1186/1471-2105-12-491 (2011).
Chen, C. J. et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 13, 1194–1202, https://doi.org/10.1016/j.molp.2020.06.009 (2020).
Buchfink, B., Xie, C. & Huson, H. D. Fast and sensitive protein alignment using diamond. Nat. Methods. 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370, https://doi.org/10.1093/nar/gkg095 (2003).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. EggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Kotera, M., Hirakawa, M., Tokimatsu, T., Coto, S. & Kanehisa, M. The KEGG databases and tools facilitating omics analysis: latest developments involving human diseases and pharmaceuticals. Methods Mol. Biol. 802, 19–39, https://doi.org/10.1007/978-1-61779-400-1_2 (2012).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29, https://doi.org/10.1038/75556 (2000).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157, https://doi.org/10.1186/s13059-015-0721-2 (2015).
Chen, T., Zhang, H. Y., Liu, Y., Liu, Y. X. & Huang, L. Q. Evenn: easy to create repeatable and editable venn diagrams and venn networks online. J. Genet. Genomics 48, 863–866, https://doi.org/10.1016/j.jgg.2021.07.007 (2021).
Shen, W., Le, S., Li, Y. & Hu, F. Q. SeqKit: s cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11, 10, https://doi.org/10.1371/journal.pone.0163962 (2017).
Edgar, R. C. Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797, https://doi.org/10.1093/nar/gkh340 (2004).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973, https://doi.org/10.1093/bioinformatics/btp348 (2009).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57, 758–771, https://doi.org/10.1080/10635150802429642 (2008).
Ynag, Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556, https://doi.org/10.1093/bioinformatics/13.5.555 (1997).
Kumar, S., Stecher, G., Suleski, M. & Hedges, B. S. TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819, https://doi.org/10.1093/molbev/msx116 (2017).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. Café 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518, https://doi.org/10.1093/bioinformatics/btaa1022 (2020).
Sun, P. C. et al. WGDI: a user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant. 15, 1841–1851, https://doi.org/10.1016/j.molp.2022.10.018 (2022).
Wang, R. Q. et al. How to survive in the world’s third poplar: insights from the genome of the highest altitude woody plant, Hippophae tibetana (Elaeagnaceae). Front. Plant Sci. 13, https://doi.org/10.3389/fpls.2022.1051587 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25382505 (2023).
Tang, H. B. et al. Synteny and collinearity in plant genomes. Science 320, 486–488, https://doi.org/10.1126/science.1153917 (2008).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25382499 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25382498 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25382500 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25382501 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25382502 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25382503 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25382504 (2023).
Chen, M., Yang, Y. & Yang, Y. Hippophae gyantsensis isolate higy01, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAUQSU010000000 (2023).
Xingyu, Y. Chromosome-level genome assembly of Hippophae gyantsensis. figshare. journal contribution. Figshare https://doi.org/10.6084/m9.figshare.23820921.v3 (2023).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. https://doi.org/10.48550/arXiv.1303.3997 (2013)
Chen, Y. et al. Accurate long-read de novo assembly evaluation with Inspector. Genome Biol 22, 312, https://doi.org/10.1186/s13059-021-02527-4 (2021).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res 46, e126, https://doi.org/10.1093/nar/gky730 (2018).
Acknowledgements
This research was supported by Regional Science and Technology Collaborative Innovation Project of Shigatse Bureau of Science and Technology(QYXTZX-RKZ2021–07), the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502), Yunling Scholar Project to Yang Yongping, Regional Science and Technology Collaborative Innovation Project of Shigatse Bureau of Science and Technology(QYXTZX-RKZ2022–01), the Major Program of National Natural Science Foundation of China (31590820, 31590823), the National Natural Science Foundation of China (31601999 and 41771123), and the 13th Five-year Informatization Plan of Chinese Academy of Sciences,Grant No. XXH13506.
Author information
Authors and Affiliations
Contributions
Designed the experiments: Y.Q.Y. and Y.P.Y. Performed the experiments: M.Y.C., D.N.Y. and S.H.Y. Analyzed the data: M.Y.C., D.N.Y., X.Y.Y., Z.Y.C. and T.Y.Y. Contributed reagents/materials/analysis tools: X.Y.Y., Z.Y.C. and T.Y.Y. Wrote the paper: M.Y.C. and D.N.Y. All Authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, M., Yang, D., Yang, S. et al. Chromosome-level genome assembly of Hippophae gyantsensis. Sci Data 11, 126 (2024). https://doi.org/10.1038/s41597-024-02909-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-02909-w
