
Abstract
Mass spectrometry-based metabolomics approaches can enable detection and quantification of many thousands of metabolite features simultaneously. However, compound identification and reliable quantification are greatly complicated owing to the chemical complexity and dynamic range of the metabolome. Simultaneous quantification of many metabolites within complex mixtures can additionally be complicated by ion suppression, fragmentation and the presence of isomers. Here we present guidelines covering sample preparation, replication and randomization, quantification, recovery and recombination, ion suppression and peak misidentification, as a means to enable high-quality reporting of liquid chromatography– and gas chromatography–mass spectrometry-based metabolomics-derived data.
Similar content being viewed by others


Main
Metabolomics, the large-scale study of the metabolic complement of the cell1,2,3, is a mature science that has been practiced for over 20 years4. Indeed, it is now a commonly used experimental systems biology tool with demonstrated utility in both fundamental and applied aspects of plant, microbial and mammalian research5,6,7,8,9,10,11,12,13,14,15. Among the many thousands of studies published in this area over the last 20 years, notable highlights5,6,7,8,10,11,16 are briefly described in Supplementary Note 1.
Despite the insight afforded by such studies, the nature of metabolites, particularly their diversity (in both chemical structure and dynamic range of abundance9,12), remains a major challenge with regard to the ability to provide adequate coverage of the metabolome that can complement that achieved for the genome, transcriptome and proteome. Despite these comparative limitations, enormous advances have been made with regard to the number of analytes about which accurate quantitative information can be acquired, and a vast number of studies have yielded important biological information and biologically active metabolites across the kingdoms of life14. We have previously estimated that upwards of 1 million different metabolites occur across the tree of life, with between 1,000 and 40,000 estimated to occur in a single species4.
However, thus far, even the most comprehensive methods cannot provide firm upper limits for metabolite number. Current capabilities for detection and quantification of metabolites fall a long way short of being comprehensive. Currently, combinations of the most comprehensive methods are able to quantify 700 of the 3,700 metabolites predicted to be present in Escherichia coli17,18, 500 of the 2,680 metabolites predicted to be present in yeast19,20, 8,000 of the 114,100 metabolites predicted to be present in humans21 and only 14,000 of the over 400,000 metabolites predicted to be present in the plant kingdom4,22. Chemical diversity, rapid turnover times and broad dynamic range in cellular abundance currently prohibit the possibility of using single-extraction and single-analysis procedures to measure all metabolites9. Consequently, many different extraction techniques and combinations of analytical methods have been developed in an attempt to achieve adequate metabolite coverage. This renders the establishment of good working practices13,15,23,24,25,26 more difficult than with RNA-seq27, for example. Furthermore, rigorous standards are needed for normalization of metabolomics data28,29. This is exacerbated by the breadth of aims associated with the measurement of metabolites, which encompass targeted metabolite analysis, metabolite profiling, flux profiling, metabolomics-scale analysis and metabolite fingerprinting techniques30,31.
Given the myriad of aims and methodologies, we argue that it is particularly important to define clear guidelines for acquisition and reporting of metabolite data because there are many potential sources of misinterpretation. This is not the first time such guidelines have been suggested, with several insightful papers published on this topic12,32 and long-established metabolome databases including MetaboLights33,34,35,36 and the Metabolome Workbench (https://www.metabolomicsworkbench.org/) also driving this field. A more detailed description of these repositories as well as of more recent developments is provided in Supplementary Note 2. Although the detailed standards set out by the Metabolomics Standards Initiative32 and these repositories are laudable and clearly represent the gold standard of metabolomics reporting, it is notable that only a small fraction of published metabolomics studies follow these standards in their entirety and submit their data to the metabolome databases. There are probably several reasons underlying this. First, few journals currently mandate that data be stored in one of the metabolomics repositories. Second, unlike the situation 20 years ago, or even when the work of the Metabolomics Standards Initiative was first published some 13 years ago32,36,37,38, metabolomics experiments often represent only one component of studies integrating a wide range of techniques. Moreover, many groups outsource their metabolomics workflow to service providers and do not always have the experience to provide the raw data or even have access to them. In parallel, requiring reviewers to comment on all aspects of multiomics studies in the absence of clear guidelines is a big ask, especially considering that many biologists lack expert competence in the area of metabolomics. Finally, and perhaps most tellingly, there is difficulty in reporting chromatogram-level information, which often requires several attempts to fulfil the criteria of the major metabolomics repositories. However, while the reporting of this information is highly useful for several purposes, it is not essential for all. As we illustrate here, evaluation of the quality of the metabolomics data presented in a paper can effectively be performed on the basis of a relatively small amount of metadata—namely, by analyzing the quality of the metabolite annotation as well as assessing the quantitative recovery of analyte peaks.
Our aim here is to present a simplified reporting workflow, with the hope of capturing more of the missing information. While nuclear magnetic resonance (NMR) and capillary electrophoresis–mass spectrometry (CE–MS) have specific advocates and have clear advantages in structure elucidation and sensitivity, respectively, we will focus here on chromatography (either gas chromatography (GC) or liquid chromatography (LC)) hyphenated to MS; we therefore focus our guidelines on such techniques, given that the majority of metabolomics studies rely on these approaches. In contrast to the suggestions of the Metabolomics Standards Initiative32,36,37,38 and the major repositories mentioned above, we provide reporting guidelines at the level of the processed data (supported by the provision of representative chromatograms allowing the assessment of metabolite identification), rather than the raw chromatograms. A similar recommendation was made to the plant research community in 2011 (ref. 39). Here we have aimed to revise and update these recommendations to (1) be more globally applicable and (2) reinforce our contention that quantification control experiments should be regarded as mandatory and can aid in determining how problematic the effects of ion suppression are in an experiment. We highlight potential sources of error and provide recommendations for ensuring the robustness of the metabolite data obtained and reported. We also present guidelines for sampling, extraction and storage, metabolite identification and reporting. We stress the need for recombination and recovery experiments aimed at checking both qualitative metabolite identifications and the quantitative recovery of these metabolites. In addition, we suggest a stricter nomenclature for metabolite annotation that would improve reporting by removing much of the ambiguity concerning the quality of metabolite annotation that is currently apparent in many metabolomics studies. The scope of our guidelines does not encompass detailed downstream computational analysis of the acquired datasets, although we note several important recent advances in this area40,41,42,43,44,45,46,47. These tools and their application are discussed in Supplementary Note 3.
We believe that such efforts are necessary to enable between-laboratory comparisons of datasets, which, as has been demonstrated for transcriptomics, provides huge statistical power and deeper biological insights and, furthermore, provides a basis for better integration with other datasets48,49.
Sampling, quenching, metabolite extraction and storage
The very first (and particularly vital) step in a metabolomics workflow (Figs. 1 and 2) is the rapid stopping, or quenching, of metabolism and extraction of the metabolites in a manner that produces a stable extract that is quantitatively reflective of the endogenous metabolite levels present in the original living cell. This is especially important in highly metabolically active systems such as cells and tissues, but less so in biofluids such as serum, plasma or urine samples12. Indeed, there is no one method to fit all cases, with specific sampling, quenching and extraction needed for each tissue type. That said, certain evaluations of quality are universally applicable, and our aim here is to provide clear instructions on how to apply them.

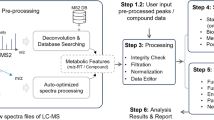
Metabolomics involves several basic steps: (1) sample preparation and extraction; (2) metabolite separation on a column (chromatography) such as by GC, LC or EC; (3) ionization of metabolites using an ion source; (4) separation by a mass analyzer as ions fly or oscillate on the basis of their mass-to-charge (m/z) ratio; and (5) detection. Metabolites can be identified on the basis of a combination of retention time (RT) and MS signature. TOF, time of flight; Q, quadrupole; IT, ion trap.

Overview chart listing the major steps and guidelines involved in typical MS-based metabolomics studies.
Quenching needs to satisfy two criteria: it should (1) completely terminate all enzyme and chemical activities and (2) avoid the perturbation of existing metabolite levels during harvesting. Details regarding specific considerations that need to be taken into account for quenching the metabolism of various species are provided in Supplementary Note 4. The efficiency of quenching can be followed either by controlled comparisons of various extraction methods38 or, alternatively, by determining the abundance of (stable isotope-labeled) standards spiked into the quenching solvent (see “Recovery and recombination experiments”). For tissues, where possible, quick excision followed by snap-freezing in liquid nitrogen is recommended, with subsequent storage of deep-frozen tissue at a constant −80 °C until the first application of extraction solvent. However, for bulky tissue, submersion in liquid nitrogen is not sufficient because the center of the tissue is cooled too slowly. In such cases, freeze-clamping, where tissue is almost instantaneously squashed flat between two prefrozen metal blocks (known as a Wohlehberger clamp), is preferred39,50.
Irrespective of the quenching method, the downstream steps of these processes also warrant caution. For example, improper freeze drying and lack of storage in sealed containers can generate artifactual geometric isomers of pigments39. Freeze drying is also unsuitable when volatile components are of interest. While the appropriate means of storage is strictly dependent on the stability of the class of targeted metabolites under study, it is not recommended to store samples between 0 and 40 °C. At these temperatures, substances can become concentrated in a residual aqueous phase39. It is therefore recommended, where necessary, to store completely dry residues for as short a time as possible before their analysis. In addition, great care must be taken to ensure that metabolism remains quenched during thawing. This is particularly pertinent for extracts containing secondary metabolites. In such extracts, degradative enzymes often retain their activities, which, if not kept in check, may result in the consumption or conversion of certain metabolites with a concomitant appearance of new compounds or breakdown products51.
Similar issues are also present with respect to both the experimental growth media and the initial extraction solvents used. Growth media often need to be removed via multiple wash steps to reduce the effects of ion suppression during the subsequent MS analysis, and the solvent used for initial extraction may need to be exchanged owing to incompatibility with the instrumentation used for the metabolite analysis. Two pitfalls are pertinent here: (1) the washing process results in the loss of metabolites and (2) solvent removal leads to concentration of the metabolites and thereby an acceleration of chemical reactions between them. Thus, considerable caution is advised in method optimization to ensure that extraction and handling methods allow adequate quantitative representation of cellular metabolites. In some instances, such as the analysis of volatile or semivolatile compounds, sample extraction and handling should only be performed on fresh material. We strongly recommend the adoption of recovery and recombination experiments (see below) when either a substantially novel metabolomics technique is introduced or a novel cell type, tissue or organism is studied.
Sample replication and randomization
An important issue is the nature and number of biological, technical and analytical replicates. Before using any new extraction protocol or analytical procedure and when working with new biological materials, it is essential to perform extensive pilot experiments to fully assess the technical variation that is necessary to design a statistically sound experiment. To avoid misunderstanding, we refer readers to the definitions of each type of replicate provided in ref. 39. While analytical replicates, that is, replicates corresponding to repeated injection of the exact same extract, are useful in assessing machine performance, technical replicates, which encompass the entire experimental procedure, allow a far more comprehensive assessment of any experimental variance in data generation39. Indeed, such analyses are essential for the establishment of a new extraction or processing procedure or a new analytical technique as well as for the optimization of a new instrument.
Biological replication is even more important and should involve at least four but preferably more replicates; the required number of replicates depends on the desired statistical power, effect size and actual variance52. Care must be taken to acquire such replicates in a highly uniform manner. For plants, this can also mean collecting samples at the same time of day and under the same environmental conditions. In many instances, a full and independent repeat of a biological experiment is advisable53. There are different stages where technical replicates can be made: at sampling, quenching, extraction and analysis, replicates can be made independently of the entire process. In our experience, the extraction step is the most critical of these. Whether technical replication is needed in support of biological replication is highly dependent on the relative magnitudes of variation; in cases in which the biological variation greatly exceeds the technical variation, it is sensible to sacrifice the latter to increase the former. With new systems, pilot experiments are highly recommended to evaluate biological and technical variation and hence determine how many samples and how many replicates are needed to achieve statistical robustness52.
Careful spatiotemporal randomization of biological samples throughout a metabolomics experiment is equally essential. If a set of samples is analyzed in a nonrandom order, treatment and control samples or time points may end up being measured under very different conditions. As a result, interpretation can be confounded by sample age or shifting instrument performance, potentially occluding biological variation between sample groups or, worse, creating artifactual differences. This is particularly important in large-scale metabolic profiling studies to characterize the natural variation of metabolism, akin to genome-wide association studies10,54,55,56. In such experiments, weeks of instrument time may be required. Clear best-practice guidelines for such large-scale studies have been presented elsewhere57,58,59,60, so we will not discuss them further here.
Irrespective of the size of the experiment, the use of quality-control samples and batch correction is also essential61. Such experimental controls help monitor instrument performance and stability and, thereby, data quality. These controls ensure that missing data or peaks with low signal-to-noise ratios do not occur. Either mixtures of authenticated metabolite samples at defined concentrations or dry-stored aliquots of a broadly shared and appropriately standardized biological extract (for example, multi-kilogram extracts of Arabidopsis, E. coli, yeast or human cell lines) can serve as broadly useful reference samples. Use of these references enhances accurate quantification and makes it possible to more effectively use the data in metabolite databases62,63,64,65,66. A pooled quality-control sample allows for evaluation (and correction) of run order and batch effects within a study, but not necessarily across experiments, as is possible with reference material.
Quantification
The aforementioned details of extraction, storage and replication are equally applicable when ensuring the accuracy of any method of metabolite quantification, including those that target single metabolites (Fig. 2). The remainder of this article will address issues that are, at least partially, restricted to untargeted metabolomics approaches. There are several essential aspects requiring consideration here.
First, it is essential to ensure that the levels of all metabolites of potential interest can be detected and, ideally, can be measured within a linear range of detection. This is most readily achieved through analyses of independent dilutions of each extract. Additionally, for experiments that begin with intact tissues, it is important to ensure complete tissue disruption. In the case of cellular studies, one must further take into consideration whether to limit the study to the endogenous cellular metabolites or also assess the exometabolome. For these controls, and many others, we provide a list of reporting recommendations in the section below on transparency in measurement, metabolite annotation and documentation.
Metabolomics data are most frequently provided as relative quantities (that is, relative quantification is performed) with respect to a reference sample. This is in contrast to NMR-based studies, which usually provide absolute concentrations (that is, absolute quantification), with peak intensities directly proportional to concentrations and directly comparable across different peaks and samples. The relative intensities of LC–MS and GC–MS peaks representing different compounds do not directly correlate to absolute concentrations. This is due to the differential ionization efficiencies of the different metabolites within a complex mixture.
To address this issue, standard curves can be used to determine how signal intensity responds as a function of analyte concentration and, moreover, the range of linearity of this relationship12. The ability to generate such curves is of course dependent on the availability of validated pure standards. While relative values are highly useful in many contexts and indeed are the only way of expressing the levels and changes in level of non-annotated analytes, absolute values have much greater utility for determining enzyme binding site occupancies, the thermodynamics of metabolic reactions12,67 and the molecular dynamics underlying the flow of atoms through a metabolic network68,69,70. A further advantage of the methods used for absolute quantification is that they can be readily adapted into a means of quality control for both quantification and the correctness of peak annotation, for example, through thermodynamics71. However, obtaining standard curves for thousands of metabolites in a complex mixture is currently not always practical. While many of the metabolite signals in such mixtures are nonlinear owing to a variety of reasons, including ion interaction, ion suppression, etc., which substantially complicates quantitation (as described in the next section), there are experimental tools allowing the extent of this problem to be quantified and reported. Quantification is particularly problematic in the case of external calibration, where quantification of standards is carried out in a far simpler mixture than that of the biological extract. Therefore, either internal quantification using isotopically labeled standards or quantification of a mixture of internal and external standards, as described below, is preferable.
A further aspect of quantification is the basis on which quantities are expressed for tissue samples. Data are often provided per gram of fresh or dry weight, while for body fluids they are often provided per volume. The case of cellular metabolomics is more complicated given that cell size is often variable; values are therefore typically provided per milligram of protein or based on cell counts. The basis on which both absolute and relative metabolite levels is provided is of fundamental importance—for example, values given on the basis of fresh weight can be dramatically influenced by the osmotic potential of the cell—yet is often not given enough consideration by the community.
Recovery and recombination experiments
Recovery experiments, in which authenticated standard compounds are added to the initial extraction solvent to assess losses during extraction, storage and handling, were vigorously championed in the 1970s to 1990s72 and can provide persuasive evidence that the data reported are a valid reflection of cellular metabolite compositions39. Recent examples exist of validated methods in microbial, plant and mammalian systems73,74,75. However, the metabolomics community has been relatively slow in adopting these control procedures. This is partially explained by the lack of commercially available standards and/or simple synthetic approaches to make standards. Indeed, for unknown analytes, this approach is by its nature impossible.
Fortunately, there is an alternative approach—extract recombination—that circumvents this practical limitation. In this approach, the extract of a novel tissue is characterized by combination with that of a well-characterized reference material such as one from E. coli, Arabidopsis or human biofluids. Such experiments not only provide information concerning the appropriateness of the extraction buffer but additionally allow an assessment of so-called matrix effects caused by ion suppression76,77,78. These experiments additionally allow a quantitative assessment of the reliability of known peaks79. A schematic representation of recovery and metabolic recombination experiments is presented in Fig. 3.

a,b, Recovery tests were performed using GC–MS (a) and LC–MS (b) peaks obtained for a mixture of extracts from Arabidopsis and lettuce leaves. The mixture was made by combining extracts from Arabidopsis (A) and lettuce (B) leaves (0.2 mg fresh weight per μl) at a 1:1 ratio. The percentage recovery was estimated using the theoretical concentration in the extract mixture: ((level in leaves (A) × A%) + (level in leaves (B) × B%))/100. Dashed lines indicate the acceptable range of 70–130%. Compounds in gray are statistically outside this range. Error bars represent ± s.e.m.
For known metabolites, we suggest that recovery or metabolic recombination experiments be carried out for each new tissue type or species. It is clear that in any metabolomics-scale study certain metabolites will have poor recovery. While this does not preclude the reporting of their values, it is important that this is documented to allow readers discretion in their interpretation. Recovery rates of 70–130% are acceptable, with anything deviating beyond this range representing a metabolite whose quantification should be subject to further testing. For example, even a 50% recovery rate—if reproducible and linear—could be deemed acceptable (Fig. 3). The importance of such control experiments is perhaps best illustrated by cases in which they were not performed. Anecdotally, there are several examples in the literature where the metabolite data reported cannot be reflective of cellular content, for example, because the zero levels reported for metabolites, if representative of cellular levels, would indicate that the cells tested were not viable.
Ion suppression
Despite the selectivity and sensitivity of MS techniques, there are considerable challenges with regard to reproducibility and accuracy when analyzing complex samples. These problems are not insurmountable but require that additional care be taken when interpreting results. Ion suppression is a general problem in LC–MS analyses due to matrix effects influencing the ionization of co-eluting analytes, affecting the precision and accuracy of quantification or preventing less abundant metabolites from being detected at all76,78,80. As mentioned above, the best method of assessing the potential impact of ion suppression is to mix two independent extracts in a recombination experiment (Fig. 3) and assess whether the metabolites detected can be quantitatively recovered51. Essentially, within this process, co-eluting analytes compete for the ionization energy, resulting in incomplete ionization. Therefore, a decreased ion count for an analyte may be due either to a decreased concentration of the analyte itself or to increased concentrations of co-eluting analytes. It is critically important to consider these effects during method validation to ensure the quality of the analysis.
While there is no universal solution to the ion suppression problem, assessing the effects of ion suppression affords greater confidence in the accuracy of the results. There are several strategies that can help minimize ion suppression77. Among these, improvements in sample preparation and chromatographic selectivity are currently the most effective. In some situations, using suitable clean-up procedures depending on sample type and analyte properties may allow removal of co-eluting components. This might involve simple dilution of extracts or the growth media from which the samples are derived51 or optimization of various steps of sample work-up, including sonication, solvent partitioning, filtration, centrifugation and protein precipitation81. In addition, solid-phase extraction (SPE) using appropriate absorbents has been demonstrated to be an effective method to reduce matrix effects. Furthermore, it is possible to adjust chromatography conditions so that the peaks of interest do not elute in regions of suppression; for example, modifying the composition of the mobile phase or gradient conditions can aid chromatographic separation and thereby improve performance.
Careful selection of the ion source and column polarity is an alternative strategy to reduce ion suppression. For example, atmospheric pressure chemical ionization (APCI) is less prone to matrix effects than electrospray ionization (ESI). In addition, using APCI can also reduce interference effects12. It has been demonstrated that ion suppression is often less severe for negatively charged compounds than for positively charged ones82. Finally, although the above-mentioned strategies may not be sufficient to completely remove the effects of ion suppression in complex samples, the extent of the problem can at least be quantified by carrying out control experiments as described in the preceding section.
Peak misidentification
The orthogonal use of chromatography (either gas or liquid based) with MS and in some cases also tandem MS (MS/MS) fragmentation patterns provides great specificity83,84. Current high-end instruments detect on the order of 10,000 or 100,000 features; however, these include a large number of adduct and isotope peaks. Bioinformatics tools for analyte identification take this into account and even use commonly observed adducts as a means of identifying analytes (discussed in detail below). Nonetheless, there are three common problems that contribute to misidentification.
First, isomers—compounds with an identical molecular formula but distinct structures—are common in nature. Important examples from primary metabolism include hexose phosphates and inositol phosphates, citrate and isocitrate, glucose and fructose, and alanine and sarcosine. High-resolution MS alone may not suffice to discriminate between these and other sets of isomers, especially when fragmentation patterns are similar, and some types of isomers may not separate well on conventional reverse-phase high-performance LC (HPLC). To improve separation, reverse-phase ion pairing chromatography, hydrophilic interaction chromatography (HILIC) and other chromatographic methods can be used; another option is chemical derivatization before chromatography12. In cases where isomers cannot be separated, this needs to be clearly stated because such compounds may have greatly different biological functions.
Second, the presence of overlapping compounds may prevent detection of some metabolites. While the increasingly high resolution of mass spectrometers has mitigated this issue to some extent, the resolving power of many current instruments is insufficient to separate ions differing in mass by less than 5 parts per million (ppm)12. This problem, however, is only acute when chromatography is also unable to separate analytes that cannot be separated on the basis of mass.
The third major hurdle (which is more relevant for LC–MS than GC–MS) is the formation of in-source degradation products. These are by-product ions of ESI due to simple loss of water, carbon dioxide or hydrogen phosphate, more complicated molecular rearrangements and the attachment of other ions. In-source degradation reduces the intensity of the metabolite parent ion, and the resulting fragment ions may confound analysis of other co-eluting compounds, for example, if they have the same molecular formula as the molecular ion of another metabolite12. We provide examples of these from our own work in Supplementary Fig. 1. These examples demonstrate the need for careful manual curation of all peak assignments, which, however, is often not feasible when annotating several hundred or thousand metabolites (Fig. 4). In ambiguous cases, the exact identification of a peak can often be best demonstrated via comparative biochemical approaches, for example, by analyzing the metabolome in known mutants that can be anticipated to lack certain metabolites24,85 or incubation of a purified peak with known enzymes or chemical treatments73. These methods can also be combined with other approaches such as using authenticated standards for isomer annotation86 and dual-labeling approaches87.

a,b, Structure elucidation workflow for data acquisition (a) and processing and annotation (b). c, Simple design for metabolic data documentation and how data can be linked to the mzTab49 tool to facilitate data representation, sharing and deposition to public repositories.
As an aside, a critical aspect of nontargeted metabolomics is peak filtering. Metabolomics datasets from such studies contain a large proportion of uninformative features that can impede subsequent statistical analysis, and there is thus a need for versatile and data-adaptive methods for filtering data before investigating the underlying biological phenomena88. A list of suggestions for the design and implementation of data filtering strategies is provided in Supplementary Note 5.
Reporting transparency
To fully exploit metabolomics data, they need to be comparable between different laboratories. Indeed, several comparative studies have been published, as we detail in Supplementary Note 6. In addition to comparability at a quantitative level, clear metabolite ontologies are also needed to ensure that metabolites are annotated in a common fashion (Supplementary Note 7).
To ensure that methods can be readily adopted by others, a wealth of detailed information is required. However, detailed descriptions of sample preparation and analytical procedures are often (at least partially) absent in publications, especially in cases where metabolomics is not the primary focus of the published work. We recommend that the following items be considered as mandatory components of any methods section for metabolomics experiments.
-
Chromatography: composition of the mobile phase, column properties, temperature, flow rate and injection volume
-
Mass spectrometry: ionization source and type of detection mode, MS method, scan number and speed, and MS/MS parameters, including resolution settings and the energy used for fragmentation (Box 1)
Extensive recommendations have been made before36,39; however, we believe that this list will need to be revisited frequently owing to improvements in instrumentation and other aspects of the metabolomics workflow. If unsure of how much methodological detail to provide, imagine that your twin is sitting on a different continent in front of similar instrumentation and has to configure the equipment in a comparable manner. Increasingly, there is software support to extract such information from raw data files converted into, for example, the mzML file format44 (Fig. 4c).
Considering the number of possible pitfalls in the annotation and quantification of metabolites in metabolomics approaches, the current general level of reporting in the literature is not entirely satisfactory (Figs. 4 and 5). Given restrictive journal word limits and the fact that scientific reports tend to be highly concise, it is perhaps not surprising that authors do not refer to compounds as ‘the metabolite that we putatively annotate as X’ within the text of their articles. That said, there is nothing to preclude highly detailed reporting of the exact nature of the annotation within the supplementary data associated with a paper, either copublished or made available through separate web resources. Databases such as MetaboLights89 and the Metabolomics Workbench90 can be used for this purpose and indeed have been adopted as a requirement for many journals.

Structure elucidation workflow of metabolite identification. MS/MS fragmentation provides information about compound structure. Metabolite annotation can be achieved using reference compounds, MS2 analysis, NMR or a photodiode array (PDA) detector for UV–visible light spectrum detection. Database searching enables molecular formula calculation. Illustrated is an example of our recommendations for reporting metabolomics data for a typical LC–MS experiment for the compound rutin (a flavonoid glycoside). Comparison of the MS and MS/MS spectra for rutin reveals a peak at 611 m/z in the MS scan and two major fragments at 611 m/z in the MS/MS scan, providing information about chemical loss of rhamnose (−146 m/z) and glucose (–162 m/z) moieties. For metabolite documentation, the current general recommended levels of reporting are shown; see Supplementary Tables 1 and 2 for further details.
We recommend a streamlined, simpler reporting approach (Fig. 5). While this is similar to that previously suggested for plant analyses39, we have updated reporting recommendations to ensure broader applicability and relevance. To simplify the adoption of these recommendations, we supply Supplementary Tables 1 and 2 as template Microsoft Excel spreadsheets. Supplementary Table 1 contains a list of simple questions regarding the reporting of metabolite data, and Supplementary Table 2 provides recommendations for metabolite annotation for typical GC–MS or LC–MS experiments. Once one is used to filling out these tables, it is our experience that it takes between 30 and 60 minutes to complete the process. In the case of large datasets consisting of hundreds to thousands of samples, which nowadays represent what is reported in a sizeable proportion of metabolomics papers, the time for upload in metabolomics repositories is thus considerably longer than the filling out of our suggested Excel tables.
Summary
In summary, we have presented here recommendations to improve the quality and cross-laboratory comparability of metabolic datasets. These range from recommendations on sampling and metabolite extraction, quantification and peak identification to guidelines on transparency in measurement and documentation, for which a data- rather than chromatogram-centric approach is suggested. We anticipate that the adoption of these recommendations will offer several advantages: (1) perusal of reported metadata will provide readers with the ability to assess the quality of the data reported and, as such, allow greater confidence in the conclusions drawn; (2) researchers will have a simple route to gain information needed to aid them in annotating their own experimental output and (3) data obtained by multiple laboratories may be compared more easily.
A recent example of comprehensive documentation of a metabolomics experiment is provided by the study of Price et al.91, who evaluated metabolite levels in understudied crop species, assembling an extensive database of the underlying data. Greater adoption of simple reporting tables such as the ones we describe here (Supplementary Tables 1 and 2) or the similar one proposed by Dorrestein and coworkers (for a comparison of these tables, see Supplementary Note 8) has the potential to elucidate general aspects of the metabolic response.
We would like to stress that the intention of the recommendations presented here is to encourage fuller and more faithful reporting of both metabolite annotations and their respective quantification. Our proposed reporting standards are not meant to be a direct replacement for the standards set by metabolome repositories. In fact, in most instances, these are entirely complementary to one another. We recommend that metabolomics practitioners follow repository standards alongside those we discuss here. There is a wealth of data reported in the literature that, for one reason or another, have not been deposited in repositories (such as MetaboLights, the Metabolomics Workbench and GNPS-MassIVE), and for such data it would be excellent if the metadata could be captured. This is important not only for possible reuse of the data but equally as a means of allowing the reader the possibility to evaluate their veracity. Expansion of such approaches, including input from both experimental and computational scientists, will facilitate the generation of pan-metabolome databases, which will undoubtedly open new horizons for metabolomics in all kingdoms of life.
We believe that more widespread adoption of these recommendations will enhance the quality of reporting of metabolite data, advance community efforts to improve the annotation of metabolomes and, finally, facilitate the exchange and comparability of metabolite data from different laboratories. These efforts will also facilitate comparison of metabolomics datasets obtained from different species, supporting the renaissance of comparative biochemistry.
References
Doerr, A. Global metabolomics. Nat. Methods 14, 32 (2017).
Fessenden, M. Metabolomics: small molecules, single cells. Nature 540, 153–155 (2016).
Oliver, S. G., Winson, M. K., Kell, D. B. & Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 16, 373–378 (1998).
Alseekh, S. & Fernie, A. R. Metabolomics 20 years on: what have we learned and what hurdles remain? Plant J. 94, 933–942 (2018).
Chevalier, C. et al. Gut microbiota orchestrates energy homeostasis during cold. Cell 163, 1360–1374 (2015). This paper demonstrates that the microbiota is a key factor orchestrating overall energy homeostasis during increased demand in mammals.
Chu, C. et al. The microbiota regulate neuronal function and fear extinction learning. Nature 574, 543–548 (2019).
Djamei, A. et al. Metabolic priming by a secreted fungal effector. Nature 478, 395–398 (2011).
Dorr, J. R. et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501, 421–425 (2013). This paper illustrates the identification of metabolite biomarkers for use in cancer diagnostics and to serve as targets for new-concept anticancer therapies.
Fernie, A. R., Trethewey, R. N., Krotzky, A. J. & Willmitzer, L. Metabolite profiling: from diagnostics to systems biology. Nat. Rev. Mol. Cell Biol. 5, 763–769 (2004).
Gieger, C. et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet. 4, e100282 (2008).
Guijas, C., Montenegro-Burke, J. R., Warth, B., Spilker, M. E. & Siuzdak, G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat. Biotechnol. 36, 316–320 (2018).
Lu, W. et al. Metabolite measurement: pitfalls to avoid and practices to follow. Annu. Rev. Biochem. 86, 277–304 (2017). A useful and comprehensive review highlighting the pitfalls encountered in metabolomics and providing guidelines for accurate metabolite measurements.
Mashego, M. R. et al. Microbial metabolomics: past, present and future methodologies. Biotechnol. Lett. 29, 1–16 (2007).
Rinschen, M. M., Ivanisevic, J., Giera, M. & Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 20, 353–367 (2019).
Van Gulik, W. M. et al. Fast sampling of the cellular metabolome. Methods Mol. Biol. 881, 279–306 (2012).
Delzenne, N. M. & Bindels, L. B. Microbiome metabolomics reveals new drivers of human liver steatosis. Nat. Med. 24, 906–907 (2018).
Guo, A. C. et al. ECMDB: the E. coli metabolome database. Nucleic Acids Res. 41, D625–D630 (2013).
Sajed, T. et al. ECMDB 2.0: a richer resource for understanding the biochemistry of E. coli. Nucleic Acids Res. 44, D495–D501 (2016).
Hautbergue, T., Jamin, E. L., Debrauwer, L., Puel, O. & Oswald, I. P. From genomics to metabolomics, moving toward an integrated strategy for the discovery of fungal secondary metabolites. Nat. Prod. Rep. 35, 147–173 (2018).
Ramirez-Gaona, M. et al. YMDB 2.0: a significantly expanded version of the yeast metabolome database. Nucleic Acids Res. 45, D440–D445 (2017).
Wishart, D. S. et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46, D608–D617 (2018). The database described is a groundbreaking, comprehensive and freely available web resource containing detailed information about the human metabolome.
Saito, K. & Matsuda, F. Metabolomics for functional genomics, systems biology, and biotechnology. Annu. Rev. Plant Biol. 61, 463–489 (2010).
Lisec, J., Schauer, N., Kopka, J., Willmitzer, L. & Fernie, A. R. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 1, 387–396 (2006).
Tohge, T. & Fernie, A. R. Combining genetic diversity, informatics and metabolomics to facilitate annotation of plant gene function. Nat. Protoc. 5, 1210–1227 (2010).
van Gulik, W. M. Fast sampling for quantitative microbial metabolomics. Curr. Opin. Biotechnol. 21, 27–34 (2010).
Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography–mass spectrometry. Anal. Bioanal. Chem. 403, 1523–1548 (2012).
Wang, Z., Gerstein, M. & Snyder, M. RNA-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Bennett, B. D., Yuan, J., Kimball, E. H. & Rabinowitz, J. D. Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat. Protoc. 3, 1299–1311 (2008).
Li, B. et al. NOREVA: normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 45, W162–W170 (2017).
Fiehn, O. Metabolomics—the link between genotypes and phenotypes. Plant Mol. Biol. 48, 155–171 (2002).
Papadimitropoulos, M. P., Vasilopoulou, C. G., Maga-Nteve, C. & Klapa, M. I. Untargeted GC–MS metabolomics. Methods Mol. Biol. 1738, 133–147 (2018).
Fiehn, O. et al. The metabolomics Standards Initiative (MSI). Metabolomics 3, 175–178 (2007). A brief report outlining the history, stature and intentions of MSI, an authorative standards initiative for metabolomics.
Haug, K. et al. MetaboLights—an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 41, D781–D786 (2013).
Salek, R. M., Haug, K. & Steinbeck, C. Dissemination of metabolomics results: role of MetaboLights and COSMOS. GigaScience 2, 8 (2013).
Steinbeck, C. et al. MetaboLights: towards a new COSMOS of metabolomics data management. Metabolomics 8, 757–760 (2012).
Sumner, L. W. et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3, 211–221 (2007).
Sansone, S. A. et al. The Metabolomics Standards Initiative. Nat. Biotechnol. 25, 846–848 (2007). This article highlights two standards and guidelines papers for MS and sample preparation by the Human Proteome Organization Proteomics Standardization Initiative (HUPO-PSI) and the Functional Genomics Experiment (FuGE) and describes how metabolomics standards should align to these.
Spicer, R. A., Salek, R. & Steinbeck, C. A decade after the Metabolomics Standards Initiative: it’s time for a revision. Sci. Data 4, 3 (2017).
Fernie, A. R. et al. Recommendations for reporting metabolite data. Plant Cell 23, 2477–2482 (2011).
Aksenov, A. A. et al. A machine learning workflow enables automatic deconvolution of GC-MS data. Nat. Biotechnol. 39, 169–173 (2021).
Aron, A. T. et al. Reproducible molecular networking of untargeted mass spectrometry data using GNPS. Nat. Protoc. 15, 1954–1991 (2020).
Blaženović, I. et al. Structure annotation of all mass spectra in untargeted metabolomics. Anal. Chem. 91, 2155–2162 (2019).
Buendia, P. et al. Ontology-based metabolomics data integration with quality control. Bioanalysis 11, 1139–1155 (2019).
Hoffmann, N. et al. mzTab-M: a data standard for sharing quantitative results in mass spectrometry metabolomics. Anal. Chem. 91, 3302–3310 (2019).
Jarmusch, A. K. et al. ReDU: a framework to find and reanalyze public mass spectrometry data. Nat. Methods 17, 901–904 (2020). This tool enables the capture of public MS-based metabolomics data and their subsequent reanalysis.
Nothias, L. F. et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 17, 905–908 (2020).
Tsugawa, H. et al. A cheminformatics approach to characterize metabolomes in stable-isotope-labeled organisms. Nat. Methods 16, 295–298 (2019).
Huan, T. et al. Systems biology guided by XCMS Online metabolomics. Nat. Methods 14, 461–462 (2017).
Patel, V. R., Eckel-Mahan, K., Sassone-Corsi, P. & Baldi, P. CircadiOmics: integrating circadian genomics, transcriptomics, proteomics and metabolomics. Nat. Methods 9, 772–773 (2012).
Palladino, G. W., Wood, J. J. & Proctor, H. J. Modified freeze clamp technique for tissue assay. J. Surg. Res. 28, 188–190 (1980).
Tohge, T. et al. From models to crop species: caveats and solutions for translational metabolomics. Front. Plant Sci. 2, 61 (2011).
Trutschel, D., Schmidt, S., Grosse, I. & Neumann, S. Experiment design beyond gut feeling: statistical tests and power to detect differential metabolites in mass spectrometry data. Metabolomics 11, 851–860 (2015).
Sanchez, D. H., Szymanski, J., Erban, A., Udvardi, M. K. & Kopka, J. Mining for robust transcriptional and metabolic responses to long-term salt stress: a case study on the model legume Lotus japonicus. Plant Cell Environ. 33, 468–480 (2010).
Chen, W. et al. Comparative and parallel genome-wide association studies for metabolic and agronomic traits in cereals. Nat. Commun. 7, 12767 (2016).
Fuhrer, T., Zampieri, M., Sevin, D. C., Sauer, U. & Zamboni, N. Genomewide landscape of gene–metabolome associations in Escherichia coli. Mol. Syst. Biol. 13, 907 (2017).
Hartiala, J. A. et al. Genome-wide association study and targeted metabolomics identifies sex-specific association of CPS1 with coronary artery disease. Nat. Commun. 7, 10558 (2016).
Alseekh, S., Wu, S., Brotman, Y. & Fernie, A. R. Guidelines for sample normalization to minimize batch variation for large-scale metabolic profiling of plant natural genetic variance. Methods Mol. Biol. 1778, 33–46 (2018).
Dunn, W. B. et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 6, 1060–1083 (2011).
Griffin, J. L. et al. Standard reporting requirements for biological samples in metabolomics experiments: mammalian/in vivo experiments. Metabolomics 3, 179–188 (2007).
Vandeputte, D., Tito, R. Y., Vanleeuwen, R., Falony, G. & Raes, J. Practical considerations for large-scale gut microbiome studies. FEMS Microbiol. Rev. 41, S154–S167 (2017).
Wehrens, R. et al. Improved batch correction in untargeted MS-based metabolomics. Metabolomics 12, 88 (2016).
Cui, Q. et al. Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol. 26, 162–164 (2008).
Nakamura, Y. et al. KNApSAcK Metabolite Activity Database for retrieving the relationships between metabolites and biological activities. Plant Cell Physiol. 55, e7 (2014).
Tautenhahn, R. et al. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat. Biotechnol. 30, 826–828 (2012).
Vinaixa, M. et al. Mass spectral databases for LC/MS- and GC/MS-based metabolomics: state of the field and future prospects. Trac-Trends Anal. Chem. 78, 23–35 (2016).
Zhu, Z. J. et al. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 8, 451–460 (2013).
Niebel, B., Leupold, S. & Heinemann, M. An upper limit on Gibbs energy dissipation governs cellular metabolism. Nat. Metab. 1, 125–132 (2019).
Jourdan, F., Breitling, R., Barrett, M. P. & Gilbert, D. MetaNetter: inference and visualization of high-resolution metabolomic networks. Bioinformatics 24, 143–145 (2008).
Pirhaji, L. et al. Revealing disease-associated pathways by network integration of untargeted metabolomics. Nat. Methods 13, 770–776 (2016).
Shen, X. T. et al. Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics. Nat. Commun. 10, 1516 (2019). This represents an important example of a tool using a metabolic reaction network that expands metabolite annotations without the need for a comprehensive standard spectral library.
Kummel, A., Panke, S. & Heinemann, M. Putative regulatory sites unraveled by network-embedded thermodynamic analysis of metabolome data. Mol. Syst. Biol. 2, 2006.0034 (2006).
Ap Rees, T. & Hill, S. A. Metabolic control analysis of plant metabolism. Plant Cell Environ. 17, 587–599 (1994).
Arrivault, S. et al. Use of reverse-phase liquid chromatography, linked to tandem mass spectrometry, to profile the Calvin cycle and other metabolic intermediates in Arabidopsis rosettes at different carbon dioxide concentrations. Plant J. 59, 826–839 (2009).
Lu, W., Bennett, B. D. & Rabinowitz, J. D. Analytical strategies for LC–MS-based targeted metabolomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 871, 236–242 (2008).
Lunn, J. E. et al. Sugar-induced increases in trehalose 6-phosphate are correlated with redox activation of ADP-glucose pyrophosphorylase and higher rates of starch synthesis in Arabidopsis thaliana. Biochem. J. 397, 139–148 (2006).
Annesley, T. M. Ion suppression in mass spectrometry. Clin. Chem. 49, 1041–1044 (2003).
Antignac, J. P., Marchand, P., Le Bizec, B. & Andre, F. Identification of ractopamine residues in tissue and urine samples at ultra-trace level using liquid chromatography–positive electrospray tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 774, 59–66 (2002).
Kebarle, P. & Tang, L. From ions in solution to ions in the gas phase—the mechanism of electrospray mass spectrometry. Anal. Chem. 65, 972A–986A (1993).
Roessner-Tunali, U. et al. De novo amino acid biosynthesis in potato tubers is regulated by sucrose levels. Plant Physiol. 133, 683–692 (2003).
Buhrman, D. L., Price, P. I. & Rudewiczcor, P. J. Quantitation of SR 27417 in human plasma using electrospray liquid chromatography–tandem mass spectrometry: a study of ion suppression. J. Am. Soc. Mass Spectrom. 7, 1099–1105 (1996).
Gerssen, A. et al. Solid phase extraction for removal of matrix effects in lipophilic marine toxin analysis by liquid chromatography–tandem mass spectrometry. Anal. Bioanal. Chem. 394, 1213–1226 (2009).
Freitas, L. G., Götz, C. W., Ruff, M., Singer, H. P. & Müller, S. R. Quantification of the new triketone herbicides, sulcotrione and mesotrione, and other important herbicides and metabolites, at the ng/l level in surface waters using liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 1028, 277–286 (2004).
De Vijlder, T. et al. A tutorial in small molecule identification via electrospray ionization–mass spectrometry: the practical art of structural elucidation. Mass Spectrom. Rev. 37, 607–629 (2018).
Dettmer, K., Aronov, P. A. & Hammock, B. D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 26, 51–78 (2007).
Tohge, T. et al. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 42, 218–235 (2005).
Shahaf, N. et al. The WEIZMASS spectral library for high-confidence metabolite identification. Nat. Commun. 7, 12423 (2016). This represents a reference metabolite spectral library developed from high-resolution MS data acquired from a structurally diverse set of 3,540 plant metabolites, providing great promise for addressing the question of comprehensivity in metabolomics.
Feldberg, L., Venger, I., Malitsky, S., Rogachev, I. & Aharoni, A. Dual labeling of metabolites for metabolome analysis (DLEMMA): a new approach for the identification and relative quantification of metabolites by means of dual isotope labeling and liquid chromatography–mass spectrometry. Anal. Chem. 81, 9257–9266 (2009).
Schiffman, C. et al. Filtering procedures for untargeted LC–MS metabolomics data. BMC Bioinformatics 20, 334 (2019).
Kale, N. S. et al. MetaboLights: an open-access database repository for metabolomics data. Curr. Protoc. Bioinformatics 53, 14.13.11–14.13.18 (2016).
Sud, M. et al. Metabolomics Workbench: an international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 44, D463–D470 (2016).
Price, E. J. et al. Metabolite database for root, tuber, and banana crops to facilitate modern breeding in understudied crops. Plant J. 101, 1258–1268 (2020). Recent exemplary documentation of a metabolomics experiment that evaluated metabolite levels in crop species, providing not only an extensive database but moreover an excellent example of how to correctly investigate understudied species.
Bino, R. J. et al. Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 9, 418–425 (2004).
Kirwan, J. A. et al. Preanalytical processing and biobanking procedures of biological samples for metabolomics research: a white paper, community perspective (for ‘Precision Medicine and Pharmacometabolomics Task Group’–The Metabolomics Society Initiative). Clin. Chem. 64, 1158–1182 (2018).
Acknowledgements
A.R.F. and S.A. are supported by the European Union’s Horizon 2020 research and innovation program, under PlantaSYST (SGA-CSA no. 739582 under FPA no. 664620). J.E. and S.S. were supported by German Research Foundation (DFG) grant numbers 210879364 and 239748522. P.D.F. is grateful for funding from the CGIAR Research Program on Roots, Tubers and Bananas (RTB) and is supported by CGIAR Fund Donors and the Biotechnology and Biological Sciences Research Council OPTICAR Project (project BB/P001742/1). R.D.H. acknowledges receipt of the Nils Foss Food Excellence Prize, which funded his time spent on this initiative. W.W. is supported by the Huazhong Agricultural University Scientific & Technological Self-Innovation Foundation (program no. 2017RC002). K.C. and M.P.S. are supported by the NIH under grant numbers 5U54HG010426-03 and 1U2CCA233311-01. H.T. acknowledges financial support from the Shanghai Municipal Science and Technology Major Project (2017SHZDZX01) and the National Natural Science Foundation of China (31821002). G.X. is supported by the National Natural Science Foundation of China (21934006). T.T. was supported by JSPS KAKENHI grants-in-aid (19H03249 and 19K06723). G.S. is supported by the NIH under grant number R35GM130385.
Author information
Authors and Affiliations
Contributions
A.R.F. and SA. wrote the manuscript with contributions and input from all authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Methods thanks Justin van der Hooft and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Allison Doerr was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Notes 1–8 and Fig. 1.
Supplementary Table 1
Metabolite reporting checklist.
Supplementary Table 2
Metabolite annotation and documentation.
Rights and permissions
About this article

Cite this article
Alseekh, S., Aharoni, A., Brotman, Y. et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat Methods 18, 747–756 (2021). https://doi.org/10.1038/s41592-021-01197-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-021-01197-1
This article is cited by
-
Lavandula angustifolia mill. for a suitable non-invasive treatment against fungal colonization on organic-media cultural heritage
Heritage Science (2024)
-
Quartet metabolite reference materials for inter-laboratory proficiency test and data integration of metabolomics profiling
Genome Biology (2024)
-
An assessment of AcquireX and Compound Discoverer software 3.3 for non-targeted metabolomics
Scientific Reports (2024)
-
Plastid ancestors lacked a complete Entner-Doudoroff pathway, limiting plants to glycolysis and the pentose phosphate pathway
Nature Communications (2024)
-
Identifying key soil characteristics for Francisella tularensis classification with optimized Machine learning models
Scientific Reports (2024)
