
Abstract
Mahogany species (family Meliaceae) are highly valued for their aesthetic and durable wood. Despite their economic and ecological importance, genomic resources for mahogany species are limited, hindering genetic improvement and conservation efforts. Here we perform chromosome-scale genome assemblies of two commercially important mahogany species: Swietenia macrophylla and Khaya senegalensis. By combining 10X sequencing and Hi-C data, we assemble high-quality genomes of 274.49 Mb (S. macrophylla) and 406.50 Mb (K. senegalensis), with scaffold N50 lengths of 8.51 Mb and 7.85 Mb, respectively. A total of 99.38% and 98.05% of the assembled sequences are anchored to 28 pseudo-chromosomes in S. macrophylla and K. senegalensis, respectively. We predict 34,129 and 31,908 protein-coding genes in S. macrophylla and K. senegalensis, respectively, of which 97.44% and 98.49% are functionally annotated. The chromosome-scale genome assemblies of these mahogany species could serve as a vital genetic resource, especially in understanding the properties of non-model woody plants. These high-quality genomes could support the development of molecular markers for breeding programs, conservation efforts, and the sustainable management of these valuable forest resources.
Similar content being viewed by others

Background & Summary
The stability of forest ecosystems is increasingly being threatened by factors such as global climate change and unrestricted anthropogenic exploitation1. Therefore, for the conservation and development of timber species, it is important to generate genomic information and decode the underlying genetic architecture and regulatory mechanisms to improve forest productivity, adaptation, resilience, and sustainability2,3. In recent years, scientists have made significant progress in sequencing and analyzing the genomes of timber tree species like Populus trichocarpa4, Eucalyptus grandis5, Tectona grandis6, Dalbergia sissoo7, and Hopea hainanensis3, which has provided valuable insights into the genetic basis of traits such as wood formation, growth, and adaptation to environmental stress2. Genomics-based approaches can be used to directly and significantly improve the productivity and adaptability of timber species. These approaches can be used to modify one or more genes in the genomes of timber species, or to identify effective genetic markers and genes for molecular breeding. Genomic research can also accelerate the generation of knowledge in systems biology, which is important for the development of computational genomics8. Computational genomics has opened up new ways of identifying genes that regulate complex traits, and through gene stacking and genome editing, customized timber species with special applications can be designed9. Forest trees are essential for maintaining biodiversity in terrestrial ecosystems and for producing fiber, fuel, and biomass10. Therefore, the importance and legitimacy of forestry studies, including genomics, will be a higher priority in the future.
Mahogany is a tropical hardwood known for its durability, stability, and beautiful reddish-brown color of its wood, and is commonly used in the manufacturing of fine furniture, cabinetry, flooring, and musical instruments11. Swietenia macrophylla, commonly known as large-leaf mahogany, is a tropical timber species in the Meliaceae family that can tolerate a wide range of soils and environmental conditions. It can grow up to 40 meters tall, have a diameter of up to two meters, and live for several centuries12. S. macrophylla is one of three species that produces genuine mahogany timber (Swietenia) and is famous for its high-quality wood, which plays an important role in the international mahogany market. The wood is used principally for making furniture, musical instruments, interior fittings and ship building13. Furthermore, S. macrophylla contains a variety of bioactive compounds such as phenols, flavonoids, terpenoids, and alkaloids, which are rich in medicinal value14,15. Overall, the study of S. macrophylla highlights the urgent need to protect this valuable and threatened species. Through better management practices, forest conservation, and the sustainable use of this resource, we can ensure the long-term survival of S. macrophylla and other important tropical hardwood species.
Khaya senegalensis is another important species of deciduous tree in the Meliaceae family that is native to Africa. The wood K. senegalensis is prized for its beauty and durability, and it is used for a variety of purposes, including carpentry, interior trim, and construction. Traditionally, the wood was also used to make dugout canoes, household implements, djembe drums, and fuel wood16,17. It is also used in traditional African folk medicine, and has been shown to be effective in treating a variety of ailments, including malaria, fever, and diarrhea. Overall, K. senegalensis is an important tree with a variety of uses. It is a valuable source of timber, and it has the potential to be used in a variety of medical applications. To date, genome sequences of several important tree species of the Meliacea family have been sequenced such as Toona sinensis18, Toona ciliata19, Azadirachta indica20, Xylocarpus rumphii, X. moluccensis and X. granatum21.
Here, we construct high-quality genomes of S. macrophylla and K. senegalensis using a combination of 10x reads and Hi-C sequencing data. We predict 34,129 (S. macrophylla) and 31,908 (K. senegalensis) protein-coding genes. We also identify 187 and 123 miRNAs, 648 and 844 tRNAs, 249 and 186 rRNAs from the S. macrophylla and K. senegalensis genomes. Although the draft genome of S. macrophylla21 has been published previously, it lacks Hi-C data, and our study elevates the genome to the chromosome-scale with a longer N50 by combining Hi-C data, resulting in a higher-quality genome assembly.
Methods
Sample collection, library construction and sequencing, genome size evaluation
The fresh young leaves of Swietenia macrophylla (HCNGB_00002344) and Khaya senegalensis (HCNGB_00002341) were collected from Ruili, Yunnan, China (24°03′04.4″N 97°56′16.9″E), and stored in the Herbarium of China National GeneBank (HCNGB) (Supplemental Figs. 1–2). DNA was extracted using CTAB (cetyltrimethylammonium bromide)22, then GEM and barcode sequences were generated based on the standard protocol (Chromium Genome Chip Kit v1, 10X Genomics, Pleasanton, USA) for S. macrophylla and K. senegalensis. The barcode libraries were followed by sequencing on the BGISEQ-500 platform to generate 150 bp read pairs23. Finally, we generated 1283.02 million reads and 192.45 Gb of raw data in S. macrophylla while K. senegalensis has 1141.22 million reads and 171. 18 Gb of raw data (Supplemental Table S1).
We also collected fresh young leaves, and branch samples from each species to collect xylem and phloem tissues, and RNA was extracted using the PureLink RNA Mini Kit (Thermo Fisher Scientific, Carlsbad, CA, USA) following the standard protocol to construct RNA libraries using the TruSeq RNA Sample Preparation Kit manual (Illumina, San Diego, CA, USA). RNA libraries were then sequenced on the BGISEQ-500 platform (paired-end, 100-bp reads or 150-bp reads) and the RNA reads were filtered to generate 241.63 million clean reads and 45.88 Gb of clean data for S. macrophylla as well as 517.49 million clean reads and 104.53 Gb of clean data for K. senegalensis (Supplemental Table S2) by the Trimmomatic24 with the parameters:ILLUMINACLIP:adapter.fa:2:30:20:8:true HEADCROP:5 LEADING:3 TRAILING:3 SLIDINGWINDOW:5:8 MINLEN:50.
For Hi-C libraries, MboI restriction enzymes were used and constructed according to the in situ ligation protocol25. The MboI-digested chromatin was end-labelled with biotin-14-dATP (Thermo Fisher Scientific, Waltham, MA, USA) and used for in situ DNA ligation. The DNA was extracted, purified, and then sheared using Covaris S2 (Covaris, Woburn, MA, USA). The DNA libraries were sequenced on a BGISEQ-500 after A-tailing, pull-down and adapter ligation to produce 100-bp read pairs which generated 1483.63 million reads and 148.36 Gb of Hi-C raw data for S. macrophylla and 1519.79 million reads and 151.98 Gb of Hi-C raw data for K. senegalensis (Supplemental Table S1).
A k-mer (k = 21) analysis was constructed using the obtained DNA sequencing reads from the 10X libraries which were filtered using SOAPnuke26 with the parameters (-l 10 -q 0. 1 -n 0. 01 -Q 2 -d–misMatch 1–matchRatio 0.4) to estimate genome sizes, proportion of repeat sequence and heterozygosity. The k-mer frequency distribution analysis was performed using the following formula:
Where Num represents the read number of reads used. Len represents the read length, K represents the k-mer length, and K_Dep refers to where the main peak is located in the distribution. The distribution of 21-kmers showed that the heterozygosity and duplication rate of the genome were respectively 1.00% and 20.14% in S. macrophylla, 0.73% and 42.60% in K. senegalensis, with genome sizes of 274.49 Mb (S. macrophylla) and 406.50 Mb (K. senegalensis) (Fig. 1 and Supplemental Table S3).

21-kmer distribution in two mahogany genomes. (a) S. macrophylla. (b) K. senegalensis. The dashed line indicates the expected K-mer depth.
Genome assembly, evaluation, and repeat annotation
To perform the genome assembly, a de novo assembly program Supernova designed to assemble diploid germline genomes using Linked-Reads (10X library sequences) was used with the default parameters and exported into fasta format using the ‘pseudohap2’ style thereby performing GapCloser27 with the parameters “-l 150” to fill the gap. The Hi-C reads were quality controlled and mapped to the genome assembly of each species using Juicer28 with default parameters. Subsequently, a candidate superscaffold-level assembly was automatically generated using the 3D-DNA pipeline with default parameters29 to correct misjoins, order, orient, and organize scaffolds from the draft assembly. The draft assembly was checked and refined manually in the Juicebox Assembly Tools30 (Fig. 2a). The transcriptome sequences were assembled using Bridger tool31 and then mapped to the scaffold assembly using BLAT software32. The 10X clean reads were preliminarily assembled into scaffold sequences of 290.21 Mb for S. macrophylla with 5.76 Mb of Scaffold N50 and 406.50 Mb for K. senegalensis with 2.53 Mb of Scaffold N50. The scaffold sequences of two mahogany species were both further anchored onto 28 pseudochromosomes, accounting for 99.38% and 98.05% of the assembled genome. The final chromosome-scale genome assembly was 288.41 Mb with a scaffold N50 of 8.51 Mb in S. macrophylla and 370.38 Mb with a scaffold N50 of 7.85 Mb in K. senegalensis (Table 1, Supplemental Tables S4-5).

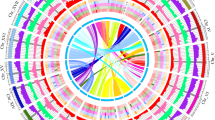
Hi-C and Circos plots of two mahogany genomes (a) Hi-C map of the S. macrophylla and K. senegalensis genome showing genome-wide all-by-all interactions. The map shows a high resolution of individual chromosomes that are scaffolded and assembled independently. The heat map colors ranging from light pink to dark red indicate the frequency of Hi-C interaction links from low to high (0–10). (b) Circos plot of S. macrophylla and K. senegalensis genome. Concentric circles from outermost to innermost show (I) chromosomes and megabase values, (II) gene density, (III) GC content, (IV) repeat density, (V) LTR density, (VI) LTR Copia density, (VII) LTR Gypsy density and (VIII) inter-chromosomal synteny (features II-VII are calculated in non-overlapping 200 Kb sliding windows).
Repeating elements were identified using a combination of homology-based and de novo approaches using default parameters. For homology-based approaches, we aligned the genome assembly with a known repeat database Repbase v. 21.0133 using RepeatMasker v. 4.0.634 for homology-based repeat element characterization. RepeatModeler v.1.0.835 and LTR Finder v. 1.0.636 were used to construct a new repeat library using genome assembly, RepeatMasker v.4.0.637 was followed, used to identify and annotate repeat elements in the genome, and finally TRF v.4. 0738 was used to tandem repeats in genomes for annotation (Table 2). We identified 85.08 Mb (29.50%) of repetitive sequences in the S. macrophylla genome and 80.85 Mb (21.83%) in the K. senegalensis genome. Most of these repeat sequences are Class I (53.57%) retro transposons, including Copia, Gypsy, LINE and SINE, accounted for 9.04%, 4.87%, 0.54%, 0.03% in S. macrophylla and 6.24%, 5.19%, 0.48%, 0.08% in K. senegalensis of the entire genome, respectively (Table 2, Supplemental Table S6).
Gene annotation, functional annotation and noncoding RNAs annotation
The MAKER-P pipeline (version 2.31)39 was used to predict protein-coding gene structures based on RNA, homologous protein and de novo prediction evidence. Clean transcriptome reads were assembled into inchworms using Trinity (version 2.0.6)40 and therefore submitted to MAKER-P as expressed sequence tags for RNA evidence. Protein sequences from the model plant or related species (Supplemental Table S7) were downloaded for two mahogany species and utilized as protein evidence for homology comparisons. In order to perform de novo prediction, multiple training sets were created for various ab initio gene predictors. The generation of a set of transcripts was initially performed by applying the genome-guided approach of Trinity40. Using PASA (version 2.0.2)41, these transcripts were then traced back to the genome, creating a collection of gene models with real gene features. For Augustus42 training, complete gene models were chosen. Genemark-ES (version 4.21)43 was self-trained with default parameters. Based on the aforementioned data, the first round of MAKER-P was run with all default parameters set to “1,” except for “est2genome” and “protein2genome”, which only produced RNA and protein-supported gene models, respectively. The gene models were then used for the training of SNAP44. The second and final rounds of MAKER-P were executed using the default parameters to generate the final gene model. The integration of protein-coding genes from S. macrophylla and K. senegalensis was successfully achieved, resulting in a total of 34129 and 32914 genes, respectively. The average gene length for S. macrophylla was determined to be 3052.92 bp, while for K. senegalensis it was 3068.00 bp. Additionally, the average lengths of exons and introns were calculated to be 215.60 bp and 402.79 bp, respectively, for S. macrophylla, and 230.06 bp and 431.15 bp, respectively, for K. senegalensis (Table 2, Supplemental Table S8).
Functional annotation of protein-coding genes was performed through the utilization of sequence similarity and domain conservation. This involved comparing the predicted amino acid sequences against publicly available databases. The initial step involved the identification of protein-coding genes by searching for optimal matches against protein sequence databases including the Kyoto Encyclopaedia of Genes and Genomes (KEGG)45, the National Centre for Biotechnology Information (NCBI), non-redundant (NR) and COG databases46, SwissProt47, and TrEMBL. This search was performed using BLASTP with a specified E-value cut-off of 1e-5. Subsequently, InterProScan 55.0 was employed to detect and classify domains and motifs using the Pfam48, SMART49, PANTHER50, PRINTS51, and ProDom52 databases. Consequently, the annotation rates for S. macrophylla and K. senegalensis were found to be 97% and 98% respectively (Table 2, Supplemental Table S9). Additionally, a combined total of 12,152 genes (equivalent to 35.61% of S. macrophylla) and 11,954 genes (equivalent to 37.46% of K. senegalensis) were jointly annotated in five functional databases (Fig. 3a).

Venn diagram and Phylogenetic position of S. macrophylla and K. senegalensis. (a) Venn diagram of S. macrophylla and K. senegalensis. (b) The phylogenetic tree constructed by IQtree with ‘-b 100’ using 317 single copy orthologues of two mahogany species and nine other representative plant species. The red nodes indicate fossil calibration nodes. Node labels represent node ages (Mya). The number of expanded gene families (+; green) and the number of contracted gene families (–; red) are shown in each branch. The numbers below the middle of each branch represent the bootstrap values.
To annotate non-coding RNAs, the ribosomal RNA (rRNA) genes were queried against the A. thaliana rRNA database using BLASTN V. 2.2.2653 with parameter (-e 1e-5 -v 10000 -b 10000). The Rfam database54 was queried for microRNAs (miRNA) and small nuclear RNA (snRNA) (tRNAscan-SE55 was also employed to scan tRNA). In this study, we successfully isolated ribosomal RNA (rRNA), microRNA (miRNA), and transfer RNA (tRNA) from S. macrophylla and K. senegalensis. The quantities obtained for S. macrophylla were 249 for rRNA, 187 for miRNA, and 648 for tRNA, while for K. senegalensis, the quantities were 630 for rRNA, 189 for miRNA, and 844 for tRNA (Table 2, Supplemental Table S10).
Genome collinearity and Circos plot construction
MCScanX1 was used to identify genomic collinearity between the two mahogany species and to obtain their pairs of colinear genes. The file of genomic collinearity generated by MCScanX was combined with the previous genome assembly and annotation results files to construct a circos plot (Fig. 2b). Here, we found that the genomes of two mahogany species share many similar structural features, including: (1) both consist of 28 chromosomes; (2) gene density and GC content show a positive correlation; (3) LTR density is negatively correlated with gene density and GC content; (4) the chromosomes of the two mahogany species show a high degree of collinearity between them, which also supports the close affinity between the two mahogany species. To show the taxonomic position of the sequenced species, the phylogenetic tree was subsequently constructed based on 317 single copy orthologues obtained from OrthoFinder v. 2.3.156 clustering (Fig. 3b). First, MAFFT v. 7.31057 was used to conduct multiple sequence alignment for single-copy orthologs protein sequences, and the alignment results were input into IQtree v. 1.6.158 with the parameters “-b 100” to construct phylogenetic tree. The tree building results were rooted and visualized using FigTree v. 1.4 (http://tree.bio.ed.ac.uk/software/figtree). Second, species divergence time was estimated by combining the MCMCTREE module of PAML v. 4.559 and the TToL5 web portal60. Finally, we used CAFÉ v. 4.2.161 to analyze the expansion and contraction events of single-copy orthologs. The S. macrophylla and K. senegalensis diverged ~13.8 Mya and were closest to the genus Citrus, which was consistent with T. sinensis18 and T. ciliate19 of the same genus. The divergent time between T. sinensis and T. ciliate was ~15.3 Mya, which overlapped with the results of Wang et al.19 In addition, these two mahogany species diverged with A. thaliana ~93.6 Mya and P. trichocarpa ~99.7 Mya, which was similar to He et al.21 A total of 1735 and 1543 gene families had expanded and contracted in the S. macrophylla genome, while 1537 and 2052 gene families had expanded and contracted in the K. senegalensis genome, respectively.
Data Records
All the genomic sequencing raw data were deposited in the Genome Sequence Archive in National Genomics Data Center (NGDC) Genome Sequence Archive (GSA) database with the accession number CRA01179362 under the BioProject accession number PRJCA01826963. The assembled scaffolds genomes were submitted to the Genome Warehouse under the accession number GWHDONZ0000000064, GWHDOOA0000000065 of S. macrophylla and K. senegalensis, respectively. The Chromosome-scale genome assemblies were also submitted to the NCBI under the accession number GCA_032401905.166, GCA_032402905.167 of S. macrophylla and K. senegalensis, respectively. The raw sequencing data and assembled genomes of S. macrophylla and K. senegalensis that support the findings of this study have also been deposited into CNGB Sequence Archive (CNSA)68 of China National GeneBank DataBase (CNGBdb)69 with accession number CNP0004053 and CNP0004052, respectively. The gene annotations, pseudogene predictions, and ncRNA files are available in the Figshare70.
Technical Validation
Genome assembly and validation of gene prediction
In order to evaluate the quality of genome assembly, we used bwa (version: 0.7.12; mode: aln)71 to align the Illumina short reads with the chromosome-level genomes, 97.43% and 97.68% of the Illumina short reads were mapped to the S. macrophylla and K. senegalensis genomes, respectively (Supplemental Table S11). BUSCO (version 3.0.1)72 was used to assess the integrity of our genome assembly, with results showing 97% (S. macrophylla), 96.2% (K. senegalensis) for scaffold-scale genomes in addition to 95.8% (S. macrophylla), 91.6% (K. senegalensis) for Chromosome-scale genomes. To assess the results of Hi-C assembly, as shown in the chromosomal interaction heatmap, the intensity of diagonal interactions within each group is higher than the intensity of non-diagonal interactions (Fig. 2a), which was consistent with the principle of Hi-C assisted genome assembly and demonstrated that the genome assembly was accurate. Taken together, the results showed that the genomes of the two mahogany species assembled in this study had a high degree of integrity.
For gene prediction, we used BUSCO (version 3.0.1) to assess the number and proportion of annotated genes from two mahogany species occupying the database of the core set of angiosperm genes (embryophyta_odb10). The results showed that S. macrophylla had 1284 genes matched back to the core gene set (93.4%), while K. senegalensis had 1268 genes (92.2%), indicating that the annotated gene sets of both mahogany species are highly complete.
Code availability
All software used in this work is in the public domain and their parameters are described in the Methods section. If a software did not mention parameters, the default parameters suggested by the developer were used.
References
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic acids research 40, e49–e49 (2012).
Neale, D. B. & Kremer, A. Forest tree genomics: growing resources and applications. Nature Reviews Genetics 12, 111–122 (2011).
Wang, S. et al. The chromosome‐scale genomes of Dipterocarpus turbinatus and Hopea hainanensis (Dipterocarpaceae) provide insights into fragrant oleoresin biosynthesis and hardwood formation. Plant Biotechnology Journal 20, 538–553 (2022).
Tuskan, G. A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). science 313, 1596–1604 (2006).
Myburg, A. A. et al. The genome of Eucalyptus grandis. Nature 510, 356–362 (2014).
Sahu, S. K. et al. Chromosome-scale genomes of commercial timber trees (Ochroma pyramidale, Mesua ferrea, and Tectona grandis). Scientific Data 10, 512 (2023).
Sahu, S. K. et al. Chromosome-scale genome of Indian Rosewood (Dalbergia sissoo). Frontiers in Plant Science 14, 1218515 (2023).
Sahu, S. K. & Liu, H. Long-read sequencing (method of the year 2022): the way forward for plant omics research. Molecular Plant 16, 791–793 (2023).
Borthakur, D. et al. Current status and trends in forest genomics. Forestry Research 2, 2–11 (2022).
Brockerhoff, E. G. et al. Forest biodiversity, ecosystem functioning and the provision of ecosystem services. Biodiversity and Conservation 26, 3005–3035 (2017).
Verissimo, A., Barreto, P., Tarifa, R. & Uhl, C. Extraction of a high-value natural resource in Amazonia: the case of mahogany. Forest ecology and Management 72, 39–60 (1995).
Gillies, A. C. M. et al. Genetic diversity in Mesoamerican populations of mahogany (Swietenia macrophylla), assessed using RAPDs. Heredity 83, 722–732 (1999).
Krisnawati, H., Kallio, M. & Kanninen, M. Swietenia Macrophylla King: Ecology, Silviculture And Productivity. (CIFOR, 2011).
Telrandhe, U. B., Kosalge, S. B., Parihar, S., Sharma, D. & Lade, S. N. Phytochemistry and pharmacological activities of Swietenia macrophylla King (Meliaceae). Sch Acad J Pharm 1, 6–12 (2022).
Moghadamtousi, S. Z., Goh, B. H., Chan, C. K., Shabab, T. & Kadir, H. A. Biological activities and phytochemicals of Swietenia macrophylla King. Molecules 18, 10465–10483 (2013).
Zhang, H., Wang, X., Chen, F., Androulakis, X. M. & Wargovich, M. J. Anticancer activity of limonoid from Khaya senegalensis. Phytotherapy Research 21, 731–734 (2007).
Arnold, R., Bevege, D. I., Bristow, M., Nikles, D. G. & Skelton, D. J. Khaya senegalensis - current use from its natural range and its potential in Sri Lanka and elsewhere in. Asia. Journal of Plant Protection 170, 1917–1930 (2004).
Ji, Y. T. et al. Long read sequencing of Toona sinensis (A. Juss) Roem: A chromosome‐level reference genome for the family Meliaceae. Molecular Ecology Resources 21, 1243–1255 (2021).
Wang, X. et al. A chromosome-level genome assembly of Toona ciliata (Meliaceae). Genome Biology and Evolution 14, evac121 (2022).
Du, Y. et al. Genomic analysis based on chromosome-level genome assembly reveals an expansion of terpene biosynthesis of Azadirachta indica. Frontiers in Plant Science 13 (2022).
He, Z. et al. Evolution of coastal forests based on a full set of mangrove genomes. Nature Ecology & Evolution 6, 738–749 (2022).
Kumar, S. S., Muthusamy, T. & Kandasamy, K. DNA Extraction Protocol for Plants with High Levels of Secondary Metabolites and Polysaccharides without Using Liquid Nitrogen and Phenol. Isrn Mol Biol 2012, 205049 (2012).
Huang, J. et al. BGISEQ-500 WGS library construction. protocols. io, 1–10 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Belaghzal, H., Dekker, J. & Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 123, 56–65 (2017).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, gix120 (2018).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 2047-2217X–2041-2018 (2012).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Dudchenko, O. et al. The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. Biorxiv, 254797 (2018).
Chang, Z. et al. Bridger: a new framework for de novo transcriptome assembly using RNA-seq data. Genome biology 16, 1–10 (2015).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome research 12, 656–664 (2002).
Jurka, J. Repbase update: a database and an electronic journal of repetitive elements. Trends in genetics 16, 418–420 (2000).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 25, 4.10. 11–14.10. 14 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–W268 (2007).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 5, 4.10. 11–14.10. 14 (2004).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573–580 (1999).
Campbell, M. S., Holt, C., Moore, B. & Yandell, M. Genome annotation and curation using MAKER and MAKER-P. Current protocols in bioinformatics 48, 4.11. 11–14.11. 39 (2014).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols 8, 1494–1512 (2013).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology 9, 1–22 (2008).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC bioinformatics 7, 1–11 (2006).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic acids research 33, 6494–6506 (2005).
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 1–9 (2004).
Aoki, K. F. & Kanehisa, M. Using the KEGG database resource. Current protocols in bioinformatics 11, 1.12.11–11.12.54 (2005).
Tatusov, R. L., Koonin, E. V. & Lipman, D. J. A genomic perspective on protein families. Science 278, 631–637 (1997).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids research 31, 365–370 (2003).
Bateman, A. et al. The Pfam protein families database. Nucleic acids research 32, D138–D141 (2004).
Letunic, I., Doerks, T. & Bork, P. SMART 6: recent updates and new developments. Nucleic acids research 37, D229–D232 (2009).
Mi, H., Muruganujan, A., Casagrande, J. T. & Thomas, P. D. Large-scale gene function analysis with the PANTHER classification system. Nature protocols 8, 1551–1566 (2013).
Attwood, T. K. et al. PRINTS and its automatic supplement, prePRINTS. Nucleic acids research 31, 400–402 (2003).
Corpet, F., Servant, F., Gouzy, J. & Kahn, D. ProDom and ProDom-CG: tools for protein domain analysis and whole genome comparisons. Nucleic acids research 28, 267–269 (2000).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S. R. Rfam: an RNA family database. Nucleic acids research 31, 439–441 (2003).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic acids research 44, W54–W57 (2016).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16, 157 (2015).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular biology and evolution 37, 1530–1534 (2020).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24, 1586–1591 (2007).
Kumar, S. et al. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol Biol Evol 39 (2022).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
NGDC Genome Sequence Archive https://bigd.big.ac.cn/gsa/browse/CRA011793 (2023).
NGDC BioProject https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA018269 (2023).
NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/64341/show (2023).
NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/64342/show (2023).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_032401905.1 (2023).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_032402905.1 (2023).
Guo, X. et al. CNSA: a data repository for archiving omics data. Database (Oxford) 2020, baaa055 (2020).
Chen, F. Z. et al. CNGBdb: China National GeneBank DataBase. Hereditas 42, 799–809 (2020).
Wang, G. Two mahogany species, Figshare, https://doi.org/10.6084/m9.figshare.23685360.v2 (2023).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Cheng, S. et al. 10KP: A phylodiverse genome sequencing plan. GigaScience 7, giy013 (2018).
Acknowledgements
This work was supported by the Major Science and Technology Projects of Yunnan Province (Digitalization, Development and Application of Biotic Resource, 202002AA100007), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB27020104). This work is part of the 10KP project (https://db.cngb.org/10kp/)73 and is also supported by China National GeneBank (CNGB; https://www.cngb.org/).
Author information
Authors and Affiliations
Contributions
H.L., S.K.S. and C.H. led and designed this project. H.L., S.K.S. and S.W., conceived the study. S.K.S., W.M., J.W., S.Z. and J.L. collected the leaf and tissue samples. S.K.S., M.L., G.W. and Y.C. contributed to the sample preparation and performed the genome and chromosome-scale assembly. S.K.S., M.L., S.W., Y.C., D.F., G.W., D.N.S., W.M., R.L. and S.W. performed annotation and comparative genomic analyses. S.K.S., G.W. and M.L. wrote the original draft manuscript. S.W., M.L., S.Z., X.X., J.L., C.H., D.N.S., Y.Z., X.L., L.L., and H.L., revised and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sahu, S.K., Liu, M., Wang, G. et al. Chromosome-scale genomes of commercially important mahoganies, Swietenia macrophylla and Khaya senegalensis. Sci Data 10, 832 (2023). https://doi.org/10.1038/s41597-023-02707-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02707-w
This article is cited by
-
Beyond NGS data sharing for plant ecological resilience and improvement of agronomic traits
Scientific Data (2024)
