
Abstract
Anisodus tanguticus is a medicinal herb that belongs to the Anisodus genus of the Solanaceae family. This endangered herb is mainly distributed in Qinghai–Tibet Plateau. In this study, we combined the Illumina short-read, Nanopore long-read and high-throughput chromosome conformation capture (Hi-C) sequencing technologies to de novo assemble the A. tanguticus genome. A high-quality chromosomal-level genome assembly was obtained with a genome size of 1.26 Gb and a contig N50 of 25.07 Mb. Of the draft genome sequences, 97.47% were anchored to 24 pseudochromosomes with a scaffold N50 of 51.28 Mb. In addition, 842.14 Mb of transposable elements occupying 66.70% of the genome assembly were identified and 44,252 protein-coding genes were predicted. The genome assembly of A. tanguticus will provide genetic repertoire to understand the adaptation strategy of Anisodus species in the plateau, which will further promote the conservation of endangered A. tanguticus resources.
Similar content being viewed by others


Background & Summary
The perennial medicinal herb Anisodus tanguticus is a member of Anisodus genus that is distributed in Qinghai–Tibet Plateau. A. tanguticus was named as “Tang Chun Na Bao” in the traditional Tibetan medicine1. Its roots were used by the local Tibetan healers to treat septic shock, ulcers, colitis, spasms and reduce pain1,2. The main active components of A. tanguticus roots are tropane alkaloids, such as hyoscyamine, anisodamine, and scopolamine3. These tropane alkaloids are the competitive, reversible antagonists of muscarinic acetylcholine receptors, and are clinically used for the treatment of motion sickness, spasticity, obstetrical analgesia, septic shock, organophosphate poisoning, Parkinson’s symptoms, etc2,4. Besides, atropine (racemic hyoscyamine) was listed as the most efficacious, safe, and cost-effective medicines for priority conditions in the World Health Organization model list of essential medicines (https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2021.02). In addition to the well-known tropane alkaloids, numerous terpenoids, indolizidine- and pyrrolidine-type alkaloids and cinnamoylphenethylamides with pharmacological activity have been isolated from A. tanguticus5,6,7,8. Due to the important medicinal value, A. tanguticus has been massively exploited and collected, resulting in the depletion of its wild resources.
In the Anisodus genus, there are four species and three varieties, such as A. tanguticus, A. luridus, A. acutangulus, and A. mairei9. These four species are mainly distributed in the plateau (mainly the Qinghai–Tibet Plateau) at altitudes ranging from 2,680 to 4,200 m, and A. tanguticus was observed to survive at a higher altitude environment than A. acutangulus9. Although the genome of A. acutangulus has been assembled to explore the evolution of tropane alkaloid biosynthesis10, few is known about the adaptation strategy of Anisodus species to overcome the adverse environment, such as the complex land conditions or the diverse climate. Recently, the chloroplast genome of A. tanguticus was sequenced to study the adaptation strategy of A. tanguticus in the Qinghai–Tibet Plateau11,12. The chloroplast genetic information accounts for only a small part of the whole genetic information of A. tanguticus, and most genetic information is deposited within the chromosomal DNA. Thus, a high-quality chromosomal-level genome is necessary to provide genetic information to understand the evolutionary process of the Anisodus genus and the adaptation strategy of Anisodus species in the plateau, which will also promote the conservation of endangered A. tanguticus resources.
In this paper, we generated a high-quality chromosomal-level genome assembly of A. tanguticus based on the Illumina short-read sequencing (182.98 Gb), Nanopore long-read sequencing (128.34 Gb) and Hi-C sequencing (136.90 Gb). The assembled genome, composed of 276 contigs, had a genome size of 1.26 Gb with a contig N50 of 25.07 Mb (Table 1). These contigs were anchored to 24 pseudochromosomes, with an anchoring rate of 97.47% and a scaffold N50 of 51.28 Mb (Table 1, Fig. 1). Of this genome assembly, 66.70% (842.14 Mb) were transposable elements with a major component of long terminal repeats (LTRs), which accounted for 44.51% (Tables 1, 2). Meanwhile, 44,252 protein-coding genes composed the final gene repertoire of A. tanguticus (Table 1). This high-quality genome will provide a genetic basis for understanding the adaptive evolution of A. tanguticus in the plateau.

The genome assembly and annotation of A. tanguticus. (a) Circular map of A. tanguticus. The 24 outer lines represent 24 pseudochromosomes (Chr1−24). The blue and red bands represent the density of transposable elements and protein-coding genes, respectively. The inner lines represent syntenic blocks in the A. tanguticus assembly. (b) Photograph of A. tanguticus. (c) The process pipeline of A. tanguticus genome assembly and annotation.
Methods
Sample collection and genomic DNA extraction
The seeds of A. tanguticus were collected from Qilian, Qinghai Province, China, and stored in the Germplasm Bank of Wild Species in Southwest China. A. tanguticus plants were cultivated in the Kunming Institute of Botany of the Chinese Academy of Sciences, Yunnan Province, China. Young leaves from an individual A. tanguticus plant were collected and then used for genomic DNA (gDNA) extraction following the modified cetyltrimethylammonium bromide (CTAB) protocol13. The purity and quality of extracted gDNA were examined by NanoPhotometer spectrophotometer (Implen, USA) and agarose gel electrophoresis. Three different tissue samples, including leaf, stem, and root, were collected from an individual cultivated A. tanguticus plant, and used for RNA extraction.
Illumina sequencing and genome survey analysis
High-quality gDNA was randomly fragmented by ultrasonic oscillation (Covaris, USA) and used for Illumina short-read sequencing. According to the protocol of TruSeq DNA Sample Preparation Guide (Illumina, USA), the sequencing libraries were constructed with 350 bp insert size. Then, these libraries were sequenced on the Illumina NovaSeq 6000 platform (Illumina, USA) with a mode of paired-end 150 bp at Benagen Technology Co., Ltd. (Wuhan, China). After removing low-quality reads, the resulting 182.98 Gb clean data were used for the survey analysis of A. tanguticus genome and the polish of preliminary assembly.
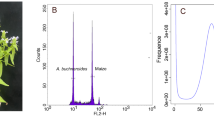
The frequencies of 19-kmer were generated by Jellyfish (version 2.2.10) based on the clean data and used for the genome evaluation by GenomeScope (version 2.0) (Fig. 2a)14,15. As a result, the genome size of A. tanguticus was estimated as 1.35 Gb, which was consistent with the genome size (~1.5 Gb) measured by flow cytometry (Fig. 2b). Meanwhile, the heterozygous ratio and the repeat content were estimated as 0.37% and 60.0%, respectively.

The evaluation of A. tanguticus genome size. (a) Genome scope profiles of 19-mer analysis. The X-axis represented the k-mer depth and the Y-axis represented the frequency of the k-mer for a given depth. (b) The flow cytometry of A. tanguticus. Endopolyploidy was observed in the genome of A. tanguticus.
Nanopore sequencing and draft genome assembly
For nanopore long-read sequencing, its libraries were constructed under the protocol of SQK-LSK110 Ligation Sequencing Kit (Nanopore, UK). The prepared libraries were loaded on flow cells (R9.4) and sequenced on the Nanopore PromethION platform (Nanopore, UK). After removing low-quality reads, a total of 128.34 Gb of clean data, composed of 8.22 million reads, were obtained. The N50 read length was 32.63 kb and the longest nanopore read length was 394.22 kb.
The preliminary assembly was generated by NextDenovo (https://github.com/Nextomics/NextDenovo) with 128.34 Gb clean nanopore data. Subsequently, Racon (version: 1.4.11)16 was used to polish the preliminary assembly with nanopore long-reads through two iterations. Pilon (version: 1.23)17 was used to polish the preliminary assembly with Illumina short-reads through two iterations. As a result, the draft genome of A. tanguticus was assembled with a total length of 1.26 Gb, composed of 276 contigs and the contig N50 was 25.07 Mb (Table 1).
Hi-C sequencing and chromosome-scale assembly
For genome scaffolding, the fresh leaves were used to construct the Hi-C libraries according to the standard library preparation protocol18. The prepared libraries were sequenced on the Illumina NovaSeq 6000 platform (Illumina, USA) with a 150-bp paired-end strategy. After the filtration of raw data, 136.90 Gb of clean data were generated.
The valid interaction pairs were identified by HiCUP (version: 0.8.0) and used to construct chromosome-scale assemblies by ALLHiC (version: 0.9.8)19,20. Finally, 97.47% of the draft genome sequences (1.23 Gb) were anchored to 24 pseudochromosomes of A. tanguticus and the final chromosome-scale assembly was composed of 131 scaffolds with a scaffold N50 of 51.28 Mb (Table 1, Fig. 3).

The Hi-C interaction heatmap of A. tanguticus genome. The dark red indicates high chromatin interactions, which were quantified based on the count of supporting Hi-C reads.
Genome annotation
Repeat sequences were identified by combining homology-based predictions and ab initio predictions. Firstly, RepeatMasker (version: 4.0.9) was used for homology-based prediction of the repeat sequences [i.e. “TE (transposable element) proteins” column in Table 2] in the genome assembly based on the Repbase database21,22. Secondly, RepeatModeler (version: 1.0.11) was used for ab initio prediction of the repetitive sequences to construct a A. tanguticus-specific repeat library23. This library was also used to annotate the repeat sequences (i.e. “De novo + Repbase” column in Table 2) of genome assembly by RepeatMasker (version: 4.0.9)21. These two repeat sequences were combined to obtain the final repeat sequences (i.e. “Combined TEs” column in Table 2), which accounted for 66.70% of the genome assembly.
Protein-coding genes were predicated by a combination of transcriptome-based prediction, ab initio predication and homologous predication. For transcriptome-based prediction, the RNA of three different tissues, including leaf, stem, and root, were used for the RNA sequencing. Stringtie (version: 2.1.4) and TransDecoder (version: 5.1.0, https://github.com/TransDecoder/TransDecoder) were used to predict the transcriptome-based genes24. GlimmerHMM (version: 3.0.4) and Augustus (version: 3.3.2) were used for the ab initio prediction25,26. Exonerate (version: 2.4.0) was used for homologous gene prediction with genes from Solanum lycopersicum (Sly), Capsicum annuum (Can), Nicotiana attenuate (Nat) and Solanum tuberosum (Stu)27. These predicated genes were integrated into 44,282 genes by MAKER (version: 2.31.10, Table 3)28. These protein-coding genes were annotated with protein sequence databases, including universal protein (Uniprot)29, protein families database (Pfam)30, gene ontology (GO)31, Kyoto encyclopedia of genes and genomes (KEGG)32, KEGG pathway database, interproscan database32, and nonredundant protein sequence (NR, https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins). 97.36% of protein-coding genes (43,112 genes) were annotated by at least one database (Table 4). In addition, 30 predicted genes with an intron less than 10 bp were designated as pseudogenes and eliminated in the gene repertoire of A. tanguticus, which led to a final gene count of 44,252.
The rRNA genes were predicated with rRNA database and the tRNA genes were predicated by tRNAscan-SE (version: 1.23)33. The non-coding RNAs were predicated by INFERNAL (version: 1.1.2) based on the Rfam database34,35. Finally, 2,758 tRNAs, 898 rRNAs, 1,821 snRNAs and 269 miRNAs were identified in A. tanguticus.
Genome evolution
175 single-copy orthologous families were clustered from A. tanguticus, A. acutangulus10, Atropa belladonna36, Datura stramonium36, S. lycopersicum37, Capsicum chinense38, N. attenuate39, Petunia inflata40, Ipomoea trifida41 and Arabidopsis thaliana by OrthoFinder (version: 2.5.2)42. These single-copy orthologous sequences were merged and aligned by MAFFT (version: 7.475)43. After the correction by Gblocks (version: 0.91b)44, the obtained sequences were used to construct the maximum likelihood tree by IQ-TREE (version: 2.0.3)45 with the best-fit model JTT + F + R3. The divergence time in the constructed phylogenetic tree was deduced by MCMCtree program (version: 4.9)46 with the divergence time of A. tanguticus and A. thaliana (111–124 Mya) from the TimeTree database (http://www.timetree.org). As a result, the divergence time between A. tanguticus and A. acutangulus was approximately 4.1 Mya (Fig. 4).

The inferred phylogenetic tree of A. tanguticus and nine other species. A. tanguticus and A. acutangulus clustered together.
Based on the analysis of constructed phylogenetic tree and clustered gene families, 1820 and 2537 gene families were expanded and contracted in the A. tanguticus genome by CAFE analysis (version: 4.2.1)47, respectively (Fig. 4). Of these, 161 expanded gene families and 42 contracted gene families were statistically significant (Table 5). The significantly expanded 161 gene families were enriched in 38 GO terms, involved in “DNA metabolic process”, “DNA integration” and “mitochondrion” (Table 6), which were probably related to strong UV radiation and low temperature in the plateau.
Data Records
The A. tanguticus genome project has been deposited in the NCBI database under BioProject accession PRJNA1018692. The genome assembly and gene annotation have been deposited at GenBank under the WGS accession JAVYJV00000000048. The genomic Illumina sequencing data were deposited in the SRA at NCBI SRR2612785049. The nanopore sequencing data were deposited in the SRA at NCBI SRR2621373550. The Hi-C sequencing data were deposited in the SRA at NCBI SRR2615288051. The transcriptomic sequencing data were deposited in the SRA at NCBI SRR26156612–SRR2615661852,53,54,55,56,57,58.
Technical Validation
Evaluation of the genome assembly
The quality of the genome assembly of A. tanguticus was evaluated based on the contiguity, completeness, and correctness. For contiguity, Hi-C interaction analysis showed apparent interactions among the 24 pseudochromosomes, which was consistent with the reported chromosomes numbers of A. tanguticus59. Moreover, 97.47% of the draft genome sequences were oriented and ordered in the 24 pseudochromosomes, with a N50 of 51.28 Mb, suggesting a high contiguity of this genome assembly. For completeness, 97.83% complete BUSCO (benchmarking universal single-copy orthologs) genes in the genome assembly of A. tanguticus were retrieved by BUSCO (version: 5.2.2) analysis with embryophyta_odb10 database60. Additionally, the fragmented and missing BUSCO genes accounted for only 0.25% and 1.92%, respectively. For correctness, all Illumina short reads were mapped to the genome assembly by BWA61, with a high map rate of 99.96% in the genome assembly. Overall, the quality of the genome assembly was assessed as high contiguity, completeness, and correctness.
Evaluation of the gene repertoire
The final gene repertoire of A. tanguticus comprised 44,252 protein-coding genes, while 38,388 or 38,128 protein-coding genes were predicted in the genome of A. acutangulus10,62. Given the phylogenetic proximity of A. tanguticus and A. acutangulus (Fig. 4), we compared the gene repertoires of these two species, focusing on both syntenic genes and non-syntenic genes. For syntenic genes, 34,447 genes in A. tanguticus genome corresponded to 33,162 genes in A. acutangulus genome (Table 7). For non-syntenic genes, 9,805 and 4,966 genes were predicated in A. tanguticus and A. acutangulus genome, respectively. The difference of gene repertoires of these two species mainly stemmed from the non-syntenic genes, which could result from the potential species-specific genes’ variation or a more detailed annotation of protein-coding gene in the A. tanguticus genome.
Code availability
The software and code used are publicly accessible. No custom programming or coding was used.
References
Chen, C., Wang, B., Li, J., Xiong, F. & Zhou, G. Multivariate statistical analysis of metabolites in Anisodus tanguticus (Maxim.) Pascher to determine geographical origins and network pharmacology. Front. Plant Sci. 13, 927336 (2022).
Ma, L. et al. Important poisonous plants in tibetan ethnomedicine. Toxins 7, 138–155 (2015).
Chen, K. et al. Long-term impact of N, P, K fertilizers in different rates on yield and quality of Anisodus tanguticus (Maxinowicz) Pascher. Plants 12, 2102 (2023).
Grynkiewicz, G. & Gadzikowska, M. Tropane alkaloids as medicinally useful natural products and their synthetic derivatives as new drugs. Pharmacol. Rep. 60, 439–463 (2008).
Zhu, H. et al. New indolizidine- and pyrrolidine-type alkaloids with anti-angiogenic activities from Anisodus tanguticus. Biomed. Pharmacother. 167, 115481 (2023).
Zhao, H.-Y. et al. Anisotanols A—D, four norsesquiterpenoids with an unprecedented sesquiterpenoid skeleton from Anisodus tanguticus. Chin. J. Chem. 39, 3375–3380 (2021).
Zhao, H.-Y. et al. New amides from the roots of Anisodus tanguticus. Biochem. Syst. Ecol. 91, 104082 (2020).
Meng, C.-W. et al. Novel indane derivatives with antioxidant activity from the roots of Anisodus tanguticus. Molecules 28, 1493 (2023).
Zheng, G. Z. & Luo, J. P. in Cryopreservation of Plant Germplasm I. Biotechnology in Agriculture and Forestry Vol. 32 (ed. Bajaj Y. P. S.) Ch. “Cryopreservation of Anisodus species” (Springer Press, 1995).
Wang, Y.-J. et al. Genomic and structural basis for evolution of tropane alkaloid biosynthesis. Proc. Natl. Acad. Sci. USA 120, e2302448120 (2023).
Zhou, D. et al. Characterization of the evolutionary pressure on Anisodus tanguticus Maxim. with complete chloroplast genome sequence. Genes 13, 2125 (2022).
Zhang, G. & Chi, X. The complete chloroplast genome of Anisodus tanguticus, a threatened plant endemic to the Qinghai-Tibetan Plateau. Mitochondrial DNA Part B-Resour. 4, 1191–1192 (2019).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15 (1997).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Vaser, R., Sovic, I., Nagarajan, N. & Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res 4, 1310 (2015).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 25, 4–10 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, II215–II225 (2003).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Apweiler, R. et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32, D115–119 (2004).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–230 (2014).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096 (2021).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
Zhang, F. et al. Revealing evolution of tropane alkaloid biosynthesis by analyzing two genomes in the Solanaceae family. Nat. Commun. 14, 1446 (2023).
Sato, S. et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Kim, S. et al. New reference genome sequences of hot pepper reveal the massive evolution of plant disease-resistance genes by retroduplication. Genome Biol. 18, 210 (2017).
Xu, S. et al. Wild tobacco genomes reveal the evolution of nicotine biosynthesis. Proc. Natl. Acad. Sci. USA 114, 6133–6138 (2017).
Bombarely, A. et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants 2, 16074 (2016).
Wu, S. et al. Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement. Nat. Commun. 9, 4580 (2018).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating Maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Yang, Z. H. PAML: a program package for phylogenetic analysis by Maximum Likelihood. Comput. Appl. Biosci. 13, 555–556 (1997).
Han, M. V., Thomas, G. W. C., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013).
Wang, Y.-J. Anisodus tanguticus isolate KB-2021, whole genome shotgun sequencing project. Genbank https://identifiers.org/ncbi/insdc:JAVYJV010000000 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26127850 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26213735 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26152880 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26156612 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26156613 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26156614 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26156615 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26156616 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26156617 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26156618 (2023).
Tu, T.-Y., Sun, H., Gu, Z.-J. & Yue, J.-P. Cytological studies on the Sino-Himalayan endemic Anisodus and four related genera from the tribe Hyoscyameae (Solanaceae) and their systematic and evolutionary implications. Bot. J. Linn. Soc. 147, 457–468 (2005).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Zhou, W. et al. A chromosome-level genome assembly of anesthetic drug-producing Anisodus acutangulus provides insights into its evolution and the biosynthesis of tropane alkaloids. Plant Commun. 5, 100680 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 32271480 and 82225043), National Key R&D Program of China (2018YFA0900600), and Yunnan Revitalization Talent Support Program “Yunling Scholar” Project.
Author information
Authors and Affiliations
Contributions
S.-X.H. conceived the project. Y.S., J.-P. H. and Y.-J.W. collected the samples and coordinated the sequencing. Y.S. and Y.-J.W. carried out the analysis. Y.-J.W., J.-P.H., Y.S. and S.-X.H. wrote and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, Y., Huang, JP., Wang, YJ. et al. Chromosome level genome assembly of endangered medicinal plant Anisodus tanguticus. Sci Data 11, 161 (2024). https://doi.org/10.1038/s41597-024-03007-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03007-7
