
Abstract
The genome of Populus davidiana, a keystone aspen species, has been sequenced to improve our understanding of the evolutionary and functional genomics of the Populus genus. The Hi-C scaffolding genome assembly resulted in a 408.1 Mb genome with 19 pseudochromosomes. The BUSCO assessment revealed that 98.3% of the genome matched the embryophytes dataset. A total of 31,862 protein-coding sequences were predicted, of which 31,619 were functionally annotated. The assembled genome was composed of 44.9% transposable elements. These findings provide new knowledge about the characteristics of the P. davidiana genome and will facilitate comparative genomics and evolutionary research on the genus Populus.
Similar content being viewed by others
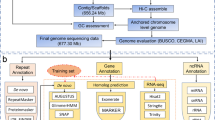
Background & Summary
Forest trees in natural populations are excellent materials for accessing the genomic architecture of evolutionary adaptation because they are mostly undomesticated and ecologically important across a wide variety of habitats and harbor abundant genetic and phenotypic variation1,2. The genus Populus (~30 species), including aspens, poplars, and cottonwoods, has a global geographic distribution throughout the Northern Hemisphere. Aspens and poplars are pioneer species with the fastest growth rates observed in temperate tree species, partly due to their characteristic heterophyllous growth3. Moreover, poplars show significant genetic variation among sections, species, individuals, and populations within the genus due to the pollen and seed airborne dispersal mechanism and their obligate outcrossing nature (dioecious)4. These traits, enhanced in interspecific hybrids, make an important contribution to meeting the global need for paper, biofuel, timber, bioremediation, and animal feed4. Due to its small genome size (less than 500 Mb), adequacy for genetic transformation, ease of propagation, and rapid growth, Populus has been established as an efficient model system for studies of forest tree species5,6.
The advance of Populus as a model system for woody perennial plants has been mainly caused by the rapid development of genomic and molecular biology resources from the Tacamahaca section of the Populus genus. This includes completion of the reference genome sequence of Populus trichocarpa (black cottonwood)7, P. euphratica (desert poplar)8, P. pruinose (sister of desert poplar)9. While draft genome sequences for two aspen species, P. tremula (European aspen)3 and P. tremuloides (American aspen)3, are available, their genome assemblies using a hybrid approach that merged 454 and Illumina short read sequencing were highly fragmented (No. of scaffolds = 216,318 for P. tremula and 164,504 for P. tremuloides)3. P. davidiana is another sibling species belonging to the same section of the genus Populus (section Populus) along with the two aspen species10,11. Previous phylogenetic studies revealed that P. tremuloides diverged earlier than the other aspen species, P. tremula and P. davidiana, due to the break-up of the Bering Land bridge12,13. After that, the uplift of the Qinghai-Tibetan Plateau and associated climate fluctuations may have driven the divergence between P. davidiana and P. tremula12. In addition, hybridization can readily occur in these aspen species, and resulting artificial hybrids exhibit heterosis for many wood characteristics14, suggesting that the speciation process has not been completed among the three aspen species13. Therefore, it is crucial to understand how different evolutionary forces have shaped the genomic landscape of differentiation along the forest tree speciation continuum. The high-quality reference genome resources from the Populus section, such as P. davidiana, will shed light on the phenomenon.
Here, we present a high-quality chromosome-level de novo genome assembly for the Asian aspen species Populus davidiana Dode. This new assembly will greatly improve genome completeness and contiguity over the previous aspen genomes. Furthermore, access to the P. davidiana genomic data set will facilitate research on the speciation continuum of Populus species and accelerate the breeding speed of forest trees by leveraging unexplored adaptive gene repositories.
Methods
Sample preparation and DNA sequencing
Fresh leaves of P. davidiana were collected from a 27-year-old female tree (Odae 19) in a clonal seedling located in Youngju (36°49′N 128°37′E; 575-m altitude) in Gyeongsangbuk-do Province, Republic of Korea (Fig. 1a). High-molecular-weight genomic DNA (gDNA) was isolated from the sample using the modified cetyltrimethylammonium bromide (CTAB) method15. The quality and quantity of the extracted DNA were then determined using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The genomic survey was performed using an Illumina paired-ended DNA library (550 bp insert), following the Illumina TruSeq DNA PCR-Free Library Prep protocol (Illumina, San Diego, CA, USA). The library was checked by Agilent 2100 Bioanalyzer High Sensitivity Kit and then sequenced on the Illumina NovaSeq6000 platform using a 150-bp paired-end strategy.

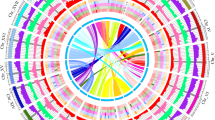
The sample and genome of P. davidiana. (a) Photograph of a 27-year-old female P. davidiana tree located in Youngju (36°49′N 128°37′E; 575-m altitude) in Gyeongsangbuk-do Province, Republic of Korea. (b) Genome characteristics of P. davidiana using GenomeScope. (c) Hi-C interaction heatmap and overview of the P. davidiana genome. The 19 assembled scaffolds are ordered by length. The x- and y-axes provide the mapping positions for the first and second reads in each read pair, respectively, grouped into bins. The color of each square indicates the number of read pairs within that bin. Grey lines have been added to indicate the borders between scaffolds. (d) The features are arranged in the order of gene density, repeat density, LTR/Gypsy, GC contents, and GC skew from outside to inside in 1 Mb intervals across the 19 chromosomes.
For HiFi sequencing, one 8 M SMRTbell DNA libraries were constructed using the following steps, according to the PacBio HiFi library construction protocol: (i) gDNA target size shearing using Megaruptor 3 (Diagenode); (ii) DNA damage repair; (iii) blunt-end ligation with hairpin adapters from the SMRTbell Express Template Prep Kit v2 (101-685-400, PacBio, Menlo Park, CA, USA); (iv) size-selection using the BluePippin Size Selection System (Sage Science, Beverly, MA, USA); and (v) binding to polymerase using the Sequel II Binding Kit v2.2 (Pacific Biosciences, Menlo Park, CA, USA). Subsequently, HiFi sequencing was performed on a PacBio Sequel II platform with the Sequel II Sequencing Kit v2.
A Dovetail Hi-C library was constructed and sequenced with the Illumina NovaSeq6000 platform, following published protocols16. Hi-C fragment libraries were prepared using the ‘Proximo Hi-C protocol’ with DpnII digestion, and the resulting libraries were sequenced using a 150-bp paired-end strategy.
Genome assembly
The original Illumina paired-ended sequencing produced 35.7 Gb and 17.8 Gb of clean data after filtering out low-quality reads containing poly-N and adapter sequences using FASTP v.0.12.6 (set to default parameters)17 (Supplementary Table 1). The trimmed sequencing reads were used to calculate the percentage of heterozygosity in the genome. First, Jellyfish v.2.2.1018 was used to compute a histogram of 19 k-mer frequencies (count -F 2 -m 19 -C -s 10 G). Then, heterozygosity was calculated using the GenomeScope v.2.0 online platform19. The platform predicted genome size of 374.7 Mb, with a heterozygosity of 1.73% (Fig. 1b). In addition, the long-read sequencing of the P. davidiana genome obtained 862,684 PacBio HiFi reads (11,636.6 Mb) representing a sequencing depth of 31.1X (Supplementary Table 1).
For de novo genome assembly, the FALCON-Unzip assembler was used with length cutoff parameters (length cutoff = 13 kb, length cutoff pr = 10 kb) and filtered subreads from SMRT Link v.5.0.0 (minimum subread length = 50 bp)20. To improve the accuracy of assembly, the Arrow algorithm was implemented using the unaligned BAM files as raw data to polish the FALCON-Unzip assembler. The de novo assembly resulted in a genome size of 498.7 Mb with a contig N50 of 2.3 Mb (Table 1). In addition, Purge Haplotigs was used to remove duplicated haplotypes as haplotigs from the whole-genome sequencing data21. The high, mid, and low cutoff read depth parameters were set to 170, 55, and 5 to remove haplotigs (default parameter). Consequently, the genome assembly contained 407.9 Mb in 484 polished contigs with an N50 of 2.74 Mb, and the GC content of the genome was 34.87% (Table 1).
The Hi-C fragment library sequencing produced 44.42 Gb (118.5X coverage) of clean data (Supplementary Table 1). The Dovetail Hi-C reads and the draft assembly were used as input data for HiRise (default parameter), a pipeline designed for scaffolding genome assemblies by utilizing proximity ligation data22. SNAP read mapper was used to align Hi-C library sequences to the draft input assembly23. Error correction was performed using Pilon24 with the short-read data, and organelle genomes were filtered out using BLAST v.2.4.025 (-max_target_seqs. 1 -evalue 0.001). A total of 259 assembled contigs were anchored onto 19 pseudochromosomes ranging from 13.1 to 51.7 Mb in length, containing 96.4% of the genome sequences (Fig. 1c; Supplementary Table 2). The final genome had N50 of 20.3 Mb, the highest among the sequenced Populus species (Supplementary Table 3). Finally, Benchmarking Universal Single-Copy Orthologs (BUSCO) v.4.1.2 was used to assess the completeness of the genome assembly (Table 2)26.
Transcriptome sequencing
Three types of tissue samples, including leaf, stem, and root, were collected from P. davidiana. The samples were immediately stored in liquid nitrogen at −80 °C until RNA extraction. Total RNAs were extracted from each sample using TRIzol reagent (Invitrogen, Waltham, MA, USA), and their purity and integrity were checked using the Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). RNA sequencing libraries were prepared according to the manufacturer’s instructions (Illumina Truseq stranded mRNA library prep kit). mRNA was purified and fragmented from total RNA using poly-T oligo-attached magnetic beads with two rounds of purification. Cleaved RNA fragments primed with random hexamers were reverse transcribed into first-strand cDNA using reverse transcriptase, random primers, and dUTP in place of dTTP. The products were purified and enriched with PCR to create the final strand-specific cDNA library. After QPCR using SYBR Green PCR Master Mix (Applied Biosystems), we combined libraries that index tagged in equimolar amounts in the pool. Finally, RNA sequencing was performed using an Illumina NovaSeq6000 system following the provided protocols for 2 × 100 sequencing. The RNA sequencing produced 15.9 Gb of raw read data (Supplementary Table 1).
Protein-coding gene annotation
The P. davidiana genome was annotated using ab initio gene prediction, custom repeat library protocols, homology search, and full-length transcript evidence. The MAKER v.2.31.8 pipeline was used for genome annotation, with three rounds of reiterative training27. Initially, the pipeline was run in ‘est2genome’ mode based on the transcriptome assembly which was generated by Trinity v2.8.5 from the RNA-seq data28. Additionally, ab initio gene prediction was performed using Augustus29 and SNAP30 (snaphmm = A.thaliana.hmm augustus_species = <BUSCO retraining model>). Finally, Exonerate v2.4.0 was implemented to polish MAKER alignments with evidence for protein-coding genes obtained from the genomes of three Populus species: P. trichocarpa, P. alba, and P. euophratica31. The best-supported gene models were selected based on the Annotation Edit Distance (AED) quality metric developed by the Sequence Ontology project32. The final genome assembly consisted of 19 pesudochromosomes and contained 31,862 protein-coding genes with an AED score less than 0.5 (Table 3). The final gene set had an average of 5.8 exons per gene, with a total length of 38.9 Mb and an average length of 1,219.6 bp.
Although P. davidiana had the highest N50 value among the sequenced Populus genomes, it had the lowest number of predicted genes (Supplementary Table 4). On the other hand, P. tremula had the second-best N50 value and the most genes among the poplar species, with 37,184 genes33. However, the gene density of P. davidiana genome was 78.1 genes per Mb (Fig. 1d; Table 3), which was not the lowest among the sequenced Populus species. P. euphratuca and P. tremuloides had the lowest and the highest gene density, respectively, with 69.82 and 96.3 genes per Mb (Supplementary Table 4). The highest density feature of P. euphratuca may be due to the relatively low genome quality8.
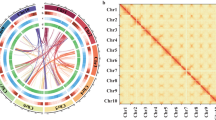
The density of genes and transcripts was analyzed based on their length distribution among different Populus species (Fig. 2). P. tremuloides, P. tremula and P. davidiana had many genes with short lengths (<1.0 kb). In contrast, genes with a length of around 1.9 kb were most abundant in P. euphratica and P. trichocarpa. The transcript length distribution was similar to the gene length distribution pattern, except for P. davidiana. It had the lowest frequency of both short- and long-length transcripts, indicating a relatively short length of CDS compared to the other Populus species (Supplementary Table 4).

The gene and transcript length distribution of P. davidiana and the other four Populus species (P. trichocarpa, P. euphratica, P. tremula, and P. tremuloides).
Functional annotation was performed using the predicted genes as queries. BLAST v.2.4.0 was run with a maximum e-value cutoff of 1e-5 against the National Center for Biotechnology Information (NCBI) UniProtKB/Swiss-Prot database25. In addition, InterProScan v.5.44.7934 and BLAST2GO-based gene ontology (GO) analysis35 were used to annotate the predicted proteins. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was also consulted for KEGG functional annotations in BLAST2GO35,36. Most CDSs (31,619 proteins, 99.2%) were annotated by the UniProt database (Supplementary Table 5). InterProScan annotated functions of 30,463 proteins (95.55%), and the other tools, including Pfam, GO, and KEGG, annotated 11,983 (37.6%), 15,966 (50.1%), and 3,039 (9.6%) proteins, respectively (Supplementary Table 5). The Third-level GO term analysis of the predicted proteome revelaed that proteins involved in cellular metabolic processes, intracellular anatomical structure, and organic cyclic compound binding were the most abundant in P. davidiana genome.
Repeat and non-coding RNA annotation
A de novo repeat library was created with the default parameters of RepeatModeler v.1.0.3, which includes RepeatScout v.1.0.537 and RECON v.1.0838. Tandem Repeats Finder v.4.0939 was used to predict repetitive sequences and classify information for each repeat, including low-complexity repeats, satellites, and simple repeats (default parameter). An LTR library was constructed with LTR_retriever40, using combined raw LTR data from LTR_FINDER and LTRharvest to identify highly accurate long terminal repeat retrotransposons (LTR-RTs)41,42. Finally, RepeatMasker v.4.0.943 was used to identify repetitive elements in the de novo repeat library and Kimura distances were calculated for all transposable element (TE) copies from each family found in the library to estimate the age of TEs44 (-lib -no_is).
Retrotransposable elements, which are known to be the dominant form of repeats in angiosperm genomes45, constituted 47.9% (195.5 Mb) of the P. davidiana genome (Fig. 3; Table 4). This is higher than those of other Populus sections, such as P. tremula (43.1%) and P. tremuloides (39.2%). Class I (retrotransposons) and Class II (DNA transposons) TEs accounted for 23.1% and 5.87% of the genome, respectively. Like other sequenced Populus genomes, LTR retrotransposons, mainly Gypsy-type and Copia-type LTRs, were predominant (22.68%), and with other DNA elements (DNAs) accounted for 4.86% of the genome. Of the repetitive elements, 10.91% could not be classified into any known families, indicating that P. davidiana, and perhaps the poplar family in general, may contain many novel repetitive or transposable elements.

Kimura distance-based copy divergence analysis of TEs in P. davidiana genomes. The graphs represent genome coverage (y-axis) for each type of TEs (DNA transposons, SINE, LINE, and LTR retrotransposons) in Kimura substitution level (CpG adjusted) illustrated on the x-axis (K-value from 0 to 50). The color chart indicates the repeat types.
Other non-coding RNAs and putative tRNA genes were identified using the Barrnap v0.9 (https://vicbioinformatics.com/software.barrnap.shtml) and tRNAscan-SE v2.0.546, respectively. Lastly, the number of rRNAs and tRNAs predicted from P. davidiana genome were 2,879 and 683, respectively.
Data Records
The P. davidiana genome project has been deposited in the NCBI database under BioProject accession PRJNA833418. The genome assembly data have been deposited at GenBank under the WGS accession JAMQGN00000000047. The sequencing reads are available at the Sequence Read Archive (SRA) under accessions from SRR24038974 to SRR24038979 (SRP430397)48. In addition, the genome, predicted transcripts and proteins, structural and functional annotation files (gff files), and results from repeat analysis had been deposited in FigShare49.
Technical Validation
The primary contigs and haplotigs of the draft FALCON-Unzip and the Purge Haplotigs-processed assemblies were evaluated using the BUSCO pipeline based on the embryophyta_odb9 database (Supplementary Table 6). Although the total number of BUSCOs was similar for both assemblies, the Purge Haplotigs haploid assembly had 12.5% more single-copy BUSCOs and 12.8% fewer duplicated BUSCOs than the draft FALCON-Unzip assembly. BUSCO assessment of the final genome assembly found that 1,587 (98.3%) of the 1,614 highly conserved orthologs were present as complete genes. This included 1,348 (83.5%) single-copy BUSCOs and 239 (14.8%) duplicated BUSCOs (Supplementary Table 6).
Code availability
We followed the developers’ instructions for the bioinformatics tools used in this study. The software and code used are publicly accessible, with the version and parameters used specified in the Methods section. No custom code was used during the compilation of the dataset.
References
Neale, D. B. & Ingvarsson, P. K. Population, quantitative and comparative genomics of adaptation in forest trees. Curr. Opin. Plant Biol. 11, 149–155 (2008).
Neale, D. B. & Kremer, A. Forest tree genomics: growing resources and applications. Nat. Rev. Genet. 12, 111–122 (2011).
Lin, Y.-C. et al. Functional and evolutionary genomic inferences in Populus through genome and population sequencing of American and European aspen. Proc. Natl. Acad. Sci. USA 115, E10970–E10978 (2018).
Stettler, R. F. Biology of Populus and its implications for management and conservation. Vol. 40337 (NRC Research Press, 1996).
Street, N., Tsai, C., Jansson, S., Bhalerao, R. & Groover, A. (Plant Genetics and Genomics: Crops and Models, eds Jansson S., Bhalerao R. …, 2010).
Wullschleger, S. D., Weston, D. J., DiFazio, S. P. & Tuskan, G. A. Revisiting the sequencing of the first tree genome: Populus trichocarpa. Tree Physiol. 33, 357–364 (2013).
Tuskan, G. A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). science 313, 1596–1604 (2006).
Ma, T. et al. Genomic insights into salt adaptation in a desert poplar. Nat. Commun. 4, 1–9 (2013).
Yang, W. et al. The draft genome sequence of a desert tree Populus pruinosa. GigaScience 6, gix075 (2017).
Eckenwalder, J. E. Biology of Populus and its implications for management and conservation. For. Sci. 7, 32 (1996).
Hamzeh, M. & Dayanandan, S. Phylogeny of Populus (Salicaceae) based on nucleotide sequences of chloroplast TRNT‐TRNF region and nuclear rDNA. Am. J. Bot. 91, 1398–1408 (2004).
Du, S. et al. Multilocus analysis of nucleotide variation and speciation in three closely related Populus (Salicaceae) species. Mol. Ecol. 24, 4994–5005 (2015).
Wang, J., Street, N. R., Park, E. J., Liu, J. & Ingvarsson, P. K. Evidence for widespread selection in shaping the genomic landscape during speciation of Populus. Mol. Ecol. 29, 1120–1136, https://doi.org/10.1111/mec.15388 (2020).
Hart, J. F., De Araujo, F., Thomas, B. R. & Mansfield, S. D. Wood quality and growth characterization across intra-and inter-specific hybrid aspen clones. Forests 4, 786–807 (2013).
Inglis, P. W., Pappas, Md. C. R., Resende, L. V. & Grattapaglia, D. Fast and inexpensive protocols for consistent extraction of high quality DNA and RNA from challenging plant and fungal samples for high-throughput SNP genotyping and sequencing applications. PloS One 13, e0206085 (2018).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 19, 1–10 (2018).
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016).
Zaharia, M. et al. Faster and more accurate sequence alignment with SNAP. arXiv preprint arXiv:1111.5572 (2011).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9, e112963 (2014).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12, 1–14 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 8, 1494–512 (2013).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 7, 1–11 (2006).
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 1–9 (2004).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 6, 1–11 (2005).
Eilbeck, K., Moore, B., Holt, C. & Yandell, M. Quantitative measures for the management and comparison of annotated genomes. BMC Bioinform. 10, 1–15 (2009).
Schiffthaler, B. et al. An improved genome assembly of the European aspen Populus tremula. bioRxiv, 805614 (2019).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 1–14 (2008).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4.10. 11–14.10. 14 (2004).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Oliver, K. R., McComb, J. A. & Greene, W. K. Transposable elements: powerful contributors to angiosperm evolution and diversity. Genome Biol. Evol. 5, 1886–1901 (2013).
Chan, P. P. & Lowe, T. M. in Gene prediction 1–14 (Springer, 2019).
Park, E. J. Populus davidiana cultivar Odae 19 isolate Odae 19, whole genome shotgun sequencing project https://identifiers.org/nucleotide:JAMQGN000000000 (2023).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRP430397 (2023).
Figshare https://doi.org/10.6084/m9.figshare.22688443 (2023).
Acknowledgements
This work was supported by the Forest Science & Technology Development Project (Project No. FG0603-2021-01-2023) of National Institute of Forest Science.
Author information
Authors and Affiliations
Contributions
E.-J.P. designed the study as the lead investigator; E.-K.B., M.-J.K. prepared the materials; K.-T.K. and S.J.L. performed the genome sequencing, assembly, annotation, and further bioinformatics analysis; and E.-K.B., M.-J.K., K.-T.K. and E.-J.P. wrote the manuscript. All authors contributed to, reviewed, and approved the final version of the manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bae, EK., Kang, MJ., Lee, SJ. et al. Chromosome-level genome assembly of the Asian aspen Populus davidiana Dode. Sci Data 10, 431 (2023). https://doi.org/10.1038/s41597-023-02350-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02350-5
This article is cited by
-
Beyond NGS data sharing for plant ecological resilience and improvement of agronomic traits
Scientific Data (2024)
