
Abstract
Adenosma buchneroides Bonati, belonging to the genus Adenosum (Plantaginaceae), is an aromatic medicinal plant and utilized in traditional Chinese medicine. It has been widely used as plant-based repellents to prevent vector-borne diseases. However, the lack of a reference genome limits the study of conservation management and molecular biology of A. buchneroides. Here, we generated a chromosome-level de novo genome assembly of A. buchneroides which is a high-quality chromosome-scale assembly of aromatic medicinal plant in Plantaginaceae. The genome has a total length of 442.84 Mb with scaffold N50 of 27.98 Mb and 95.55% of the genome assigned to 14 chromosomes. BUSCO assessment yielded a completeness score of 97.2%. Furthermore, we predicted 24,367 protein-coding genes, and 95.79% of them was functionally annotated. The chromosome-scale genome of A. buchneroides will be a significant resource for understanding the genetic basis and evolution of active components biosynthesis, which will facilitate further study and exploit of A. buchneroides.
Similar content being viewed by others

Background & Summary
The genus Adenosum (Plantaginaceae) comprises 26~29 species and is native to the tropical eastern Asia and tropical Oceania, with essential oils from most of the species and traditionally used for herbal medicine. Adenosma buchneroides Bonati, one taxa of the genus Adenosum, is a well-recognized aromatic medicinal plant long favored by the Aini people in southwest of China as an insect repellent1,2,3. As mentioned in pharmacopoeia and traditional herbal medicine books, the whole plant of A. buchneroides has multiple pharmaceutical activities, such as anti-rheumatic, dissipate stasis, analgesia, and diminishing swelling4. The essential oil of A. buchneroides was used for the treatment of gastro-intestinal disorders, respiratory disorders and heptatitis4,5, and showed strong mosquito repellent activity1 and positive insecticidal activity against Callosobruchus maculatus6. The medicinal value of essential oil in A. buchneroides is attributed to its abundant active ingredients including γ-terpinene (40.26%), carvacrol (34.98%), p-cymene (6.60%), α-terpinene (4.05%) and carvacrol methyl ether (3.42%)7. There is currently a greater requirement for plant-based repellents to prevent vector-borne diseases, such as dengue, malaria, etc8. Up to now, many efforts on the regulation mechanism of aromatic component biosynthesis has been made in the genus Thymus (Lamiaceae). Though most recently, several pseudo-chromosome level genomes of Plantaginaceae were published9,10,11. The molecular basis and evolution of those components biosynthesis in A. buchneroides (Plantaginaceae) are rarely reported due to the lack of a high-quality reference genome.
Here, we generated a chromosome-scale assembly of A. buchneroides, deciphered by integrating PacBio, Illumina and Hi-C sequencing technologies. Approximately 404.03 Mb genome was assembled with the scaffold N50 length of 27.98 Mb. A total of 386.05 Mb (95.55%) of the assembled sequences were anchored to 14 pseudo-chromosomes. The genome contains 24,367 protein-coding genes, and 95.79% of them were annotated. In addition, we identified 597 miRNAs, 1,018 tRNAs, 5,202 rRNAs, and 339 snRNA. The genome assembly of A. buchneroides is a valuable genetic resource of aromatic medicinal plant. The results provided new insights into the molecular basis and evolution of aromatic component biosynthesis, and laid a foundation for molecular breeding and genetic conservation of A. buchneroides.
Methods
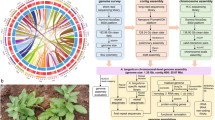
Flow cytometry-based genome size estimation. The seeds of A. buchneroides were obtained from Mengla county of Yunnan Province, China. Seeds were germinated in a greenhouse and grown to maturity (Fig. 1A). Fresh young leaves of A. buchneroides were collected and immediately transferred to a pre-chilled Petri dish and chopped by a razor blade in 1.5 mL ice-cold Otto I consisting of 0.1 M citric acid, 0.5% Tween-20 with pH = 2.0-3.012. The resulting suspension was thoroughly mixed and filtered through a 40 µm nylon mesh. Following incubation at room temperature for 20 min, staining solution consisting of 1 mL of Otto II solution (0.4 M Na2HPO4∙12H2O with pH = 8.0-9.0), 50 µg mL−1 propidium iodide (PI) and 50 µg mL−1 RNase A and 2 µL mL−1 β-mercaptoethanol, was added to the suspension. And then samples were kept in the dark for 30 min with occasional mixing. Flow cytometry analysis was performed in a BD FACSAria Fusion flow cytometer (BD Biosciences). Maize (2.3 Gb)13 was used as standard reference sample with known genome sizes. We determined that the genome size of A. buchneroides is approximately 439.55 ± 6.76 Mb (Fig. 1B).

Morphology and genome size estimation of A. buchneroides. (A) Morphology of A. buchneroides. (B) Flow cytometry-based estimation. (C) 17-kmer distribution estimation.
Sequencing library construction and preliminary genome survey. High-quality genomic DNA was extracted from fresh young leaves of A. buchneroides using CTAB (cetyl trimethylammonium bromide) method. The qualified genomic DNA was broken to the target fragment (350 bp) by ultrasonic shock, and then was used to construct the short-read sequencing libraries using Illumina TruSeq® Nano DNA library preparation kit (Illumina, San Diego, CA, USA). Next, paired-end sequencing was conducted on the Illumina HiSeq platform (Illumina, CA, USA), which finally generated 44.78 Gb of raw data, which covered the genome ~101.12-fold-coverage (×) (Table 1). After removing contaminants, low-quality reads and adapters by fastp software14, clean reads were subjected to KmerGenie15 for the optimal k-mer size analyzed. Then, Jellyfish16 was used to analyze the k-mer counts, which were used to estimate the genome size, proportion of repeat sequence and heterozygosity. From the 17-kmers distribution, we predicted that the genome size is 442.84 Mb, which is almost identical to the estimated 439.55 ± 6.76 Mb by flow cytometer. The heterozygosity and repeat ratio of A. buchneroides genome were predicted to be 0.28% and 58.17%, respectively (Fig. 1C). PacBio libraries were constructed using the SMRTbell template preparation kit following the manufacturer’s standard instructions, subsequently was sequenced using Single-Molecule Real Time (SMRT) sequencing on a PacBio Sequel II platform (Pacific Biosciences). In total, 40.12 Gb raw data, accounting for ~ 90.60× of the entire genome, were generated (Table 1). For Hi-C analysis, fresh leaf of A. buchneroides fixed with formaldehyde was used to construct library according to the protocol of Belton et al.17. The library was then sequenced on Illumina HiSeq platform, which generated 50.86 Gb raw data, accounting for ~ 114.85 × of the genome.
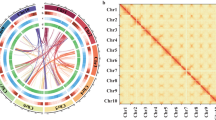
De novo genome assembly. The pipelines overview of A. buchneroides chromosome-level genome assembly and annotation was shown as in Fig. 2. PacBio long reads were de novo assembled using HiCanu v.2.218 and Hifiasm v.0.1319, followed with polishing using NextPolish20. After removing low quality reads and contaminants, the high-quality Hi-C reads were used to cluster, order and orient the scaffold onto pseudo-chromosomes using the ALLHiC software v.0.9.1221. The Juicebox v. 20100822 was used to manually adjust the chromosome segmentation boundary and any wrong assembly. We preliminary assembled the PacBio long reads into 161 contigs of 404.02 Mb with N50 of 21.63 Mb, and the longest contig was 30.82 Mb (Table 1). Using Hi-C technology, these contigs were further anchored onto 14 pseudo-chromosomes, accounting for 95.55% of the assembled genome (Fig. 3A). The somatic cells of A. buchneroides contained 28 chromosomes by the cytological observation method (Fig. 3B). Finally, the chromosome-scale genome assembly of A. buchneroides was 404.03 Mb with a scaffold N50 of 27.98 Mb (Table 1).

The pipelines overview of A. buchneroides chromosome-level genome assembly and annotation.

Chromosome information of A. buchneroides. (A) Hi-C interaction heatmap of A. buchneroides genome. Hi-C interaction matrix showing the pairwise correlations among 14 pseudomolecules. (B) The karyotype of A. buchneroides.
RNA sequencing. Root, stem, leaf and flower tissue of the A. buchneroides were collected for RNA extraction. Total RNA was extracted from each tissue respectively using a standard Trizol protocol (Invitrogen, USA), and then used for libraries construction. After libraries construction followed the manufacture’s guideline, the transcriptomes were sequenced on Illumina HiSeq X Ten platform. In total, 20.12 Gb RNA-seq data were generated (Table 1). These RNA-seq data were used for whole-genome protein-coding gene prediction.
Repeat annotation. A combination of ab initio and homology-based approaches to identify the repetitive sequences. We first used LTR_FINDER v.1.0523, RepeatScout v.1.0524 and RepeatModeler v.2.0.125 to build a de novo repeat sequences library of A. buchneroides genome. Then, RepeatMasker v.4.1.026 was used to search for known and novel repetitive elements by mapping sequences against the de novo repeat library and the Repbase v.19.0627 database. Finally, a total of 236.86 Mb of A. buchneroides genome was identified as repetitive sequences, which accounted for 58.62% of the assembled genome. Specifically, four classes of transposable elements (TEs) including long terminal repeats (LTRs), long interspersed nuclear elements (LINEs), DNA elements (DNAs) and short interspersed nuclear elements (SINEs) were identified. Most of these TEs were LTRs, accounted for 49.81% of the A. buchneroides genome, followed by DNAs (4.75%), LINEs (0.34%) and SINEs (0.001%) (Table 2).
Protein-coding genes prediction and functional annotation. Prediction of protein-coding genes was based on ab initio gene predictions, homology-based predicitions and transcriptome-based predictions. The ab initio prediction was performed by Genscan v.3.128, Augustus v.3.129, GlimmerHMM v.1.230, GeneID v.1.431, and SNAP32 (v.2013-02-16). For homology-based prediction, BLAST v.2.10.133 and Genewise software v.2.4.134 were used to annotation the gene models in A. buchneroides using amino acid sequences from Antirrhinum majus, Thymus quinquecostatus, and Arabidopsis thaliana genome. For RNA-Seq-based prediction, RNA-Seq data were assembled against reference transcripts using Hisat v.2.0.435 and Stringtie v.1.3.336. Then, the no-reference transcripts were assembled de novo using Trinity v.2.1.137. The results of gene prediction from three approaches were saved in GFF3 files, and then set the weight values for each annotation method. All the predicted gene structures were integrated into consensus set with EVidenceModeler v1.1.138. Finally, 24,367 gene models were predicted after integrating results of the three aforementioned methods (Table 3).
For protein-coding gene functional annotation, we aligned the predicted protein-coding gene sequences against public functional databases using BLAST (E-value 1E-5), including Swissprot, NR, KEGG, InterPro, GO and Pfam. As a result, 23,341 of protein-coding genes (95.79%) were annotated (Table 4).
Non-coding RNA annotation. We annotated four types of non-coding RNAs (ncRNAs) that were not translated into proteins, including transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), microRNA (miRNAs) and small nuclear RNAs (snRNAs). The tRNAs with high confidence were predicted using tRNAscan-SE v.1.3.139. The homology searching was used to predict rRNAs against plant rRNA database. Furthermore, miRNAs and snRNAs were annotated by aligning the assembled genome against the to Rfam40 database using Infernal software v.1.1.241. Finally, we totally identified 597 miRNAs, 5,202 rRNAs, 1,018 tRNAs and 339 snRNAs in A. buchneroides genome (Table 5).
Comparative genomics and phylogenetic analyses. Orthologues is critical for comparative genomics and phylogenetic analysis, and was predicted in our study. For orthologous and paralogous gene families clustering, orthologous genes of A. buchneroides and other 10 representative plant species, namely A. majus, T. quinquecostatus, Callicarpa americana, Buddleja alternifolia, Solanum tuberosum, Solanum lycopersicum, Vitis vinifera, A. thaliana, Oryza sativa, and Amborellaceae, were analyzed through all-versus-all protein sequence similarity searches (E-value cutoff of 1E-7) using OrthoMCL software v.2.0.942. We obtained the longest transcript per locus for orthologous cluster. As a result, clustering protein-coding sequences yielded 27,275 ortholog groups, including 7,145 common orthologs and 1,578 common single-copy orthologs. In A. buchneroides, there were 269 unique paralogs (Fig. 4A). Then, we further compared the orthologous genes among the four species including A. buchneroides, A. majus, B. alternifolia and A. thaliana. As shown in Fig. 4B, 10,151 ortholog genes were shared by the four species. There were 12,335 shared ortholog genes clusters between A. buchneroides and A. majus. However, there were 12,179 shared ortholog genes cluster between A. buchneroides and B. alternifolia. The result suggested that there was a closer relationship between A. buchneroides and A. majus than between A. buchneroides and B. alternifolia. Additionally, A. buchneroides had fewer unique gene families (400) than that in A. majus (1,248) in the comparison among the four species. These species-specific genes in the unique families may have close relationship with species-specific characters, and are worthy of further investigation.

Comparative genomics and phylogenetic analyses. (A) Classification and statistics of common and lineage-specific genes in A. buchneroides and other representative plant species. (B) Venn diagram of orthologous genes shared among A. buchneroides and three other species. (C) Phylogenetic analysis, gene family expansion/contraction analyses and divergence time estimations. Inferred divergence times are denoted at each node in black. Gene family expansion and contraction are indicated in green and red, respectively.
We performed alignment of conserved single-copy orthologs shared by A. buchneroides and other 10 representative plant species with MUSCLE v3.8.3143. Based on these alignment, a maximum likelihood (ML) phylogenetic tree was constructed using RAxML v.8.2.1244. The result showed that A. buchneroides and A. majus clustered together, while the T. quinquecostatus, C. americana and B. alternifolia formed another cluster. These results indicated there was a closer relationship between A. buchneroides and A. majus than between A. buchneroides and B. alternifolia, in line with the result of gene family analysis. Then, we used the Bayesian related molecular clock approach in MCMCtree program with the PAML Package45 to estimate divergence time. The divergence times were calibrated with the TimeTree database46. The divergence time was as follows: A. buchneroides-A. majus, 30.1 million years ago (mya); Thymus quinquecostatus-Callicarpa americana, 33.6 mya; A. buchneroides-B.alternifolia, 52.8 mya. The divergence time between A. buchneroides and A. majus (30.1 mya) was more recent compared with the divergence time of A. buchneroides and B.alternifolia (52.8 mya). Gene families that had undergone expansion and contraction in the 11 sequenced species were determined using CAFE v3.147 with a p value threshold = 0.05. In total, 17 and 169 gene families expanded and contracted in the A. buchneroides genome, respectively (Fig. 4C).
Whole-genome duplication analysis. To identify the whole-genome duplication (WGD) events in the A. buchneroides genome, we used MCScanX48 to calculated four-fold degenerated sites (4DTv) for all gene pairs. As illuminated in Fig. 5, A. buchneroides and A. majus exhibited characteristic peaks at approximately 0.20 and 0.28, respectively. The homologs of A. buchneroides with A. majus had a peak at 0.34. The results indicated a WGD event for A. buchneroides after divergence from A. majus (Fig. 5).

Distribution of 4DTv among A. buchneroides (Abuc), A. majus (Amaj), V. vinifera (Vvin) and A. thaliana (Atha) in intra- and intergenomic comparisons.
Identification of genes involved in the biosynthetic pathways of terpenoids. Previous studies reported that the medicinal value of essential oil in A. buchneroides was attributed to its abundant active ingredients, especially terpenoids, such as γ-terpinene and cavacrol1,2,5. Based on the KEGG database and the suggested biosynthesis pathways, we used a combined method of homolog searching and functional annotation to identify candidate genes for terpenoids biosynthesis (Fig. 6A). In total 70 genes in the present genome, which encoded 18 enzymes, were identified to be involved in terpenoid biosynthesis. To further explore the classification and function prediction of terpene synthases (TPSs), TPS proteins sequences in Arabidopsis were used as query to search against the protein database of A. buchneroides, A. majus and T. quinquecostatus using BLASTP program with e-value >10−5. All candidate proteins were further confirmed via SMART/Pfam analysis. And then all predicted TPSs were aligned with CLUSTAL. A Maximum-Likelihood (ML) phylogenetic tree was constructed by MEGA X v.10.1.749, with the bootstrap values of 1000 replicates. The phylogenetic trees was imported to iTOL for visualization50.

The putative biosynthetic pathway of terpenoids and gene family analysis of terpene synthases (TPSs). (A) The terpenodis biosynthesis pathway. (B) Phylogenetic analysis of TPSs from four genomes. The TPS genes of A. buchneroides, T. quinquecostatus, A. thaliana and A. majus were showed in red, blue, green and black font, respectively.
Data Records
The genome sequencing data, chromosomal assembly, genome annotations and RNA-Seq data had been deposited at the Genome Warehouse in National Genomics Data Center (NGDC), Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation51, under BioProject accession number PRJCA017315. The genome sequencing data had been deposited in the Genome Sequence Archive (GSA) of NGDC under the accession number CRA011236. The genome assembly and annotation data had been deposited in Genome Assembly Sequences and Annotations (GWH) of NGDC under accession number GWHCBPZ00000000. The genome assembly and annotations and the information of identified genes involved in terpenoid biosynthesis shown in Fig. 6 had been deposited at the figshare database52.
Technical Validation
The genome assembly was evaluated using Benchmarking Universal Single-Copy Orthology (BUSCO) software v.4.0.553. The results revealed the retrieval of 97.25% of the complete single-copy genes, of which 8.13% were duplicated. In addition, 0.3% of BUSCO genes were fragmented, and 2.45% were missing from the genome. The BUSCO results indicated a high genome assembly completeness of A. buchneroides (Table 6, Fig. 7).

BUSCO analysis (A) and short Illumina reads mapping results (B).
Core Eukaryotic Genes Mapping Approach (CEGMA, v.2.5)54 employs highly conserved core eukaryotic genes (CEGs) to assess the extent of comprehensive gene coverage. The CEGMA analysis showed that the assembled genome complete recalled 238 (95.95%) of the 248 highly conserved CEGs (Table 6).
The filtered short Illumina reads were aligned back to evaluate assembly integrity and sequencing uniformity using Burrows-Wheeler Aligner (BWA) software55. Approximately 99.36% of the short reads mapped to the genome, and genome coverage is approximately 99.84%. By using SAMtools software56, we found that the ratios of heterozygous and homozygous single nucleotide polymorphisms (SNPs) were 0.001% and 1.7e-05%, respectively, indicating that the assembly had high single-base-level accuracy (Table 6).
Code availability
All pipeline and software used in this study were performed to data analysis according to the manuals and protocols. The parameters and the version of the software are described in the Methods section. If no detailed parameters are mentioned for a software, the default parameters were used.
References
Ma, Y. et al. Bioassay-guided isolation of active compounds from Adenosma buchneroides essential oil as mosquito repellent against Aedes albopictus. J. Ethnopharmacol. 231, 386–393 (2019).
Gou, Y., Fan, R., Pei, S. & Wang, Y. Before it disappeared: ethnobotanical study of fleagrass (Adenosma buchneroides), a traditional aromatic plant used by the Akha people. J. Ethnobiol. Ethnomed. 14, 0–79 (2018).
Shen, et al Ethnobotany of fleagrass (Adenosma Buchneroides Bonati), a traditional cultivated plant of the Hani people, Xishuangbanna, Yunnan, China. In: The Museum, 1990, vol. 1. Belém: Ethnobiology: implications and applications: proceedings of the First International Congress of Ethnobiology; 1988.
China’s State Administration of traditional Chinese medicine,. t.C.M.M.e.b. Chinese Materia Medica. Shanghai Science and Technology Press (1998).
Wang, C. et al. A review of the aromatic genus Adenosma: Geographical distribution, traditional uses, phytochemistry and biological activities. J. Ethnopharmacol. 275, 114075 (2021).
Li, H. et al. Effect of 25 plant essential oils against Callosobruchus maculatus. In: Proceedings of the 7th International Working Conference on Stored-product Protection, Beijing, China, pp. 849–851 (1998).
Xu, Y., Cheng, B. Q., Yu, Z. & Ding, J. K. A preliminary study on the new perfume plant Adenosma buchneroides Bonati. In: The 7th Proceedings of the Seminar on Fragrance and Flavor China, Hangzhou, China, pp. 26–29 (2008).
Sukumar, K., Perich, M. J. & Boobar, L. R. Botanical derivatives in mosquito control: a review. J. Am. Mosq. Control Assoc. 7, 210–237 (1991).
Lyu, S. et al. Genome assembly of the pioneer species Plantago major L. (Plantaginaceae) provides insight into its global distribution and adaptation to metal-contaminated soil. DNA Res. 30, 1–14 (2023).
Herliana, L. et al. A chromosome-level genome assembly of Plantago ovata. Sci. Rep. 13, 1528 (2023).
Zhu, S. et al. The snapdragon genomes reveal the evolutionary dynamics of the S-locus supergene. Mol. Bio. Evol. 40, msad080.
Otto, F. DAPI staining of fixed cells for high-resolution flow cytometry of nuclear DNA. Meth. Cell Biol. 33, 105–110 (1990).
Schnable, P. et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 326, 1112–1115 (2009).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Chikhi, R. & Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics. 30, 31–37 (2013).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Belton, J. M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58, 268–276 (2012).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 18, 170–175 (2021).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efcient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, 265–268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identifcation of repeat families in large genomes. Bioinformatics. 21, 351–358 (2005).
Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0. http://www.repeatmasker.org (2008–2015).
Tempel, S. Using and understanding RepeatMasker. Methods Mol. Biol. 859, 29–51 (2012).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 19, 215–225 (2003).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-fnders. Bioinformatics. 20, 2878–2879 (2004).
Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Curr. Protoc. Bioinformatics. 18, e56 (2007).
Johnson, A. D. et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 24, 2938–2939 (2008).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, 1–22 (2008).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Griffithsjones, S. et al. Rfam: Annotating Non-Coding RNAs in complete genomes. Nucleic Acids Res. 33, 121–124 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30, 1312–1313 (2014).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819 (2017).
Han, M. V., Thomas, G. W., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Kumar, S. et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, 242–245 (2016).
Huang, H. Genome assembly and annotation of the Adenosma buchneroides. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 50, D27–D38 (2022).
Huang, H. Genome assembly of the Adenosma buchneroides. figshare https://doi.org/10.6084/m9.figshare.23259164 (2023).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate coregenes in eukaryotic genomes. Bioinformatics. 23, 1061–1067 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience. 10, giab008.
Acknowledgements
This work was supported by the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502), Biological Resources Programme, CAS (KFJ-BRP-007-019), the Yunnan key laboratory for wild plant resources of Kunming Institute of Botany, CAS (E03K781261), and digitalization, development and application of biotic resource (202002AA100007).
Author information
Authors and Affiliations
Contributions
H.H., Y.W. and S.P. conceived and designed the study. H.H. performed data analysis and drafted the manuscript. C.W. prepared plant samples. All authors read, edited, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, H., Wang, C., Pei, S. et al. A chromosome-level reference genome of an aromatic medicinal plant Adenosma buchneroides. Sci Data 10, 660 (2023). https://doi.org/10.1038/s41597-023-02571-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02571-8
This article is cited by
-
Beyond NGS data sharing for plant ecological resilience and improvement of agronomic traits
Scientific Data (2024)
