
Abstract
Lithocarpus, with >320 species, is the second largest genus of Fagaceae. However, the lack of a reference genome limits the molecular biology and functional study of Lithocarpus species. Here, we report the chromosome-scale genome assembly of sweet tea (Lithocarpus polystachyus Rehder), the first Lithocarpus species to be sequenced to date. Sweet tea has a 952-Mb genome, with a 21.4-Mb contig N50 value and 98.6% complete BUSCO score. In addition, the per-base consensus accuracy and completeness of the genome were estimated at 60.6 and 81.4, respectively. Genome annotation predicted 37,396 protein-coding genes, with repetitive sequences accounting for 64.2% of the genome. The genome did not undergo whole-genome duplication after the gamma (γ) hexaploidy event. Phylogenetic analysis showed that sweet tea diverged from the genus Quercus approximately at 59 million years ago. The high-quality genome assembly and gene annotation resources enrich the genomics of sweet tea, and will facilitate functional genomic studies in sweet tea and other Fagaceae species.
Similar content being viewed by others


Background & Summary
Lithocarpus is the second largest genus of the Fagaceae family, comprising more than 320 species1. Species belonging to the genus Lithocarpus, commonly known as stone oaks, are dominant canopy trees in the tropical and subtropical forests of East Asia2. Lithocarpus polystachyus Rehder (2n = 2x = 24; syn. Lithocarpus litseifolius [Hance] Chun) is an evergreen plant widely distributed in south China and the adjacent southeast Asian countries3. The L. polystachyus is commonly known as “sweet tea”, because its leaves have a sweet taste when brewed. The leaves of L. polystachyus also have medicinal properties and have long been used as herbal tea to prevent and manage diabetes.
The leaves of L. polystachyus contain high concentration of dihydrochalcones (DHCs), which is the main source of its sweet taste. DHCs is a class of minor flavonoids (e.g. trilobatin and neohesperidin), which have been reported to act as flavor sweeteners and bitterness blockers4,5,6. DHCs such as phloretin, phlorizin, and sieboldin have also been demonstrated to play important roles in human health by providing a wide range of beneficial effects against diabetes, cardiovascular, cancer, and free radical-involving diseases7,8,9,10. In addition, phloretin exhibits strong broad-range bactericidal and fungicidal activities, and sieboldin is a powerful multipotent antioxidant9,11. DHCs have been isolated from many medicinal plants belonging to different families, but their contents vary significantly both among and within species12,13,14,15. Four DHCs (phloretin, phlorizin, trilobatin, and sieboldin) have been reported in sweet tea, and the biosynthesis pathway of the first three DHCs have been proposed14,16. However, our knowledge of DHC biosynthesis and regulatory mechanisms is limited in sweet tea. Specifically, candidate genes and transcription factors involved in the DHCs biosynthesis pathway remain to be investigated.
Here, we assembled a high-quality chromosome-scale genome of sweet tea, the first ever in the genus Lithocarpus, using PacBio HiFi and Hi-C data. The assembled sweet tea genome had a total length of 952 Mb, with a contig N50 of 21.4 Mb and a complete BUSCO score of 98.6%. A total of 922.6 Mb (96.9%) of the sequences were anchored to the 12 chromosomes. Genome annotation predicted 37,396 protein-coding genes and 597.5 Mb repetitive sequences. The high-quality sweet tea genome provides a valuable resource for exploring key genes and molecular regulatory mechanisms involved in the biosynthesis of important compounds, including DHCs, and will further serve as a strong foundation for trait improvement in this species.
Methods
Plant material and sequencing
A healthy sweet tea tree growing in the South China National Botanical Garden (accession number: 19940074) was selected for de novo genome assembly (Fig. 1). Young leaves were collected from the selected individual for whole-genome sequencing. The leave, stem, and root were collected for RNA-sequencing (RNA-seq) for the transcriptome assembly. All samples were immediately flash-frozen in liquid nitrogen after harvest, and stored at −80 °C for subsequent nucleic acid extraction.

The sweet tea tree sequenced in this study. (a) The sequenced tree planted in South China National Botanical Garden (accession number: 19940074) (b) The fruits of sequenced tree.
Genomic DNA and total RNA were isolated from the leaves using DNeasy Plant MiniKit (Qiagen, Germany) and RNAprep Pure Plus Kit (Tiangen, China), respectively. The mRNA was purified from total RNA using poly-T oligo-attached magnetic beads for subsequent sequencing. To perform short-read sequencing for genome survey, transcriptome assembly, and transcriptomic profiling, libraries with an insert size of ~350 bp was constructed and sequenced on the Illumina NovaSeq 6000 platform to generate 150-bp paired-end reads. To perform de novo genome assembly, a 15–20-kb PacBio HiFi library was prepared using the SMRTbell Express Template Preparation Kit 2.0 (Pacific Biosciences, USA) and sequenced on the PacBio Sequel IIe platform to produce PacBio HiFi long reads. To generate the high-throughput chromatin conformation capture (Hi-C) data, DNA was isolated from the leaves and fixed with paraformaldehyde. The genomic DNA was then enzymatically digested with DnpII, generating fragments with sticky ends. These sticky ends were repaired by ‘A’ or ‘C’ deoxynucleotides with biotin by using DNA polymerase. Subsequently, the DNA fragments were ligated together to form chimeric circles using DNA ligase. The ligated DNAs were then uncrosslinked, purified, and sheared into 300–500 bp in size. Finally, the Hi-C sequencing library was sequenced on the Illumina NovaSeq 6000 platform, generating 150-bp paired-end reads.
A total of 32.8 Gb PacBio HiFi long reads (~37.6 × coverage), 118.8 Gb Hi-C reads (~135.9 × coverage), 44.3 Gb (~51 × coverage) paired-end Illumina reads, and 21.5 Gb RNAseq reads were generated for the genome assembly, genome survey, and transcriptome assembly.
Genome survey
The 21-bp K-mers with Illumina reads were counted using Jellyfish v1.1.1117, with default parameters. The genome size, the level of heterozygosity, and repeat content were estimated using GenomeScope v1.018. The estimated genome was 874 Mb in length and a heterozygosity of 1.5% (Fig. 2).

K-mer (21-mer)-based estimation of genome characters of sweet tea.
De novo genome assembly
The HiFi long reads were initially assembled into contigs using hifiasm v0.18.6-r51319 with default parameter to generate one primary haplotype-collapsed assembly and a pair of partially phased assemblies. The primary assembly was selected for further scaffolding because it was much more contiguous than the partially phased assemblies. To anchor the contigs onto scaffolds, the Hi-C reads were mapped on to the contigs using Juicer v2.020 with DpnII (GATC) as the restriction enzyme site, and scaffolds were generated using the 3D-DNA (v201008) pipeline21 with the following parameters: “-m haploid -i 150000 -r 0--editor-repeat-coverage 5”. Based on the chromosome number of sweet tea (n = 12) determined previously22,23 and the interaction information of Hi-C reads generated with the 3D-DNA pipeline, the chromosome segmentation boundaries and assembly errors were manually checked and adjusted using Juicebox v1.11.0824.
The total length of the final sweet tea genome assembly was 952.3 Mb, which is slightly larger than the genome size estimated by K-mer analysis (Fig. 2 and Table 1), and smaller than the size measured by flow cytometry (ca. 1,149 Mb)22. The contig and scaffold N50 values of the sweet tea genome were 21.4 and 78.6 Mb, respectively, which are comparable with those of recently published Fagaceae genome assemblies (Table 2). A total of 922.6 Mb (96.9%) of the sequences were anchored to the 12 chromosomes (Table 1). The Hi-C interaction map showed a strong intrachromosomal interactive signal along the diagonal (Fig. 3).

Genome-wide chromatin interaction heatmap of sweet tea based on Hi-C data.
Identification and characterization of repetitive elements
Tandem duplications were identified using TRF 4.0925, and both structurally intact and fragmented transposable elements (TEs) were annotated using EDTA (Extensive de-novo TE Annotator) v1.9.626. Overlapping regions of each class of repetitive elements were counted only once when calculating their total size. The divergence (K) of intact LTRs identified was estimated by Kimura two-parameter distance (K2P)27. The insertion time was calculated by the formula: T = K/(2 × r), where r refers to a substitution rate of 1.3 × 10–8 per site per year28.
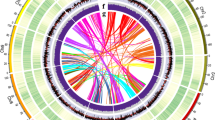
A total of 597.5 Mb (62.74%) of the assembled sequences were annotated as TEs, with LTR (33.49%), TIR (14.27%), and Helitron (10.82%) being the three most abundant TE superfamilies (Fig. 4 and Table 3). We found most of the LTRs have been accumulated recently over a short time span with the peak of 0.3 million years ago (Ma), suggesting an expansion event (Fig. 5). In addition, TEs were unevenly distributed along the genome, with greater accumulation in regions with low gene density (Fig. 4).

Genomic features of sweet tea. The tracks from the outer to the inner circle represent the 12 chromosomes (Chr1–Chr12), gene density, trnasposable element (TE; Copia, Gypsy, TIR, and Helitron) density, tandem repeat density, and GC content. The connecting lines in the center of the Circos plot indicate the syntenic gene blocks.

The distribution of insertion time of intact LTRs in sweet tea. Ma, million years ago.
Gene prediction and functional annotation
Protein-coding genes in the sweet tea repeat-masked genome were predicted by applying MAKER v3.01.0429 to the combined results of RNA-seq-based prediction, protein-homology-based prediction, and ab initio prediction. Trinity v2.14.030 was used to de novo assemble the transcriptome for RNA-seq-based gene prediction. HiSat2 v2.2.131 was used to align the RNA-seq reads against the sweet tea genome, and then Trinity and StringTie v2.2.132 were used to assemble the genome-guided transcriptomes. Then, the de novo and genome-guided transcriptome assemblies were merged and refined using CD-HIT-EST v4.8.133, generating the transcript-based gene prediction. Plant protein sequences from the Swiss-Prot database (https://www.uniprot.org/downloads) and annotated proteins from Arabidopsis thaliana Araport1134 and Populus trichocarpa v4.135 were used for protein-homology-based prediction. AUGUSTUS v3.4.036,37 with BUSCO single-copy genes as training data, GeneMark-ES v4.69_lic38 with a self-training algorithm, and SNAP v2006-07-2839 with the output of MAKER as training data were used for ab initio prediction. MAKER was run for a total of three rounds to obtain high-quality gene models.
To further improve the gene prediction, genes predicted by MAKER were integrated into consensus gene models using EVidenceModeler v1.1.140 and then further polished using PASA v2.5.241. The longest transcript of each predicated gene model was considered as the representative gene models. The completeness of gene models was assessed by searching the gene content of the embryophyta_odb10 database using BUSCO v5.4.342.
Functional annotations were performed using different tools: InterProScan v5.56-89.043 for predicting potential protein domains; KofamKOALA v1.3.044 for determining KEGG ortholog assignments; and eggNOG-mapper v2.1.945 for inferring orthology assignments. In addition, BLASTP v2.13.0 + was used to search the homologous sequences in Nr, Swiss-Prot, and TrEMBL databases. The GO (Gene ontology) terms of the genes were extracted from the eggNOG and InterPro entries. Transcription factors were annotated using PlantTFDB v5.046 and PlanTFcat47.
A total of 37,396 protein-coding genes were predicted, with 97.1% of the complete BUSCO genes covered (Table 1). The average length of genic regions, coding sequences (CDSs), and intron sequences was 4,903 bp, 1,163 bp, and 948 bp, respectively. The average length of genes and introns in sweat tea was comparable to that of Fagaceae species (Table 2). Among the predicted protein-coding genes, 36,096 (96.5%) were annotated in functional databases (Tables 1), and 3,200 were identified as transcription factor (TF) genes belonging to 99 different families.
Comparative genomic analysis
To investigate the syntenic relationships between the protein-coding genes of sweet tea and those of the other Fagaceae species, collinear blocks between sweet tea and the representative species of each genus or section were identified based on protein sequences using MCScan implemented in jcvi v1.2.748. The synonymous substitution rate (Ks) was estimated using KaKs_Calculator v3.049 based on paralogous gene pairs extracted from the collinear blocks.
The syntenic gene blocks showed 1:1 syntenic patterns between sweet tea and other Fagaceae species (Fig. 6a), suggesting a conserved genome structure in Fagaceae. The Ks distribution pattern of the sweet tea syntenic genes presented the same signature of Ks peaks only at ~1.2 as that observed in the other Fagaceae and Vitis vinifera genomes, indicating that the genome of sweet tea did not undergo WGD after the gamma (γ) hexaploidy event (Fig. 6b).

Syntenic anasis between sweet tea and other Fagaceae species. (a) Syntenic blocks between sweet tea and other Fagaceae species. (b) Distribution of the synonymous substitution (Ks) rates of paralogous gene pairs within syntenic blocks. The blue dashed line represents the 1.2 of Ks. (c) Phylogenetic tree of sweet tea and 11 other Fagaceae species. The numbers in square brackets indicate the 95% confidence intervals of the divergence time, and two fossil calibrations are indicated in blue. The red solid circle represents support lower than 100%, based on 1,000 bootstrap replicates.
Phylogenetic analyses
We conducted phylogenetic analyses to infer the relationship between sweet tea and other Fagaceae species. To do that, we utilized OrthoFinder v2.5.450 to identify orthologous genes between 12 Fagaceae species (Table 2). A total of 3,498 single-copy orthologous genes identified by OrthoFinder were aligned using MAFFT v7.50551, trimmed using trimAl v1.4.rev1552, and then concatenated together. Based on the concatenated alignment, a maximum likelihood phylogenetic tree was constructed using IQ-TREE v2.0.553,54. The divergence times among Fagaceae species were estimated using MCMCTree in the PAML v4.9j package55, based on four-fold degenerate sites. Two fossil calibrations were used to constrain the age of nodes: (1) the split between the Fagus genus and the rest of the Fagaceae species at 82–81 Ma56, and (2) the divergence between the Castanopsis and Castanea genera at 53–52 Ma57. The phylogenetic analyses showed that sweet tea was sister to the genus Quercus, with strong bootstrap support (100%). Calibration of the phylogenetic tree showed that sweet tea diverged from the genus Quercus ~59 Ma (95% HPD: 41.05–77.38 Ma) (Fig. 6c).
Data Records
The genome assembly have been deposited in the GenBank database of NCBI with accession number JAWTZU00000000058. The raw sequence data have been deposited in the Genome Sequence Archive (GSA) in National Genomics Data Center (NGDC) database (https://ngdc.cncb.ac.cn/) under the accession number CRA01239759. The genome annotation files and synteny data were deposited in the Figshare database60.
Technical Validation
Genome completeness was assessed by searching the gene content of the embryophyta_odb10 database with Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.4.342; the quality of repetitive genomic regions was assessed using the LAI vbeta3.2 program (Ou et al., 2018); per-base consensus accuracy (QV) and k-mer completeness was estimated with Merqury v1.361 using PacBio HiFi long reads with a K-mer value of 20-bp; and PacBio HiFi long reads were mapped on to the genome using minimap2 v2.24-r112262 to calculate the mapping rate. The telomeric sequences in the sweet tea genome assembly were identified using quartet v1.1.363 with “-c plant”.
The sweet tea genome assembly showed a high degree of completeness and accuracy at the chromosome scale as indicated by the following statistics (Table 1): (1) the complete BUSCO score was 98.6%, which suggests high-gene space completeness of the assembly; (2) the LTR Assembly Index (LAI) was 21.5 for the assembly, which suggested it was a high quality genome64; (3) the values of QV and k-mer completeness were estimated as 60.6 and 81.4, respectively, based on the analysis of 20-mer spectra from the PacBio HiFi long reads; (4) 99.88% of PacBio HiFi long reads were aligned to the sweet tea genome assembly, with an average coverage depth of ~32 × along the 12 chromosomes (Fig. 7); and (5) telomeric sequences were detected at both ends of two chromosomes (Chr5 and Chr10).

Coverage of HiFi long reads mapped across the 12 chromosomes of sweet tea.
Code availability
No custom code was used for this study. All data analyses were conducted using published bioinformatics software with default settings, unless otherwise specified.
References
Huang, C., Zhang, Y., Bartholomew, B., Wu, Z. & Raven, P. Flora of China: Cycadaceae through Fagaceae (Science Press, Missouri Botanical Garden, Beijing, St Louis, MO, 2000).
Chen, X., Kohyama, T. S. & Cannon, C. H. Associated morphometric and geospatial differentiation among 98 species of stone oaks (Lithocarpus). PLoS One 13, e0199538 (2018).
Cheng, J. et al. Population structure and genetic diversity of Lithocarpus litseifolius (Fagaceae) assessed using microsatellite markers. Nord. J. Bot. 34, 752–760 (2016).
Tomás-Barberán, F. A., Borrego, F., Ferreres, F. & Lindley, M. G. Stability of the intense sweetener neohesperidine dihydrochalcone in blackcurrant jams. Food Chem. 52, 263–265 (1995).
Tomás-Barberán, F. A. & Clifford, M. N. Flavanones, chalcones and dihydrochalcones – nature, occurrence and dietary burden. J. Sci. Food Agric. 80, 1073–1080 (2000).
Wang, Y. et al. Biosynthesis of the dihydrochalcone sweetener trilobatin requires phloretin glycosyltransferase2. Plant Physiol. 184, 738–752 (2020).
Figtree, G. A. et al. Plant-derived estrogens relax coronary arteries in vitro by a calcium antagonistic mechanism. J. Am. Coll. Cardiol. 35, 1977–1985 (2000).
Ehrenkranz, J. R. L., Lewis, N. G., Ronald Kahn, C. & Roth, J. Phlorizin: A review. Diabetes Metab. Res. Rev. 21, 31–38 (2005).
Dugé de Bernonville, T. et al. Dihydrochalcones: Implication in resistance to oxidative stress and bioactivities against advanced glycation end-products and vasoconstriction. Phytochemistry 71, 443–452 (2010).
Szliszka, E., Czuba, Z. P., Mazur, B., Paradysz, A. & Krol, W. Chalcones and dihydrochalcones augment TRAIL-mediated apoptosis in prostate cancer cells. Molecules 15, 5336–5353 (2010).
Gaucher, M. et al. Histolocalization and physico-chemical characterization of dihydrochalcones: Insight into the role of apple major flavonoids. Phytochemistry 90, 78–89 (2013).
Gutierrez, B. L., Arro, J., Zhong, G.-Y. & Brown, S. K. Linkage and association analysis of dihydrochalcones phloridzin, sieboldin, and trilobatin in Malus. Tree Genet. Genomes 14, 91 (2018).
Yang, J., Huang, Y., Yang, Z., Zhou, C. & Hu, X. Identification and quantitative evaluation of major sweet ingredients in sweet tea (Lithocarpus polystachyus Rehd.) based upon location, harvesting time, leaf age. J. Chem. Soc. Pak. 40, 158–164 (2018).
Wang, Y.-K. et al. Dihydrochalcones in sweet tea: Biosynthesis, distribution and neuroprotection function. Molecules 27, 8794 (2022).
Rivière, C. in Studies in Natural Products Chemistry Vol. 51 (ed Rahman, A. U.) Ch. 7, 253–381 (Elsevier: Amsterdam, Netherlands, 2016).
Lou, W., Mu, X., Liu, J., Xun, M. & Hu, Y. Study on the differences of metabolites and their bioactivities of Lithocarpus under different processing methods. Food Biosci. 54, 102817 (2023).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Chen, S.-C., Cannon, C. H., Kua, C.-S., Liu, J.-J. & Galbraith, D. W. Genome size variation in the Fagaceae and its implications for trees. Tree Genet. Genomes 10, 977–988 (2014).
Chokchaichamnankit, P., Chulalaksananukul, W., Phengklai, C. & Anamthawat-Jonsson, K. Karyotypes of some species of Castanopsis, Lithocarpus and Quercus (Fagaceae) from Khun Mae Kuang Forest in Chiang Mai province, northern Thailand. Thai For. Bull. (Bot.) 0, 38–44 (2014).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Ma, J., Devos, K. M. & Bennetzen, J. L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 14, 860–869 (2004).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Cheng, C.-Y. et al. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 89, 789–804 (2017).
Tuskan, G. A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604 (2006).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Keller, O., Kollmar, M., Stanke, M. & Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763 (2011).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506 (2005).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354 (2021).
Aramaki, T. et al. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. Eggnog-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Tian, F., Yang, D.-C., Meng, Y.-Q., Jin, J. & Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 48, D1104–D1113 (2020).
Dai, X., Sinharoy, S., Udvardi, M. & Zhao, P. X. PlantTFcat: An online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinform. 14, 321 (2013).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Zhang, Z. KaKs_Calculator 3.0: Calculating selective pressure on coding and non-coding sequences. Genom. Proteom. Bioinform. 20, 536–540 (2022).
Emms, D. M. & Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Grímsson, F., Grimm, G. W., Zetter, R. & Denk, T. Cretaceous and Paleogene Fagaceae from North America and Greenland: Evidence for a late Cretaceous split between Fagus and the remaining Fagaceae. Acta Palaeobotanica 56, 247–305 (2016).
Wilf, P., Nixon, K. C., Gandolfo, M. A. & Cúneo, N. R. Eocene Fagaceae from Patagonia and Gondwanan legacy in Asian rainforests. Science 364, eaaw5139 (2019).
NCBI GenBank, https://identifiers.org/ncbi/insdc:JAWTZU000000000 (2023).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA012397 (2023).
Hui, L. et al. Chromosome-scale genome assembly of sweet tea (Lithocarpus polystachyus Rehder). figshare https://doi.org/10.6084/m9.figshare.24297544 (2023).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Lin, Y. et al. quarTeT: A telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 10, uhad127 (2023).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126–e126 (2018).
Wang, W.-B. et al. Chromosome-scale genome assembly and insights into the metabolome and gene regulation of leaf color transition in an important oak species, Quercus dentata. New Phytol. 238, 2016–2032 (2023).
Sork, V. L. et al. High-quality genome and methylomes illustrate features underlying evolutionary success of oaks. Nat. Commun. 13, 2047 (2022).
Ai, W. et al. A chromosome-scale genome assembly of the Mongolian oak (Quercus mongolica). Mol. Ecol. Resour. 22, 2396–2410 (2022).
Han, B. et al. A chromosome-level genome assembly of the Chinese cork oak (Quercus variabilis). Front. Plant Sci. 13, 1001583 (2022).
Fu, R. et al. Genome-wide analyses of introgression between two sympatric Asian oak species. Nat. Ecol. Evol. 6, 924–935 (2022).
Zhou, X. et al. A chromosome-scale genome assembly of Quercus gilva: Insights into the evolution of Quercus section Cyclobalanopsis (Fagaceae). Front. Plant Sci. 13, 1012277 (2022).
Wang, J. et al. Chromosome-level genome assembly provides new insights into Japanese chestnut (Castanea crenata) genomes. Front. Plant Sci. 13, 1049253 (2022).
Sun, Y., Lu, Z., Zhu, X. & Ma, H. Genomic basis of homoploid hybrid speciation within chestnut trees. Nat. Commun. 11, 3375 (2020).
Huang, W.-C. et al. A chromosome-scale genome assembly of Castanopsis hystrix provides new insights into the evolution and adaptation of Fagaceae species. Front. Plant Sci. 14, 1174972 (2023).
Sun, Y. et al. Chromosome-scale genome assembly of Castanopsis tibetana provides a powerful comparative framework to study the evolution and adaptation of Fagaceae trees. Mol. Ecol. Resour. 22, 1178–1189 (2022).
Mishra, B. et al. A chromosome-level genome assembly of the European beech (Fagus sylvatica) reveals anomalies for organelle DNA integration, repeat content and distribution of SNPs. Front. Genet. 12, 691058 (2022).
Acknowledgements
We thank Dr. Kai-Hua Jia for sharing the updated version of Quercus variabilis genome sequence and annotation files. We thank anonymous reviewers and Associate Editor for their insightful comments. This study was supported by Guangdong Province Basic Research Flagship Project (2023B0303050001), and project from Guangdong Provincial Key Laboratory of Applied Botany (2023B1212060046).
Author information
Authors and Affiliations
Contributions
B.W. and H.L. conceived and designed the study. H.L., R.Z., B.-F.Z., Z.S., X.-Y.C. and J.G. prepared the materials and analyzed the data. H.L. prepared the results and wrote the manuscript. B.W. edited and improved the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, H., Zhang, R., Zhou, BF. et al. Chromosome-scale genome assembly of sweet tea (Lithocarpus polystachyus Rehder). Sci Data 10, 873 (2023). https://doi.org/10.1038/s41597-023-02791-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02791-y
