
Abstract
Animals that lived at high altitudes have evolved distinctive physiological traits that allow them to tolerate extreme high-altitude environment, including higher hemoglobin concentration, increased oxygen saturation of blood and a high energy metabolism. Although previous investigations performed plenty of comparison between high- and low-altitude mammals at the level of morphology, physiology and genomics, mechanism underlying high-altitude adaptation remains largely unknown. Few studies provided comparative analyses in high-altitude adaptation, such as parallel analysis in multiple species. In this study, we generated high-quality small RNA sequencing data for six tissues (heart, liver, spleen, lung, kidney and muscle) from low- and high-altitude populations of four typical livestock animals, and identified comparable numbers of miRNAs in each species. This dataset will provide valuable information for understanding the molecular mechanism of high-altitude adaptation in vertebrates.
Measurement(s) | miRNA |
Technology Type(s) | small RNA sequencing assay |
Factor Type(s) | geographic location • altitude • age • weight |
Sample Characteristic - Organism | Bos taurus • Bos grunniens • Ovis aries • Sus scrofa • Gallus gallus |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.9805424
Similar content being viewed by others

Background & Summary
Organisms encounter constant environmental changes, which occurred throughout their lifetime and across generations. As organisms move into a new habitat, natural selection helps facilitating evolution of phenotypes that are better equipped to survive in the new environment. As a classic example of adaptation to a novel environment, high-altitude adaptation involves multifaceted adaptations, including at the level of morphology1,2, genetics3,4 and physiology5,6,7. Previous studies investigated genetic mechanisms of high elevation adaptation, and identified numerous genes that are under selection. It was reported that persistent changes in gene expression may be induced by long-term environmental changes, and can respond rapidly to sudden external challenges8.
microRNAs (miRNAs) are a class of ~22 nucleotide (nt) RNAs that post-transcriptionally regulate gene expression through translational inhibition or mRNA degradation9. It was reported that miRNAs play important roles in cold acclimation10,11, hypoxia stress12,13 and ultraviolet radiation stress14,15. For example, miR-32 is induced by cold, and directly targets transducer of ERBB2, 1 (Tob1) to activate p38/MAPK signaling to promote brown adipocyte function and trans-activates white fat browning through fibroblast growth factor 21 (FGF21) in mice16. miR-210, a so-called ‘hypoxamir’ regulated by HIF-1α, has been demonstrated to target a variety of genes involved in the cell cycle, mitochondrial metabolism, angiogenesis, DNA damage repair, and cell survival17. Thus, it is reasonable to suppose the important roles of microRNAs in high-altitude adaptation.
To reveal the potential roles of miRNAs in high-altitude adaptation, we generated high-quality small RNA sequencing data for six tissues (heart, liver, spleen, lung, kidney, muscle) from high- and low-altitude populations of three species: chicken, sheep and pig. In addition, we also generated small RNA sequencing data from low-altitude cattle and high-altitude yak populations (Fig. 1). Detailed sample information is listed in Table 1. In total, 141 samples were analyzed in this study, including 11 samples that were published18. Each population was represented by two or three deep-sequenced biological replicates.

Graphical representation of animal distribution, sample collection and the process of RNA extraction, small RNA sequencing and data analysis.
Using a detection threshold of ≥4 reads across >40% samples in each species, we detected 2,036 mature miRNAs. Briefly, we identified comparable amounts of mature miRNA genes and precursor miRNAs for each species (Table 2). The unbiased miRNA annotation for each species prominently improved the uniformity of microRNA count in domesticated animals compared with miRbase21.0, which facilitated comparative analysis of miRNA evolution in domesticated animals.
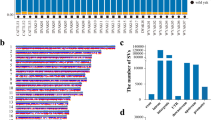
In addition, one-to-one orthologous miRNA genes were also identified based on evolutionary relationships. Overall, we identified 53 orthologues among vertebrates, 116 orthologues in Artiodactyla and 171 orthologues in ruminants (Fig. 2), consistent with a previous study which reported ~100 miRNA orthologues in mammals9. The detailed information of one-to-one orthologous miRNA genes for each pair of species is listed in figshare19 (The detailed information about one to one miRNA orthologue, figshare https://doi.org/10.6084/m9.figshare.c.4584113).

The number of one to one miRNA orthologues identification across four species. (a) Number of miRNA orthologues identified in each pair of species. (b) Number of miRNA orthologues across three species or four species.
Methods
Sample collection and RNA isolation
Three adult females for each population of the five species from distinct altitudes were used (Table 1). These populations have been confirmed to live at corresponding altitudes for their lifetime. Animals were humanely killed to ameliorate suffering. Upon excision from the corpse, a piece of tissue fragments from six organs including heart, liver, spleen, lung, kidney and skeletal muscle(longissimus muscle for pig, cattle, yak and sheep, and pectoral muscle for chicken) were immediately placed frozen in liquid nitrogen and then stored at −80 °C until use. Total RNA was extracted using the standard Trizol (Takara, Japan) protocol.
Small RNA library preparation, sequencing, and miRNA annotation
Small RNA libraries were constructed using the Illumina TruSeq Small RNA Sample Prep kit. Libraries were assessed using the Agilent 2200 TapeStation and sequenced on the Illumina HiSeq. 2500 platforms. Initial bioinformatics analysis (base calling) was performed with CASAVA 1.8 (Illumina, USA) to generate raw reads (in FASTQ form). Next, raw reads were subjected to a series of stringent filters (such as removing low-quality reads, repeated sequences, and adaptor sequences) and processed sequences were then mapped to the corresponding reference genome [i.e., chicken (Galgal4), pig (Suscrofa 10.2), cattle (UMD3.1), goat (CHIR 1.0), and sheep (Oar v3.1)] for each species with stringent criteria (<1 mismatch along the whole sequence) using Bowtie20. Then, mapped reads were submitted to miRDeep2.021 to detect miRNAs for each species with default parameters. Mature miRNA sequences of chicken and all annotated mammalian species in miRbase21 were selected as reference. To identify miRNAs with ≥4 reads across >40% samples were retained for further analysis. Notably, our previous study identified comparable numbers of miRNA species and similar expressional patterns in bovine genome and yak genomes18, therefore, we selected the bovine genome as the reference genome in this study.
Orthologous miRNA identifications
To identify miRNA orthologues, we first performed an all-against-all BLATST22 analysis of miRNA precursors for a specific evolutionary taxonomy. All BLAT matches with >75% of both precursors aligned, an identity of ≥70% and an e-value < 10−5 were retained, retained matches were clustered using hcluster (http://treesoft.svn.sourceforge.net/viewrc/treesoft/trunk/hcluster). Clustered families with only one member for each species were first defined as “natural one-to-one” orthologues. For those families with more than one member in any species, we performed liftOver23 of each reference genome on human miRNA coordinates to assistant with extraction of one-to-one orthologues.
Data Records
Small RNA-seq data of heart and lung for yak/cattle which we published before18 were available in the NCBI Gene Expression Omnibus (GEO) under accession number GSE8783324, and the small RNA-seq data of 126 remaining samples (excluded four samples with mapping rate <65%) were under accession number GSE12441825. The detailed information of 126 samples is uploaded to figshare19 (Metadata of samples submitted to the NCBI Gene Expression Omnibus, figshare https://doi.org/10.6084/m9.figshare.c.4584113).
Technical Validation
Sequencing quality control
As shown in Fig. 2, the quality of the small RNA sequencing was checked by analyzing the raw reads, clean reads, and mapping rate for each sample. Briefly, total of 1.66 Gb raw data were obtained. After stringent filtering, 1.60 Gb high-quality reads were remained and an average proportion of 80.89% high-quality reads can be mapped to the respective genomes (Fig. 3 and Sequencing quality control for 141 transcriptomes, figshare https://doi.org/10.6084/m9.figshare.c.4584113). Notably, four samples with mapping rate <65% were excluded to ensure data quality.

Overview of small RNA-Seq data. (a) Distribution of raw reads. (b) Distribution of clean reads, and (c) mapping rate of each sample for each species.
Reproducibility validation
To examine the reproducibility of biological replicates, we calculated the Pearson’s correlation for each pair of biological replicates. The majority of biological replicates showed high Pearson’s correlation coefficient (Fig. 4a). Meanwhile, we evaluated between-replicate variation with MvA plots for all pairs. Consistent with Pearson’s correlation analysis, most of biological replicates showed minor variation (Fig. 4b and MvA plots for examining between-replicates variation, figshare https://doi.org/10.6084/m9.figshare.c.4584113). Principal component analysis also showed that the majority biological samples could be grouped together (Principal component analysis of each tissue for each species, figshare https://doi.org/10.6084/m9.figshare.c.4584113). These results highlight the strong experimental confidence of this dataset.

Assessment of reproducibility across biological replicates. (a) Pearson’s correlation coefficient of biological replicates for each species. (b) MvA plots for examining between-variation of heart in yak/cattle group. The Y-axis represents a log (base 2) fold-change in expression changes and the X-axis indicates the log of the average gene expression level.
Usage Notes
Because of its ability to identify novel miRNAs and accurately determine quantitative expression, small RNA sequencing technique has made it possible to obtain large datasets26. High-altitude adaptation has recently become a topic of interest, and attracted many specialists in fields such as genetics and molecular biology. We believe that this dataset will provide valuable groundwork for understanding the molecular mechanisms of high-altitude adaptation in vertebrates. These data could also provide an opportunity to comprehensively compare the similarity and difference of the mechanisms underlying high-altitude adaptation across multiple domesticated animals. In addition, our laboratory profiled the mRNA transcriptome for corresponding samples, which facilitates more precise investigation of the interaction between miRNAs and mRNA genes associated with vertebrate high-altitude adaptation27 Epigenetic modification vary from one individual to another and could be affected by sex, age, weight, habitat, nutrition and so on, thus it is necessary to be cautious to interpret the mechanism of high altitude adaptation using this dataset.
References
Brutsaert, T. D., Soria, R., Caceres, E., Spielvogel, H. & Haas, J. D. Effect of developmental and ancestral high altitude exposure on chest morphology and pulmonary function in Andean and European/North American natives. American Journal of Human Biology the Official Journal of the Human Biology Council 110, 435–455 (1999).
Bailey, D. M., Bärtsch, P., Knauth, M. & Baumgartner, R. W. Emerging concepts in acute mountain sickness and high-altitude cerebral edema: from the molecular to the morphological. Cellular & Molecular Life Sciences 66, 3583–3594 (2009).
Yu, L. et al. Genomic analysis of snub-nosed monkeys (Rhinopithecus) identifies genes and processes related to high-altitude adaptation. Nature genetics 48, 947 (2016).
Li, M. et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nature genetics 45, 1431 (2013).
Storz, J. F. et al. Evolutionary and functional insights into the mechanism underlying high-altitude adaptation of deer mouse hemoglobin. Proceedings of the National Academy of Sciences of the United States of America 106, 14450–14455 (2009).
Naeije, R. Physiological Adaptation of the Cardiovascular System to High Altitude. Progress in Cardiovascular Diseases 52, 456–466 (2010).
Scott, G. R. et al. Molecular Evolution of Cytochrome c Oxidase Underlies High-Altitude Adaptation in the Bar-Headed Goose. Molecular Biology & Evolution 28, 351 (2011).
Lópezmaury, L., Marguerat, S. & Bähler, J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nature Reviews Genetics 9, 583–593 (2008).
Berezikov, E. Evolution of microRNA diversity and regulation in animals. Nature Reviews Genetics 12, 846–860 (2011).
Tao, C. et al. Changes in white and brown adipose tissue microRNA expression in cold-induced mice. Biochemical & Biophysical Research Communications 463, 193–199 (2015).
Zhen, L. et al. Identification of cold-responsive miRNAs in rats by deep sequencing. Journal of Thermal Biology 66, 114–124 (2017).
Rupaimoole, R. et al. Hypoxia Mediated Downregulation of miRNA Biogenesis Promotes Tumor Progression. Nature. Communications 5, 5202 (2015).
Serocki, M. et al. miRNAs regulate the HIF switch during hypoxia: a novel therapeutic target. Angiogenesis 21, 1–20 (2018).
Bell, A. et al. Detection of a MicroRNA molecular signature of ultraviolet radiation in the superficial regions of melanocytic nevi on sun-exposed skin. Modern Pathology 31, 1744–1755 (2018).
Li, X., Qin, Y., Zhao, J., Min, R. & Zhang, J. Inhibitory effects of microRNA-133b on ultraviolet-induced apoptosis of lens epithelial cells and its mechanism. Journal of Experimental Ophthalmology 35, 977–983 (2017).
Ng, R. et al. miRNA-32 Drives Brown Fat Thermogenesis and Trans-activates Subcutaneous White Fat Browning in Mice. Cell Reports 19, 1229–1246 (2017).
Xin, H., Quynh-Thu, L. & Giaccia, A. J. MiR-210–micromanager of the hypoxia pathway. Trends in Molecular Medicine 16, 230–237 (2010).
Guan, J. et al. Comparative analysis of the microRNA transcriptome between yak and cattle provides insight into high-altitude adaptation. Peerj 5, e3959 (2017).
Long, K. et al. Small non-coding RNA transcriptome of four high-altitude vertebrates and their low-altitude relatives. figshare. https://doi.org/10.6084/m9.figshare.c.4584113 (2019).
Langmead, B. Aligning Short Sequencing Reads with Bowtie. Current protocols in bioinformatics 32, 11–7 (2010).
Friedländer, M. R., Mackowiak, S. D., Li, N., Chen, W. & Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic acids research 40, 37–52 (2011).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology 215, 403–410 (1990).
Kuhn, R. M. et al. The UCSC genome browser and associated tools. Briefings in bioinformatics 14, 144–161 (2012).
Gene Expression Omnibus, https://identifiers.org/geo:GSE87833 (2017).
Gene Expression Omnibus, https://identifiers.org/geo:GSE124418 (2019).
Luo, S. MicroRNA Expression Analysis Using the Illumina MicroRNA-Seq Platform. (Humana Press, 2012).
Tang, Q. et al. Comparative transcriptomics of 5 high-altitude vertebrates and their low-altitude relatives. GigaScience 6, gix105 (2017).
Acknowledgements
This work was supported by grants from the National Key R & D Program of China (2018YFD0500403 and 2018YFD0501204), the National Natural Science Foundation of China (31872335, 31601918 and 31772576), the Sichuan Province & Chinese Academy of Science of Science & Technology Cooperation Project (2017JZ0025), the Science & Technology Support Program of Sichuan (2016NYZ0042 and 2017NZDZX0002), the Earmarked Fund for China Agriculture Research System (CARS-35-01A).
Author information
Authors and Affiliations
Contributions
K.L., S.F., J.M., M.L. and X.L. conceived and designed the experiments and the analytical strategy. J.Z. and L.L. performed animal work and prepared biological samples. M.M. and X.W. constructed the cDNA library and performed sequencing. K.L., S.F., J.M., Q.T. and L.J. designed the bioinformatics analysis process. K.L., S.F. and J.M. wrote the paper. M.L. and X.L. revised the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Long, K., Feng, S., Ma, J. et al. Small non-coding RNA transcriptome of four high-altitude vertebrates and their low-altitude relatives. Sci Data 6, 192 (2019). https://doi.org/10.1038/s41597-019-0204-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0204-5
This article is cited by
-
Identification and expression profile of microRNA in seven tissues of the Golden snub-nosed monkey (Rhinopithecus roxellanae)
Molecular Genetics and Genomics (2020)
