
Abstract
Mitophagy impairment and oxidative stress are cardinal pathological hallmarks in Parkinson’s disease (PD), a common age-related neurodegenerative condition. The specific interactions between mitophagy and reactive oxygen species (ROS) have attracted considerable attention even though their exact interplay in PD has not been fully elucidated. We highlight the interactions between ROS and mitophagy, with a focus on the signalling pathways downstream to ROS that triggers mitophagy and draw attention to potential therapeutic compounds that target these pathways in both experimental and clinical models. Identifying a combination of ROS inhibitors and mitophagy activators to provide a physiologic balance in this complex signalling pathways may lead to a more optimal outcome. Deciphering the exact temporal relationship between mitophagy and oxidative stress and their triggers early in the course of neurodegeneration can unravel mechanistic clues that potentially lead to the development of compounds for clinical drug trials focusing on prodromic PD or at-risk individuals.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative condition identified as one of the leading causes of disability, particularly in countries with rapidly growing aging populations and with a prevalence that is predicted to double in the next 20 years1,2,3. A selective loss of dopaminergic (DA) neurons from the pars compacta of the substantia nigra (SN) and the formation of Lewy bodies constitute the primary pathological hallmarks of PD. Clinically, patients frequently present with tremor, rigidity, postural instability and bradykinesia accompanied by nonmotor symptoms (such as fatigue, depression, and dementia)1. The etiology of PD has been extensively investigated and is likely due to a combination of genetic, epigenetic and environmental factors. Clinical, postmortem and animal model studies suggest that mitochondrial dysfunction, oxidative stress, impaired proteasomal system, impaired autophagy/mitophagy, and neuroinflammation dysregulation1,4,5 contribute to the onset and development of PD. Available therapies are largely symptomatic, and disease-modifying therapies are still lacking, highlighting the necessity of therapy targeting key areas of PD pathogenesis6. There is growing evidence implicating that mitophagy impairment7,8 and oxidative stress9,10 are critical factors of neurodegeneration over time in PD.
Mitophagy
Mitophagy refers to autophagy that selectively degrades damaged mitochondria. There exists a multi-tiered, highly integrated network of mitochondrial quality control (MQC) which regulates various cellular processes to maintain the structural and functional integrity of mitochondria in response to various stressors (oxidative stress, mutagenic stress, and proteotoxicity etc). Various MQC pathways are dysregulated in PD, including mitochondrial regeneration (biogenesis11, protein import12), mitochondrial dynamics (fission, fusion)13,14 mitochondria-derived vesicles15 and mitochondrial removal (mitophagy)7,16.
An essential component of mitochondrial homeostasis, mitophagy can be defined as the selective degradation of mitochondria, with mitochondria enveloped by autophagosomes before being degraded in the lysosome17,18. Given that this breakdown of mitochondria via autophagosome formation is crucial for neuronal health, compromised mitophagy has been implied in PD, and associated with accelerated neurodegeneration19,20. Based on their relevance to PD pathogenesis, mitophagy pathways can be categorised into Parkin-dependent and Parkin-independent pathways, with the latter largely comprising of receptor-mediated mitophagy, lipid-mediated mitophagy and ubiquitin-mediated mitophagy.
Parkin/PINK1-mediated mitophagy
Mechanisms of Parkin/PINK1-mediated mitophagy
Mutations in PINK1 (a serine/threonine kinase)21 and Parkin (an E3 ubiquitin ligase)22 are the most common causes of recessive forms of PD. PINK1 and Parkin are the key components in the Parkin/PINK1-mediated mitophagy23,24. Under basal conditions, PINK1 translocates to the inner mitochondrial membrane (IMM) for cleavage by mitochondrial proteases25. Subsequently, truncated PINK1 is targeted to the proteasome for eventual degradation via an N-end rule pathway26. Mitochondrial stress or damage causes the collapse of mitochondrial membrane potential, preventing this manner of translocation of PINK1 which instead accumulates on the outer mitochondrial membrane (OMM)25,27 and undergoes autophosphorylation28. In this initiation stage, PINK1 phosphorylates OMM conjugated ubiquitins at Serine 65, driving Parkin to be recruited from cytosol to the mitochondria29,30. Phosphorylated ubiquitin may both activate Parkin and act as an anchor for Parkin on mitochondria where Parkin is additively activated by PINK1 phosphorylation24,29,30.
The phosphorylation of ubiquitin and Parkin triggers a series of structural remodelling for Parkin, converting it from a dormant enzyme with self-inhibited conformation to an active and promiscuous E3 ligase which mediates ubiquitination of OMM proteins non-selectively29,31,32,33. Among the Parkin substrates, Miro were ubiquitinated and degraded to arrest mitochondrial motility, priming mitochondria for autophagic clearance34. Ubiquitin attached to the substrates on the damaged mitochondria can be phosphorylated by PINK1 to recruit and activate more Parkin and form a positive feedback loop to propel the cascade of mitophagy29,31,35. Subsequently, the ubiquitinated substrates serve as a degradation signal to be recognized by autophagy receptors, including OPTN and NDP5223. This occurs concomitantly with Parkin/PINK1-dependent activation of TBK1 which phosphorylates and recruits OPTN and NDP52 to the depolarized mitochondria, achoring them to the forming autophagosome coated with LC323,36,37. The process is achieved via the development of an isolation membrane surrounding the mitochondria via an ATG8-dependent positive feedback loop37.
The damaged mitochondria are engulfed by the autophagosome which subsequently fuses with lysosome containing acidic hydrolases37,38,39. Mitophagy is completed as the autophagosome-lysosome fusion leads to the degradation of mitochondria38,40 (Fig. 1). In addition, several Parkin-independent mitophagy pathways have been identified, which will be discussed later, having the potential to be utilized to compensate for the dysfunction of Parkin-mediated mitophagy.

In Parkin-dependent mitophagy, PINK1 is stabilized to the OMM and autophosphorylated following mitochondrial depolarization. This is followed by the phosphorylation of ubiquitin and Parkin, with Parkin being recruited towards OMM and activated, leading to the broad ubiquitination of OMM substrates. Mitophagy is executed via autophagy receptor proteins (OPTN, NPD52) which link the impaired mitochondria to autophagosome. In Parkin-independent mitophagy, this linking of mitochondria to autophagosome is achieved in the absence of Parkin, with three pathways and their key players, including but not limited to: receptor-mediated mitophagy (FUNDC1, AMBRA1, BNIP3/NIX), lipid-mediated mitophagy (Cardiolipin, Ceramide), and ubiquitin-mediated mitophagy (SIAH1, MUL1).
Genetic and pathological evidence of Parkin/PINK1-mediated mitophagy
Parkin and PINK1 mutations disrupt their physiologic functions on mitophagy. Extensive studies in cultured cells have not only unravelled the molecular mechanism underlying Parkin/PINK1 pathway discussed above, but also revealed that over half of tested PD-related mutations in Parkin and PINK1 are associated with defect in mitophagy27,41,42,43. Furthermore, there are in vivo evidence showing that mitophagy dysfunction impacts DA neuronal health and contributes to PD pathogenesis. Phosphorylation of ubiquitin at Ser65, a hallmark of PINK1 activation during mitophagy was increased in a Mutator mouse with mitochondrial dysfunction, and loss of endogenous Parkin leads to specific DA neurodegeneration and L-DOPA reversible motor deficit44. Studies using Drosophila model expressing mitophagy probes demonstrated Parkin/PINK1-dependent mitophagy activation in DA neurons in an age-dependent manner45. Another study demonstrated that depletion of deubiquitinase UPS30 in fly may rescue Parkin/PINK1-dependent mitophagy and counteract PD-like symptoms of paraquat-induced decreased dopamine level and motor dysfunction46. In addition to Parkin and PINK1 which play a fundamental role in mitophagy, other PD causative genes, including α-synuclein, LRRK2 and GBA, may contribute to PD pathogenesis also through their impact on mitophagy which will be discussed below.
Abnormality in PINK1-dependent mitophagy induced by aging can be inferred with the accumulation of PINK1 activity biomarker, pS65-Ub found in elderly human subjects47. Further pathological evidence of altered mitophagy was found in aging PD brain samples where there were increased mitochondrial matrix protein, ATP synthase subunit β (ATP5β), and OMM protein, Miro. Upregulation of Miro may increase mitochondrial motility and compromise the initiation of mitophagy. Therefore, aberrant accumulation of ATP5β and Miro in postmortem brain indicates that dysfunctional mitophagy is associated with PD16. PD patient platelets, compared to aged-matched controls, exhibited downregulated mitophagy accompanied by reduced LC3II and MsrB2, which affects Parkin methionine oxidation48. These pathological and clinical findings are indicative of defective mitophagy in PD.
Evidence of Parkin/PINK1-mediated mitophagy from patient iPSC-derived neurons
Although mutations in Parkin or PINK1 were found to compromise mitophagy in cultured cells and animal models, direct pathological evidence is still lacking if the same mutations would impair mitophagy and if such defect is the key factor for the loss of DA neurons in vivo or in PD patients49. Recent advancements in stem cell and gene editing technology may help address these questions. The impact of disease-causing mutations in Parkin and PINK1 on mitophagy are being examined in more physiologically related setting with the aid of induced pluripotent stem cell (iPSC)-derived neurons from PD patients. Parkin translocation to mitochondria was arrested in DA neurons derived from PD patients carrying PINK1 mutation50. The expression of several mitophagy-related genes was altered in DA neurons derived from Parkin knockout isogenic iPSCs51. Aberrant accumulation of phospho-ubiquitin and aggregated mitochondria have been demonstrated in neurons differentiated from iPSCs carrying Parkin or PINK1 mutations. Of note, the majority of phospho-ubiquitin was detected in TH-positive neurons, but not in TH-negative neurons, highlighting the cell type-specific mitophagy defects52. In idiopathic PD, impaired mitochondrial clearance was also observed, but not in all the iPSC clone-derived neurons, which could be attributed to the heterogeneity of PD pathogenesis53. In addition to phospho-ubiquitin, abnormalities of other mitophagy-related proteins have been recorded utilizing patient iPSC-derived neurons. It was found that Miro, a substrate of Parkin, was resistant to proteasomal degradation induced by mitochondrial depolarization, interferencing mitochondrial mobility and initiation of mitophagy in PD patients with LRRK2 G2019S mutation and sporadic PD patients7. Mutation in α-synuclein also causes Miro protein accumulation, inhibiting mitophagy in PD patient neurons16. Consistently, co-localization of mitochondria with lysosomes and clearance of mitochondria were significantly reduced in iPSC-derived neurons carrying Miro mutation54. mt-Keima is a mitochondria-targeted fluorescent protein, which is sensitive to pH changes but resistant to degradation mediated by lysosomal proteases. When mitophagy occurs, mt-Keima is engulfed by acidic lysosome where the excitation of mt-Keima shifts to a longer wavelength compared with mitochondrial physiological environment. The ratio change in excitation wavelengths of mt-Keima has been utilized to quantify mitophagy54,55. With the aid of mt-Keima, mitophagy defects were confirmed in iPSC-derived neurons with Parkin/PINK1 mutations by independent groups56,57. However, the fidelity of the iPSC-derived neuron model system to reveal the mitophagy impairment should be interpreted cautiously. Inhibition of PINK1-dependent mitophagy dramatically compromises iPSC reprogramming58. In addition, mitophagy mediates the metabolic switch between glycolysis and mitochondrial oxidative phosphorylation, which is critical during cell differentiation59. It is therefore possible that compensatory mechanism may have been developed during the conversion process of patient somatic cells to neurons. Alternatively, cells with substantial mitophagy defects may have failed to be transformed into neurons. Thus, the mitophagy dysfunction revealed in the patient iPSC-derived neurons may not faithfully recapitulate neuronal mitophagy status in the patient brain. Despite the limitations, studies in iPSC-derived neurons provides unequivocal evidence of mitophagy defects in PD.
Parkin-independent mitophagy
Accumulating evidence suggests that Parkin is not canonically essential for all mitophagy processes. Receptor-mediated mitophagy centers around the ability of LC3-interacting region (LIR) motif that are identified in certain protein receptors to allow binding to LC3 for mitophagy induction, regardless of the presence of Parkin60,61,62. AMBRA1 utilizes LIR motif to facilitate interaction with LC3, and mitophagy induction under conditions lacking Parkin61. FUNDC1, initiates mitophagy independent of Parkin, whereby evidence points towards its LIR domain association with LC3 as well as MARCH5 and ULK1 interactions for inducing hypoxia-related mitophagy62,63. Bcl-2/adenovirus E1B 19kD-interacting protein 3 -like proteins (BNIP3L), also known as NIX is an outer mitochondrial membrane protein equipped with an LIR near its N-terminus to perform Parkin-independent mitophagy64,65. A study using PD patient cells identified that despite carrying non-functional Parkin, BNIP3L/NIX-mediated mitophagy had maintained mitochondrial function, suggesting the PD-relevant neuroprotection66. A slew of other emerging proteins acting as receptor-mediated mitophagy interactors have been added on to this growing list, including STX1767, FKBP868, Bcl2-L-1339 and PHB269. Cardiolipin is a lipid that triggers mitophagy through similar mechanisms mentioned in the previous receptors, externalizing from IMM to OMM under mitochondrial stress and binding to LC3, which upon reaching a critical threshold, trigger the induction mitophagy in a neuronal setting70. The sphingolipid ceramide, capable of anchoring autophagolysosomes to mitochondrial membranes via LC3 interactions71 has also been suggested in parkin-independent, lipid-mediated mitophagy. Ubiquitin-mediated mitophagy was identified through PINK1’s ability to trigger mitophagy through NDP52 and optineurin regardless of Parkin’s presence, rewriting Parkin’s role from an essential regulator to a signal amplifier23. Other E3 ligases aside from Parkin, such as MUL1 was found to interact with mitofusin (Mfn) during mitophagy induction at damaged mitochondria, and in Parkin-independent pathway leading to mitochondrial ubiquitination and mitophagy72,73. Another E3 ligase, SIAH1 is recruited to mitochondria by Synphilin-1 to promote PINK1-dependent mitophagy via mitochondrial ubiquitination in a Parkin-independent manner74. Collectively, these observations suggest a complex mitophagy pathway involves an intricate compensatory network that responds to various homeostatic and pathological conditions (Fig. 1). Although there is little evidence of Parkin-independent mitophagy being involved in PD, these alternative mitophagy mechanism, such as MUL1- or NIX-involved mitophagy as discussed above, may be utilized to correct the mitophagy defects to rescue neurodegeneration in PD.
Reactive oxygen species
Reactive oxygen species (ROS) are highly reactive derivatives of molecular oxygen generated as by-products of cellular respiration. Common ROS include superoxide anion radical (O2●−), hydroxyl radicals (OH●−), and hydrogen peroxide (H2O2). ROS are normally sustained at low levels under redox homeostasis conditions75. Maintaining ROS within balance is supported by antioxidant systems, with endogeneous enzymatic antioxidants, such as superoxide dismutase (SOD)76, catalase (CAT)77,78, glutathione peroxidase (GPx)79 and thioredoxin (Trx)80, as well as nonenzymatic antioxidants including vitamins such as vitamin C81, vitamin E82 which collectively counter ROS overproduction within cells directly or indirectly83. ROS are predominantly the result of excessive electron slippage in the electron transport chain (ETC) exceeding the various antioxidant systems, and capable of readily oxidizing other molecules (proteins, DNA and lipids), causing a wide-range of effects84,85,86,87. ROS oxidizing effects on proteins include protein carbonylation, the most well-documented form of protein oxidation, whereby protein side chains are targeted, resulting in loss of protein functions84. ROS are the primary endogenous damaging agent of DNA, with these oxidative reactions incurred through oxidation of DNA bases85. ROS compromise the structural and dynamic properties of lipid membranes by altering their fluidity and permeability86. Lipid peroxidation is the common term used to describe the chain reaction induced by ROS targeting lipids leading to key breakdown molecules malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE)87. These intermediary compounds may react with DNA and proteins to form adducts in a neurodegenerative setting88,89. Lipid peroxidation by-product HNE triggers AMPK/mTORC pathway90 and JNK signal transduction pathway91 subsequently leading to autophagy activation92,93. Cardiolipin, a phospholipid and important mitophagy mediator in the inner membrane of the mitochondrial lipid bilayer, was identified to be specifically susceptible to lipid peroxidation94,95. Oxidation of Cardiolipin leads to the generation of reactive lipid mediators, including HNE and epoxide-containing cardiolipin94,95. The mitochondrial network as a whole is a critical target of 4-HNE, with neuronal autophagy flux identified to be modulated by 4-HNE in a concentration-dependent manner likely through direct modification of autophagy proteins96. Considering the effects of lipid peroxidation on autophagy, cardiolipin and mitochondrial protein qualities, it stands to reason the possibility that ROS-induced lipid peroxidation could eventually regulate mitophagy. Despite the classic dogma of ROS and reactive nitrogen species (RNS) being widely considered as harmful to cellular structures, their role is dynamic, serving pleiotropic intracellular signal transducers of inter-connected processes, including development processes97,98, metabolic networks99 and autophagy100.
Evidence of ROS in PD
ROS are signalling molecules that maintain several physiologic functions. When the physiologic compensation fails to keep ROS at appropriate levels, the resulting oxidative stress will damage cellular macromolecules and promote cell death101, leading to neurodegeneration. Various in vitro and in vivo studies in PD models have shown ROS-induction are critical in the pathophysiologic mechanisms of PD genetic mutations (PINK1 and Parkin102, DJ-1103 LRRK2104 etc) and mitochondrial dysfunction105. Furthermore, dopamine auto-oxidation promotes neurotoxicity in DA neurons10. In post-mortem studies, there is evidence of reduced mitochondrial electron transport chain activity, and an increase in iron in SN compared to age-matched controls106,107. Iron is known to raise ROS production through generating hydroxyl radicals, leading to α-synuclein-mediated reactive species formation in SN via Fenton reaction and redox-active iron accumulation in neuromelanin granules in SN25. DA neurons in the SN are more susceptible to excess ROS generation as a result of their distinct cellular structure (relatively increased size and complexity) and biochemical activities (unique Ca2+ pacemaking activity)108,109.
Evidence of ROS in mitophagy
There is considerable evidence suggesting a key role of ROS in mitophagy. Light-induced activation of mitochondria-targeted photosensitizer may cause selective ROS-mediated damage to a subset of mitochondria and subsequently trigger mitophagy in both cell lines110 and rodent neurons111. In vitro studies have demonstrated that mild and transient oxidative stress can trigger mitophagy but not non-selective autophagy40. Downregulation of mitochondrial fusion can spatially isolate damaged mitochondria for efficient removal by mitophagy, highlighting the selectivity of oxidative conditions in a ROS signalling cascade that dominantly triggers selective mitophagy40. Our own studies found that inhibition of ROS burst attenuated mitophagy at a couple of stages, i.e. PINK1-dependent Parkin translocation to mitochondria102 and elimination of mitochondria through autophagy112. Interestingly, chronic low-dose treatment of mitochondrial uncoupler failed to stimulate ROS upsurge and Parkin translocation to mitochondria despite bringing PINK1 protein to a comparable level as acute high-dose treatment. Given the indispensable role of PINK1 in the initiation of mitophagy, there is the possible modification of ROS on PINK1 in the process of mitophagy102.
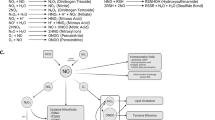
The bridges between ROS and Mitophagy
Being the primary source of cellular ROS and the central focus of mitophagy, mitochondria are the crux of a number of signalling pathways linking ROS and mitophagy112,113. ROS are a double-edged sword, possessing bidirectional impacts on autophagic flux. Generally, excessively high levels of ROS specifically trigger general autophagy over mitophagy40. In contrast, moderate levels of ROS may trigger mitophagy likely through the activation of specific signalling pathways and redox signalling40,114 with mitophagy in turn possessing neuroprotective effect on disease progression66,115. Collectively, whilst ROS as a signalling molecule and protective mitophagy are physiologically critical, prolonged dysregulation of mitophagy flux7,16 and/or excessive ROS levels9,108 are often detrimental for neuronal status, including within PD context. Identifying the bridges linking ROS and mitophagy is crucial for establishing their roles in PD pathogenesis and postulate potential therapeutic avenues using these interplays.
Signaling pathways
NF-κB
Nuclear factor kappaB (NF-κB) represents a family of five transcription factors, including NF-κB1 (p50/p105), NF-κB2 (p100/p52), RelA (p65), RelB, and c-Rel116. Critical in inflammation, NF-κB family members upon activation through canonical and/or noncanonical pathways, may rapidly translocate to the nucleus and mediate transcription of target genes by binding to κB enhancer117. Substantial evidence suggests that ROS (H2O2) acting upstream of this master regulator of redox balance, NF-κB, releases its NF-κB inhibitor (IκB) via H2O2 oxidation leading to NF-κB activation118. Recently, it was found that NF-κB may promote mitophagy through inducing p62 expression to attenuate mitochondrial damage triggered by NLRP3-inflammasome activator. Macrophage death was aggravated by inhibition of mitophagy through ablation of p62, or pathologically compromised ‘NF-kB-p62-mitophagy’ pathway. The same study suggested that NF-κB, through this anti-inflammatory pathway, directly affects Parkin-mediated mitophagy119. A study by Duan et al. identified several NF-κB-binding sites within the PINK1 promoter and demonstrated that NF-κB overexpression or administration of NF-κB activator upregulated PINK1 at transcription level120. TRAF6, an E3 ligase that acts as a transducer of the NF-κB pathway, and its related NF-κB activation may activate PINK1 cytosolic form to promote non-selective mitophagy through stabilization by enhanced Lys-63-linked ubiquitination121. In addition, NF-κB may promote RIPK1 translocation to the mitochondria where it forms a complex with PINK1 and phosphoglycerate mutase family member 5 (PGAM5) that stabilizes and activates PINK1, eventually inducing mitophagy122.
p38 MAPK
p38 MAPK belongs to the family of mitogen-activated protein kinases (MAPKs) and is responding to stress stimuli, including inflammatory cytokines and oxidative stress123. Under the canonical pathway of activation, ROS signalling by oxidative stress specifically oxidize antioxidant protein thioredoxin (TRX) and disassociate it away from the critical component ASK-1, allowing for ASK-1 dimerization and autophosphorylation, activating p38 pathway123. MAPK14, one of the four p38 isoforms, and its upstream signalling pathways are identified to be required in mammalian cells for both starvation- and hypoxia-induced mitophagy, but not macroautophagy, demonstrating the selectivity of this pathway124. A recent study by Qu et al. identified that inhibition of MAPK/p38 compromised the redistribution of Parkin in Parkin/PINK1-dependent mitophagy, with ROS identified to be a critical mediator upstream of this mitophagy pathway112. Additionally, our own study identified that inhibition of p38 signalling pathway halted mitophagy progression driven by ROS even after Parkin relocated to mitochondria, further corroborating the hypothesis of MAPK having a significant influence on upregulating ROS-mediated mitophagy113.
mTOR
Mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase, and it inhibits autophagy through a complex interplay and mTOR dysfunction can lead to apoptosis of DA neurons in PD animal models125. ROS may regulate autophagy/mitophagy through the PI3K/AKT/mTOR signalling pathway126,127. The role of MTORC1 in autophagic removal of mitochondria is confirmed with TSC/MTORC1 identified to be essential regulators for downstream mitophagy induction128. Additionally, there is a possible influence on FoxO regulated Parkin/PINK1 axis, with inhibition of mTORC1 identified to precede FoxO-dependent mitophagy upregulation129.
Nrf2-related pathways
Nrf2 (nuclear factor erythroid 2-related factor 2) is a redox-regulated transcription factor, a core element in the Nrf2-Antioxidant Response Element (ARE) related pathways. Their regulation can be either Kelch-like ECH-associated protein 1 (Keap1)-dependent or Keap1-independent (via phosphorylation by Casein kinase II, protein kinase C, glycogen synthase kinase 3β etc)130. These pathways are identified to mediate oxidative stress response and are established to be dysregulated in aging and neurodegenerative diseases, including PD130. Nrf2 pathway activation in PD was observed at a systemic level, likely to counteract oxidative stress131. Oxidative stress may deprive Keap of its ability to ubiquitinate Nrf2 by modifying key cysteinyl residues leading to nucleus transport of Nrf2. Accumulated Nrf2 subsequently activates a body of antioxidant enzymes, acting as a critical master regulator for a broad set of oxidative stress responses118. Oxidative stress triggered Nrf2 binds to ARE located in the p62 promoter, with p62 possibly activating Nrf2 and driving its own transcription in a positive feedback cycle132. p62-mediated mitophagy inducer (PMI) disrupts the Nrf2-Keap1 interaction and induces mitophagy independently of Parkin/PINK1133. Alternatively, another study suggests that p62, in an Nrf2-dependent manner, recruits two subunits of a cullin-RING ubiquitin (Keap1 and Rbx1) to mitochondria, promoting mitochondrial ubiquitination and subsequently mitophagy in a parkin-independent manner134.
SIRT
Sirtuins, are a family of nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylases, which respond to ROS-activated NAD+ function to regulate numerous antioxidant and redox signalling pathways through transcription factors (including Nrf2, p53, NF-κB, FOXO, PGC-1a) and several molecules of antioxidant response element (ARE)135. Sirtuins relevancy in PD context includes a variety of SIRTs appearing to alter mitophagy in PD models in addition to their expression dysregulated in PD patient samples. For instance, decreased SIRT3 was found in the fibroblasts from PD patients and SIRT5 was accumulated in idiopathic PD fibroblast cells, with both these sirtuins being mitochondrial proteins136. The relationship between sirtuins (SIRT1-7) and ROS, is uniquely complex and contested, with current evidence indicating that SIRT4 conditionally upregulates or suppresses ROS; SIRT1, SIRT3 and SIRT5 function against ROS; SIRT2, SIRT6, and SIRT7 mediates critical oxidative stress genes and mechanisms135. SIRT1 influences Parkin translocation to mitochondria and is closely tied to alterations NAD + /NADH ratio in studies observing SIRT1 influence on mitophagy137. In addition, SIRT2-mediated mitophagy regulated via ATG32 is identified to be essential in α-synuclein toxicity in yeast samples138, implicating this ROS-mitophagy interplay in PD context. SIRT3, known for downregulating ROS through modulation of several critical enzymes, was identified to be key for inducing Parkin/PINK1-mediated mitophagy, likely through SIRT3 ability to interact and deacetylate both PINK1 and Parkin directly or indirectly via FOXO3a139. Interestingly, another study identified SIRT3 effect of inducing mitophagy by upregulating BNIP3 expression, this mitophagy modulating activity further tied with ERK-CREB signalling pathway140 (Fig. 2).

ROS function upstream of intracellular signalling pathways which involve several key mediators (NF-κB, p38 MAPK, mTOR, Nrf2, SIRT) to regulate mitophagy.
PD-related mutations/genes
Parkin/PINK1
Mutations in the parkin and PINK1 account for the most common causes of autosomal recessive early-onset Parkinson disease (EOPD). Early studies exploring functions of Parkin and PINK1 revealed that defects in Parkin or PINK1 result in enhanced ROS production in mouse brain141 or patient fibroblast142, suggesting that Parkin and PINK1 may confer protection to neurons by attenuating ROS-related neurotoxicity. The mechanism of this protection at least partially lies in the role of Parkin/PINK1-mediated mitophagy that is progressively unravelled and refined. Overexpressed parkin in cultured cell lines can eliminate the entire mitochondrial network in cells, eradicating detrimental factors released from damaged mitochondria143. Although dramatic mitophagy is not practicable in neurons, evidence of ROS inhibition through mitophagy in a physiological context has been garnered. Parkin/PINK1-dependent mitophagy was shown to attenuate NLRP3 inflammasome activation and mitochondrial ROS production, eventually reducing apoptosis in epithelial cells and renal injury in mice144. AMPKα2 may interact with phosphorylated PINK1 and trigger Parkin recruitment and subsequent mitophagy, leading to reduced ROS production and apoptosis of cardiomyocytes145. Inhibition of PINK1 accumulation on mitochondria by morphine led to mitophagy defect and excessive ROS accumulation in spinal cord neurons146.
α-synuclein
Mutations in the gene encoding α-synuclein (SNCA) cause autosomal-dominant PD through point mutations, including A53T, E46K, H50Q etc, as well as copy number variations (duplication or triplication)147. Neurodegenerative effects of α-synuclein in PD have been increasingly tied to α-synuclein reciprocal relationship with mitochondrial dysfunction148, with a possible link to PINK1-related autophagy/mitophagy16,149. α-synuclein role in ROS includes its accumulation on mitochondria via TOM20 leading to an excessive generation of ROS, with pathogenic a-synuclein-TOM20 interaction confirmed in PD post-mortem samples150. α-synuclein delays mitophagy by targeting the N-terminus of Miro, leading to excessive and abnormal Miro accumulation on the mitochondrial surface16. Upregulated production of ROS and reactive nitrogen species (RNS) are present in iPSCs carrying α-synuclein A53T mutation relative to isogenic control lines151. α-synuclein E46K mutation was also identified to result in oxidative stress accumulation in SN DA neurons, likely increasing neuronal vulnerability towards mitochondrial impairment by mitochondrial toxins152. Both A53T and E46K mutations promoted mitophagy through increasing α-synuclein accumulation on mitochondria, primarily through cardiolipin externalization to the OMM153.
DJ-1
DJ-1 gene has been identified to be mutated in autosomal recessive PD. DJ-1 protein acts as a neuroprotective factor by directly eliminating hydrogen peroxide, while cells expressing DJ-1 carrying PD-related mutations are sensitive to oxidative effects10,154. Interestingly, as a sensor for oxidative stress, DJ-1 protein is susceptible to the formation of adducts with PD-relevant dopamine155. This relationship is theorized as a key susceptibility factor within PD-relevant A9 neurons in SN region that contain dopamine susceptible to ROS-induced adduct formation by DJ-1, further supported by in vivo evidence of elevated α-synuclein aggregates in DJ-1 deficient mice leading to the increased oxidized form of DA10,156. Additionally, a previous study identified that DJ-1 overexpression rescued DA neurons against neurotoxicity, including oxidative stress, by enhancing extracellular signal-regulated protein kinase 1/2 (ERK1/2)-dependent mitophagy115. This is in addition to the likely reciprocal interplay with Parkin/PINK1, as DJ-1 deficiency was identified to, in a ROS-dependent manner, promote Parkin recruitment and mitophagy, while mitochondrial DJ-1 level being regulated by Parkin/PINK1103. Hence, while the exact mechanism is yet to be fully elucidated, DJ-1 is believed to influence Parkin/PINK1 mitophagy in a ROS-dependent manner.
LRRK2
LRRK2, is a multifunctional enzymatic (kinase, GTPase etc), scaffolding protein157. LRRK2 G2019S is a common genetic cause for familial and sporadic PD in caucasian populations157, whereas in Asian population, LRRK2 S1647T, R1628P, and G2385R variants are associated with increased risk and lowered onset age of PD158. iPSC-derived neurons carrying G2019S mutation was shown to upregulate the expression of key genes related to oxidative stress-response and α-synuclein levels159. Our group have shown that quenching ROS through either genetic manipulation (expressing peroxiredoxin-3)160 or administration of chemical antioxidant161 effectively rescued PD phenotypes in neuronal and Drosophila models, suggesting that ROS play a crucial role in LRRK2 pathogenesis. LRRK2 mutations possess a complicated relationship with mitophagy, as the same G2019S mutations have demonstrated various mechanisms of mitophagy alterations, from increasing mitophagy through histone deacetylase activation136, or in contradiction, to decreasing mitophagy162 probably through compromised Miro removal7, sirtuin activity163 or aberrant RAB10 phosphorylation164. Regardless of the uncertainty of specific regulation, which is likely context-dependent, it can be agreed that LRRK2 mutations, particularly G2019S, result in aberrant mitophagy within the PD context, likely contributing to its increased mitochondrial vulnerability and overall neurodegeneration156.
GBA
Glucocerebrosidase (GBA1) gene mutation is associated with a 20- to 30-fold increased PD development risk165. L444P mutation in GBA has been established to induce mitochondrial dysfunctions, including altered mitophagy activation, autophagy flux and ROS levels166. Specifically, L444P mutation in GBA has been identified to inhibit two critical areas related to mitophagy, namely mitochondrial priming and autophagic removal of the organelle167. Mitophagy induction is identified to enhance transcription factor EB (TFEB) expression leading to increased GBA1 expression. The involvement of mitophagy in lysosomal biogenesis suggests a positive feedback loop, with GBA mutations inducing dysfunctions within this system167. In addition, dysfunctional mitophagy and excessive oxidative stress have been identified in post-mortem tissue of PD patients with GBA mutations167, further linking the effects of GBA on ROS-mitophagy interplay in PD context.
In our review, the selection of genes we focus on reflect the most well-documented PD-related genes with regards to how they are functionally linked to mitophagy and ROS. However, genome-wide association study (GWAS)168,169,170 have revealed a host of relatively novel risk loci and their genes, with limited but emerging evidence that some could be implicated in both mitophagy and ROS, including VPS13C171 and SREBF1172. We speculate that evidence of genetic variants in PD relevant context could increase in the coming years.
We have highlighted a few PD genes that are associated with both aberrant ROS accumulation and defective mitophagy. The body of the evidence above clearly proves that the abnormalities in these two aspects are universal and critical in PD. However, more studies are warranted to determine which factor is the major culprit for PD pathogenesis and how the entanglement of these two factors contribute to PD pathogenesis. Therefore, both factors have to be taken into account to deliver effective therapy when designing a strategy to tackle PD.
In addition, although the overwhelming evidence supports the agonistic role of ROS in mitophagy through activating various signalling pathways, there are reports showing antioxidative molecule(s) and/or compound(s) may promote mitophagy. For instance, KH176, a chemical derivative of a water-soluble form of vitamin E, enhanced mitophagy in neurons from subjects with or without Parkin mutation, suggesting its pro-mitophagy effect in a Parkin-independent manner173. KH176 exerts its anti-oxidative effects through direct interaction with peroxiredoxins174, while peroxiredoxin 3175 and peroxiredoxin 6176 have been found to be involved in mitophagy. Another study showed that mitochondria-targeted antioxidant, MitoQ rescued mitophagy, mitochondrial dysfunction and apoptosis through Nrf2 and PINK1 rather than its direct antioxidative effect177. Thus, the paradoxical pro-mitophagy effect of antioxidant may be attributed to its specific effects on mitophagy-related signalling pathways or molecules instead of its role in suppressing ROS. In addition, ROS may directly oxidize ATG3 and ATG7 to prevent LC3 lipidation, eventually impairing autophagy178. Given that Atg3179 and Atg7180 also play indispensable roles in mitophagy, it is therefore conceivable that antioxidants may suppress the oxidation of these autophagy-related proteins to promote mitophagy in a context-dependent manner. The success of antioxidant in promoting mitophagy indicates the potential of the therapeutic strategy for PD by promoting mitophagy while inhibiting ROS.
Therapeutic avenues utilizing the ROS-mitophagy link
Clinical trials throughout the decades of antioxidants (creatine181, vitamin E182, coenzyme Q10183, etc) in PD have not provided conclusive evidence that they are neuroprotective. One possible reason we postulate for the failure of antioxidants could be that blockage of signalling pathway necessary for mitophagy activation by undue inhibition of ROS is unlikely to be optimal against neurodegeneration. Promoting mitophagy by direct induction of ROS may not be ideal strategy either given the high volume of evidence showing the detrimental effects of ROS on cellular health, on account of its ability to exacerbate oxidative stress damage in PD9.
Existing compounds/drugs with effects on ROS and mitophagy
Targets in the downstream signalling of ROS which can simultaneously activate protective mitophagy, while minimizing the harm of ROS may be potentially utilized in PD therapeutics. Here, we highlight some promising chemical compounds/drugs that target the various signalling pathways that may be involved in mitophagy. These chemical compounds/drugs have been approved or under investigation for other diseases, so that the safety concern may be circumvented.
Melatonin (N-acetyl-5-methoxytryptamine) has been shown to increase phosphorylation of Akt and NF-κB, leading to PINK1-dependent protective mitophagy184. Melatonin may also upregulate NRF2-induced mitophagy to protect against neuronal apoptosis in subarachnoid haemorrhage (SAH)185. The ROS levels, initially increased by high glucose conditions and necessary in order to stimulate mitophagy via Akt and NF -κB pathways, were subsequently attenuated by melatonin-induced PINK1-dependent mitophagy184. Melatonin has been well-documented to function against ROS and RNS, but may have pro-oxidant capabilities under specific conditions186. Existing evidence points toward melatonin’s role as an antioxidant showing systemic therapeutic benefits in PD-relevant clinical trials (dyskinesia, sleep disorder)187, which combined with amphiphilic nature allowing for BBB diffusion further encourages therapeutic focus revisiting melatonin as a mitophagy agonist with NF-κB and Nrf2 activating effect mentioned prior187.
Pioglitazone, an anti-diabetic drug for Type 2 diabetes mellitus (T2DM) under the category of thiazolidinediones (TZDs), increases PINK1 expression via NF-κB activation and enhances mitophagy. It protects against mitochondrial dysfunction induced by toxins and reduces ROS in a chronic kidney disease study188,189. Pioglitazone also improved phenotypes (impaired locomotion, DA neurodegeneration) in rat PD model190. Clinical cohort studies showed a 30% reduced risk of PD among diabetes treated with TZDs191,192.
Rapamycin, an FDA-approved compound, induces autophagy by binding and inhibiting mTORC1193, and has been tested as a disease-modifying agent in experimental models of PD and neurodegenerative diseases194. It also promotes Parkin/PINK1 mitophagy, likely via p62, to restore mitochondrial homeostasis195,196. Rapamycin’s effect on ROS appears to be conditional. It may downregulate intracellular ROS via Nrf2 pathway, glutathione and SOD197,198. Nevertheless, it is also able to increase ROS levels, probably through c-Jun and endoplasmic reticulum (ER) stress pathway in certain circumstances199. Metformin, an mTOR inhibitor, activates mitophagy via signalling pathways including AMPK-Nrf2 as well as SIRT3 pathway200,201. It has been shown to upregulate mitophagy in a recent randomized controlled clinical trial in type 2 diabetics202. Metformin has been found to downregulate ROS production203,204. A longitudinal cohort study in an aging population with diabetes demonstrated a lower incidence of neurodegenerative diseases in those on long-term metformin therapy205. Spermidine, a polyamine with anti-aging properties, induces mitophagy through several signalling pathways, including modulation of the mTOR axis via ATM-dependent Parkin/PINK1 pathways206,207. Of note, PD gene PARK9 encoded protein ATP13A2 is a lysosomal exporter of polyamine (putrescine, spermidine, and spermine)208. Spermine is pumped by ATP13A2 into the cytosol and subsequently absorbed by mitochondria. The polyamine transport activity of ATP13A2 is responsible to counter the mitochondrial oxidative stress209. Mitochondrial defects observed in C. elegans carrying a mutation in CATP-6 (a C. elegans ortholog of ATP13A2) was rescued via mitophagy induction210. In another study, spermidine was able to rescue behavioural deficits in the PD C. elegans model via the Parkin/PINK1 equivalent pathways207. Taken together, these studies suggest that spermidine may have neuroprotective effect on PD with ATP132A being a key component. Despite spermidine-generated ROS being a possible upstream signal in activating the latter ATM pathway206, spermidine appears to act as direct ROS scavengers, resulting in inhibition of mitochondrial ROS production211,212. Salidroside, a plant extract, has been shown to protect DA neurons in PD models through enhancing mitophagy with its bioactive effects on DJ-1/Nrf2 pathway being the likely mechanism213. Consistently, studies revealed neuroprotective effects of salidrosides in pre-clinical trials in Alzheimers disease, stroke and PD. A number of these studies emphasized its safety and substantial bioactive effects on the regulation of oxidative stress214.
Tomatidine, a natural compound with antiaging properties activates Nrf2-SKN-1 pathway, resulting in the C. elegans homologue induction of PINK-1/BNIP3/Nix-mitophagy that is suspected to preserve cell function by clearing damaged or dysfunctional mitochondria215. Tomatidine may activate mitophagy via Nrf2-SKN-1 pathway or TRAF6. Meanwhile, it may have a negligible or mild stimulating effect on ROS production215,216. Although evidence is limited, Ursolic and oleanolic acids, natural triterpenoid compounds widely found in plants, have been shown to induce Parkin-independent mitophagy, possibly concurrent with activation of AKT/mTOR and Nrf2/ARE pathways, with the ROS induction suspected to be an upstream factor for mitophagy induction for both triterpenes217.
Honokiol, an agonist of SIRT3 known for anti-inflammatory and antitumor effects, promotes mitophagy and mitochondrial dynamics in vitro in an SIRT3-dependent manner via the AMPK-PGC-1α signalling pathway, and this neuroprotection has been validated in vivo218. Honokiol’s effect of downregulating ROS in this study218 is consistent with existing in vitro and in vivo evidence highlighting its inhibitory effect on ROS production, likely in an SIRT3-dependent manner219,220.
Liraglutide, an anti-diabetic drug, is a glucagon‑like peptide‑1 receptor agonist that elevates the levels of SIRT1 in a dose-dependent manner and is able to trigger autophagy. It can augment Parkin expression, leading to Parkin-mediated mitophagy activation221. The SIRT1/Parkin/mitophagy pathway activation is protective, downregulating cellular oxidative stress221. Similar observations of ROS inhibition by liraglutide has been consistently observed222,223. Liraglutide shows good BBB permeability and neuroprotective effects in PD animal models224. We await the results of its ongoing trial in idiopathic PD, which should be completed soon. (ClinicalTrials.gov Identifier: NCT02953665).
We discuss here the compounds/drugs that regulate mitophagy with emphasis on ROS-related signalling pathway(s). As for the modulators of mitophagy reviewed elsewhere, most of the targets are localized on mitochondria225,226. Therefore, modulation of these targets may have profound impacts on mitochondrial functions, including ROS homeostasis. For instance, Ubiquitin-specific protease 30 (USP30) removes ubiquin attached by Parkin to the OMM substrates, attenuating subsequent mitophagy. On the other hand, reduction in USP30 level rescues mitophagy caused by Parkin/PINK1 mutation. Based on its influence on mitophagy, USP30 inhibitors have been developed to promote mitophagy as a potential therapeutic option for PD227,228. However, USP30 is also involved in the regulation of the import of intramitochondrial proteins, including subunits of electron transport chain proteins such as Complex-I229. Manipulation of USP30 may cause aberrant ROS generation by interfering the proper functions of the respiratory chain. Thus, the effects of USP30 or other mitophagy regulators on ROS production have to be assessed and the benefit of neuroprotection through mitophagy and the potential detrimental effect of ROS dysregulation has to be weighed.
Maximizing the potential therapeutic benefits of these compounds in Table 1 requires factoring in their effects on ROS levels. Among them, a few compounds may potentially upregulate ROS, having a negative impact on cell survival. When applied in PD, these drugs should be coupled with ROS inhibitors (general antioxidants or mitochondrial-targeted antioxidants) necessary to negate any harmful oxidative effects. Most compounds listed have apparently null effects or downregulate ROS levels, whereby their use in combination with ROS inhibitors may be considered to synergistically improve therapeutic performance230. We reason that mitophagy activation in PD relevant therapeutics should simultaneously avoid provoking oxidative stress, while maintaining their neuroprotective effects of downstream signalling on mitochondrial homeostasis, particularly mitochondrial turnover through autophagy-lysosomal pathway.
Some of these compounds are currently undergoing pre-clinical and clinical trials (i.e. Liraglutide, Salidroside, and Melatonin) in PD, further encouraging the efforts in drug repurposing. Therapeutic usage of these compounds could implemented in a targeted manner or in combined multidrug regime with existing pharmacological (carbidopa-levodopa, pramipexole etc) and non-pharmacological approaches (physical, occupational, speech therapies etc)6.
Drug repurposing, utilizing various degrees of pre-existing clinical efforts covering efficacy and safety efforts, offsets the high cost and lengthy timeline of de novo drug discovery231. However, drug repurposing comes with issues that should be taken into consideration, including but not limited to intellectual property considerations and high chance to fail in clinical trials231,232. The ROS inhibitors as mentioned earlier (creatine181, vitamin E182) have yet to be proved as effective repurposing drugs in PD treatment.
Potential target-based screening for mitophagy activator
Alternatively, potential therapeutic compounds for PD may be discovered de novo from the perspective of mitophagy. A recent study was conducted using an imaging-based drug screening in patient iPSC-derived neurons carrying Parkin or PINK1 mutation. Among the 320 compounds screened, 73 hits were found to promote mitophagy with two candidates attenuating ROS and cell death53. Large-scale screenings are needed to identify more potential compounds which can be further validated and taken to clinical trials. It should be noted that neurons are usually cultured in B27 supplemented neurobasal medium, which contains antioxidant to promote neuronal survival233. Potential oxidative stress caused by compounds may not be revealed in the cultured neurons. Additionally, animal model may have different tolerance and compensative mechanism for mitophagy impairment49 and ROS stress234,235. Target-based screening with stringent validation of mitophagy activator may help to efficiently identify promising drug candidate. The screening of mitophagy activator targeting ROS-related signalling pathways in the presence of certain antioxidant in mitophagy-intact or mitophagy-defective iPSC-derived neurons may yield compounds that potentially reverse the pathogenesis of PD. The compounds identified in mitophagy-defective neurons represent the potential drug candidates which may promote alternative mitophagy to rescue PD caused by mitophagy defects. The candidates that can promote mitophagy in the presence of antioxidant may trigger mitophagy by activating certain signalling pathway while eliminating potential ROS-related detrimental effects. With the efficient target-based screening, more efforts could be made to evaluate the effects of the hits on PD phenotypes in multiple model systems, including animal and human midbrain organoid236 PD model before taken into clinical trial together with the antioxidant (Fig. 3).

Existing compounds/ drugs could be utilized to induce mitophagy while their direct/indirect effects on ROS are evaluated. Alternatively, compound screening of mitophagy activator targeting ROS-related signalling pathways, may yield hits that potentially reverse the pathogenesis of PD. The hits of the screening should be validated in multiple PD model systems given the limitations of the current disease models. The drug candidates identified from either strategy will be recommended to enter potential clinical trial with antioxidant therapy to aid removal of potential ROS-related detrimental effects.
Challenges and potential solutions
Therapeutic trials studying the potential of activating ROS downstream signalling pathway(s) to boost mitophagy face some challenges (Table 2). First, it is important to pinpoint mitophagy defects in relevant biological samples. In vitro culture of DA neurons or midbrain organoid derived from patient iPSCs may provide a useful tool to study mitophagy in vitro with the aid of constantly developing fluorescence dyes to readily detect mitophagy status or single cells sequencing to identify the cell populations and signalling pathways implicated in the mitophagy. Furthermore, it is of note that pioglitazone failed to halt PD progression in trials, but lowered the risk of PD, suggesting that mitophagy activation in rescuing neurodegeneration is more likely to be useful in early stages of neurodegeneration, as opposed to clinically diagnosed PD patients whereby numerous cascades of cellular dysfunctions rendering neurodegeneration are inevitable and irreversible. Thus, mitophagy activation may be more effective in the preclinical/prodromal state. Hence clinical trials should focus on asymptomatic genetic carriers or those at risk or those with prodromic symptoms. A comprehensive analysis of genetic background (known genes related to PD) and longitudinal clinical studies of blood, urine and CSF biomarkers and clinical manifestation (including symptoms of anosmia, REM disorder) will help to stratify clinical trial subjects and to monitor their progression.
In conclusion, the dynamic and complex interplay between mitophagy and excessive ROS plays an important pathophysiologic role in both sporadic and familial PD. Identifying the relationship between these processes and their triggers early in the course of neurodegeneration will provide novel mechanistic clues that can potentially lead to the development of drugs that target specific pathways in this network. Proper selection of specific subsets of subjects for longitudinal clinical trials will enhance the chance of a favourable therapeutic outcome.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study
References
Jankovic, J. & Tan, E. K. Parkinson’s disease: etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 8, 795–808 (2020).
Ou, Z. et al. Global Trends in the Incidence, Prevalence, and Years Lived With Disability of Parkinson’s Disease in 204 Countries/Territories From 1990 to 2019. Front. Public Heal. 9, 776847 (2021).
Abbas, M. M., Xu, Z. & Tan, L. C. S. Epidemiology of Parkinson’s Disease—East Versus West. Mov. Disord. Clin. Pract. 5, 14–28 (2018).
Giráldez-Pérez, R. M., Antolín-Vallespín, M., Muñoz, M. D. & Sánchez-Capelo, A. Models of α-synuclein aggregation in Parkinson’s disease. Acta Neuropathol. Commun. 2, 1–17 (2014).
Torres-Odio, S. et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J. Neuroinflammation 14, 154 (2017).
Armstrong, M. J. & Okun, M. S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA - J. Am. Med. Assoc. 323, 548–560 (2020).
Hsieh, C. H. et al. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 19, 709–724 (2016).
Liu, H. et al. Aberrant mitochondrial morphology and function associated with impaired mitophagy and DNM1L-MAPK/ERK signaling are found in aged mutant Parkinsonian LRRK2(R1441G) mice. Autophagy 17, 3196–3220 (2021).
Musgrove, R. E. et al. Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular α-synuclein transfer. J. Clin. Invest. 129, 3738–3753 (2019).
Burbulla, L. F. et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357, 1255–1261 (2017).
Chen, C. et al. Investigation of mitochondrial biogenesis defects in single substantia nigra neurons using post-mortem human tissues. Neurobiol. Dis. 134, 104631 (2020).
Franco-Iborra, S. et al. Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson’s disease. Cell Death Dis. 9, 1122 (2018).
Awad, A. J. et al. VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Physiol. Behav. 176, 139–148 (2017).
Portz, P. & Lee, M. K. Changes in Drp1 Function and Mitochondrial Morphology Are Associated with the α-Synuclein Pathology in a Transgenic Mouse Model of Parkinson’s Disease. Cells 10, 885 (2021).
Wang, W. et al. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 22, 54–63 (2016).
Shaltouki, A., Hsieh, C. H., Kim, M. J. & Wang, X. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 136, 607–620 (2018).
Ni, H. M., Williams, J. A. & Ding, W. X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4, 6–13 (2015).
Liu, J., Liu, W., Li, R. & Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 8, 712 (2019).
Palikaras, K., Lionaki, E. & Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022 (2018).
Palikaras, K., Daskalaki, I., Markaki, M. & Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Therapeutics 178, 157–174 (2017).
Valente, E. M. et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160 (2004).
Kitada, T. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 (1998).
Lazarou, M. et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 (2015).
Koyano, F. et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166 (2014).
Jin, S. M. et al. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942 (2010).
Yamano, K. & Youle, R. J. PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769 (2013).
Matsuda, N. et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221 (2010).
Okatsu, K. et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3, 1010–1016 (2012).
Wauer, T., Simicek, M., Schubert, A. & Komander, D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524, 370–374 (2015).
Okatsu, K. et al. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 209, 111–128 (2015).
Kazlauskaite, A. et al. Binding to serine 65‐phosphorylated ubiquitin primes Parkin for optimal PINK 1‐dependent phosphorylation and activation. EMBO Rep. 16, 939–954 (2015).
Tanaka, A. et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380 (2010).
Gegg, M. E. & Schapira, A. H. V. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2. Autophagy 7, 243–245 (2014).
Wang, X. et al. PINK1 and Parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906 (2011).
Ordureau, A. et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 56, 360–375 (2014).
Heo, J. M., Ordureau, A., Paulo, J. A., Rinehart, J. & Harper, J. W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 60, 7–20 (2015).
Padman, B. S. et al. LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat. Commun. 10, 1–13 (2019).
Parzych, K. R. & Klionsky, D. J. An overview of autophagy: morphology, mechanism, and regulation. Antioxid. Redox Signal. 20, 460–473 (2014).
Murakawa, T. et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 6, 7527 (2015).
Frank, M. et al. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta - Mol. Cell Res. 1823, 2297–2310 (2012).
Narendra, D. P. et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298 (2010).
Sekine, S. et al. Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1. Mol. Cell 73, 1028–1043.e5 (2019).
Yi, W. et al. The landscape of Parkin variants reveals pathogenic mechanisms and therapeutic targets in Parkinson’s disease. Hum. Mol. Genet. 28, 2811–2825 (2019).
Pickrell, A. M. et al. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 87, 371–381 (2015).
Cornelissen, T. et al. Deficiency of parkin and PINK1 impairs age-dependent mitophagy in drosophila. Elife 7, 1–14 (2018).
Bingol, B. et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375 (2014).
Fiesel, F. C. et al. (Patho‐)physiological relevance of PINK 1‐dependent ubiquitin phosphorylation. EMBO Rep. 16, 1114–1130 (2015).
Lee, S. H. et al. Mitochondrial MsrB2 serves as a switch and transducer for mitophagy. EMBO Mol. Med. 11, e10409 (2019).
McWilliams, T. G. et al. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 27, 439–449.e5 (2018).
Seibler, P. et al. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 31, 5970–5976 (2011).
Shaltouki, A. et al. Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem cell Rep. 4, 847–859 (2015).
Shiba-Fukushima, K. et al. Evidence that phosphorylated ubiquitin signaling is involved in the etiology of Parkinson’s disease. Hum. Mol. Genet. 26, 3172–3185 (2017).
Yamaguchi, A. et al. Identifying Therapeutic Agents for Amelioration of Mitochondrial Clearance Disorder in Neurons of Familial Parkinson Disease. Stem cell Rep. 14, 1060–1075 (2020).
Berenguer-Escuder, C. et al. Impaired mitochondrial-endoplasmic reticulum interaction and mitophagy in Miro1-mutant neurons in Parkinson’s disease. Hum. Mol. Genet. 29, 1353–1364 (2020).
Liu, Y.-T. et al. Mt-Keima detects PINK1-PRKN mitophagy in vivo with greater sensitivity than mito-QC. Autophagy 17, 3753–3762 (2021).
Kumar, M. et al. Defects in Mitochondrial Biogenesis Drive Mitochondrial Alterations in PARKIN-Deficient Human Dopamine Neurons. Stem cell Rep. 15, 629–645 (2020).
Suzuki, S. et al. Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem. Biophys. Res. Commun. 483, 88–93 (2017).
Vazquez-Martin, A. et al. Mitophagy-driven mitochondrial rejuvenation regulates stem cell fate. Aging (Albany NY). 8, 1330–1352 (2016).
Esteban-Martínez, L. et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 36, 1688–1706 (2017).
Liu, L., Sakakibara, K., Chen, Q. & Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 24, 787–795 (2014).
Strappazzon, F. et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 22, 419–432 (2015).
Chen, Z. et al. Mitochondrial E3 ligase MARCH 5 regulates FUNDC 1 to fine‐tune hypoxic mitophagy. EMBO Rep. 18, 495–509 (2017).
Wu, W. et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 15, 566–575 (2014).
Novak, I. et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51 (2010).
Naeem, S., Qi, Y., Tian, Y. & Zhang, Y. NIX compensates lost role of parkin in cd-induced mitophagy in HeLa cells through phosphorylation. Toxicol. Lett. 326, 1–10 (2020).
Koentjoro, B., Park, J. S. & Sue, C. M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 7, 1–11 (2017).
Xian, H., Yang, Q., Xiao, L., Shen, H. M. & Liou, Y. C. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun. 10, 2059 (2019).
Bhujabal, Z. et al. FKBP8 recruits LC3A to mediate Parkin‐independent mitophagy. EMBO Rep. 18, 947–961 (2017).
Wei, Y., Chiang, W. C., Sumpter, R., Mishra, P. & Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 168, 224–238.e10 (2017).
Chu, C. T. et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205 (2013).
Sentelle, R. D. et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 8, 831–838 (2012).
Yun, J. et al. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife 2014, e01958 (2014).
Villa, E., Marchetti, S. & Ricci, J. E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 28, 882–895 (2018).
Szargel, R. et al. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum. Mol. Genet. 25, 3476–3490 (2016).
Rahal, A. et al. Oxidative stress, prooxidants, and antioxidants: The interplay. Biomed Res. Int. 2014, 761264 (2014).
Van Raamsdonk, J. M. & Hekimi, S. Superoxide dismutase is dispensable for normal animal lifespan. Proc. Natl Acad. Sci. USA. 109, 5785–5790 (2012).
Vetrano, A. M. et al. Characterization of the oxidase activity in mammalian catalase. J. Biol. Chem. 280, 35372–35381 (2005).
Singhal, A., Morris, V. B., Labhasetwar, V. & Ghorpade, A. Nanoparticle-mediated catalase delivery protects human neurons from oxidative stress. Cell Death Dis. 4, e903–e909 (2013).
Mason, R. P. et al. Glutathione peroxidase activity is neuroprotective in models of Huntington’s disease. Nat. Genet. 45, 1249–1254 (2013).
Yang, Y. et al. Thioredoxin activity confers resistance against oxidative stress in tumor-infiltrating NK cells. J. Clin. Invest. 130, 5508–5522 (2020).
Liu, Y., Liu, C. & Li, J. Comparison of vitamin c and its derivative antioxidant activity: Evaluated by using density functional theory. ACS Omega 25467–25475 https://doi.org/10.1021/acsomega.0c04318 (2020).
Azman, N. H. E. N., Goon, J. A., Ghani, S. M. A., Hamid, Z. & Ngah, W. Z. W. Comparing palm oil, tocotrienol-rich fraction and α-tocopherol supplementation on the antioxidant levels of older adults. Antioxidants 7, 74 (2018).
He, L. et al. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 44, 532–553 (2017).
Chang, R. L. et al. Protein structure, amino acid composition and sequence determine proteome vulnerability to oxidation‐induced damage. EMBO J. 39, 1–21 (2020).
Azzouz, D., Khan, M. A. & Palaniyar, N. ROS induces NETosis by oxidizing DNA and initiating DNA repair. Cell Death Discov. 7, 113 (2021).
Yusupov, M. et al. Effect of head group and lipid tail oxidation in the cell membrane revealed through integrated simulations and experiments. Sci. Rep. 7, 5761 (2017).
Ayala, A., Muñoz, M. F. & Argüelles, S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 360438 (2014).
Wataya, T. et al. High molecular weight neurofilament proteins are physiological substrates of adduction by the lipid peroxidation product hydroxynonenal. J. Biol. Chem. 277, 4644–4648 (2002).
Citron, B. A. et al. Membrane lipid peroxidation in neurodegeneration: Role of thrombin and proteinase-activated receptor-1. Brain Res. 1643, 10–17 (2016).
Dolinsky, V. W. et al. Resveratrol prevents the prohypertrophic effects of oxidative stress on lkb1. Circulation 119, 1643–1652 (2009).
Haberzettl, P. & Hill, B. G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol. 1, 56–64 (2013).
Kim, J., Kundu, M., Viollet, B. & Guan, K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 (2011).
Klein, S. R. et al. C-Jun N-terminal kinases are required for oncolytic adenovirus-mediated autophagy. Oncogene 34, 5295–5301 (2015).
Zhong, H., Lu, J., Xia, L., Zhu, M. & Yin, H. Formation of electrophilic oxidation products from mitochondrial cardiolipin in vitro and in vivo in the context of apoptosis and atherosclerosis. Redox Biol. 2, 878–883 (2014).
Liu, W., Porter, N. A., Schneider, C., Brash, A. R. & Yin, H. Formation of 4-hydroxynonenal from cardiolipin oxidation: Intramolecular peroxyl radical addition and decomposition. Free Radic. Biol. Med. 50, 166–178 (2011).
Dodson, M. et al. Regulation of autophagy, mitochondrial dynamics, and cellular bioenergetics by 4-hydroxynonenal in primary neurons. Autophagy 13, 1828–1840 (2017).
Han, Y. et al. Ca2+ -Induced Mitochondrial ROS Regulate the Early Embryonic Cell Cycle. Cell Rep. 22, 218–231 (2018).
Bazopoulou, D. et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 576, 301–305 (2019).
Peralta, D. et al. A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol. 11, 156–163 (2015).
Chen, Y., Azad, M. B. & Gibson, S. B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 16, 1040–1052 (2009).
Sies, H. & Jones, D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21, 363–383 (2020).
Xiao, B. et al. Reactive oxygen species trigger Parkin/PINK1 pathway–dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 292, 16697–16708 (2017).
Joselin, A. P. et al. ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 21, 4888–4903 (2012).
Angeles, D. C. et al. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum. Mutat. 32, 1390–1397 (2011).
Ahn, E. H. et al. Mitochondrial dysfunction triggers the pathogenesis of Parkinson’s disease in neuronal C/EBPβ transgenic mice. Mol. Psychiatry 26, 7838–7850 (2021).
Schapira, A. H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 7, 97–109 (2008).
Genoud, S. et al. Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics 9, 1447–1455 (2017).
Trist, B. G., Hare, D. J. & Double, K. L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 18, e13031 (2019).
Poewe, W. et al. Parkinson disease. Nat. Rev. Dis. Prim. 3, 1–21 (2017).
Wang, Y., Nartiss, Y., Steipe, B., McQuibban, G. A. & Kim, P. K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 8, 1462–1476 (2012).
Ashrafi, G., Schlehe, J. S., LaVoie, M. J. & Schwarz, T. L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 206, 655–670 (2014).
Xiao, B. et al. Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis. 8, 1–12 (2017).
Qu, F. et al. Manipulation of Mitophagy by ‘All-in-One’ nanosensitizer augments sonodynamic glioma therapy. Autophagy 16, 1413–1435 (2020).
Zhang, C. et al. Oxidative stress-induced mitophagy is suppressed by the miR-106b-93-25 cluster in a protective manner. Cell Death Dis. 12, 209 (2021).
Gao, H. et al. DJ-1 protects dopaminergic neurons against rotenone-induced apoptosis by enhancing ERK-dependent mitophagy. J. Mol. Biol. 423, 232–248 (2012).
Oeckinghaus, A. & Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 1, 1–14 (2009).
Morgan, M. J. & Liu, Z. G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 21, 103–115 (2011).
Sies, H., Berndt, C. & Jones, D. P. Oxidative Stress: Annual Review of Biochemistry. Annu. Rev. Biochem. 86, 715–748 (2017).
Zhong, Z. et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 164, 896–910 (2016).
Duan, X. et al. Upregulation of human PINK1 gene expression by NFκB signalling. Mol. Brain 7, 1–10 (2014).
Lim, G. G. Y. et al. Cytosolic PTEN-induced putative kinase 1 Is stabilized by the NF- κB pathway and promotes non-selective mitophagy. J. Biol. Chem. 290, 16882–16893 (2015).
Hawk, M. A. et al. RIPK1-mediated induction of mitophagy compromises the viability of extracellular-matrix-detached cells. Nat. Cell Biol. 20, 272–284 (2018).
Cuadrado, A. & Nebreda, A. R. Mechanisms and functions of p38 MAPK signalling. Biochemical J. 429, 403–417 (2010).
Hirota, Y. et al. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 11, 332–343 (2015).
Zhou, Q. et al. Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2, and autophagy pathways. Sci. Rep. 6, 1–12 (2016).
Kim, J. H. et al. Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ. 25, 1921–1937 (2018).
Ding, R. et al. ROS-AKT-mTOR axis mediates autophagy of human umbilical vein endothelial cells induced by cooking oil fumes-derived fine particulate matters in vitro. Free Radic. Biol. Med. 113, 452–460 (2017).
Ebrahimi-Fakhari, D., Saffari, A., Wahlster, L. & Sahin, M. Using tuberous sclerosis complex to understand the impact of MTORC1 signaling on mitochondrial dynamics and mitophagy in neurons. Autophagy 13, 754–756 (2017).
Bartolomé, A. et al. MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell. Biol. 37, e00441-17 (2017).
Fão, L., Mota, S. I. & Rego, A. C. Shaping the Nrf2-ARE-related pathways in Alzheimer’s and Parkinson’s diseases. Ageing Res. Rev. 54, 100942 (2019).
Petrillo, S. et al. Systemic Activation of Nrf2 Pathway in Parkinson’s Disease. Mov. Disord. 35, 180–184 (2020).
Jain, A. et al. p62 / SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-driven Gene Transcription. J. Biol. Chem. 285, 22576–91 (2010).
East, D. A. et al. PMI: A ΔΨm independent pharmacological regulator of mitophagy. Chem. Biol. 21, 1585–1596 (2014).
Yamada, T. et al. Mitochondrial Stasis Reveals p62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 28, 588–604.e5 (2018).
Singh, C. K. et al. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 28, 643–661 (2018).
Yakhine-Diop, S. M. S. et al. Impaired Mitophagy and Protein Acetylation Levels in Fibroblasts from Parkinson’s Disease Patients. Mol. Neurobiol. 56, 2466–2481 (2019).
Di Sante, G. et al. Loss of sirt1 promotes prostatic intraepithelial neoplasia, reduces mitophagy, and delays park2 translocation to mitochondria. Am. J. Pathol. 185, 266–279 (2015).
Sampaio-Marques, B. et al. SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 8, 1494–1509 (2012).
Zhou, Z. D. & Tan, E. K. Oxidized nicotinamide adenine dinucleotide-dependent mitochondrial deacetylase sirtuin-3 as a potential therapeutic target of Parkinson’s disease. Ageing Res. Rev. 62, 101107 (2020).
Li, R. et al. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 18, 229–243 (2018).
Palacino, J. J. et al. Mitochondrial Dysfunction and Oxidative Damage in parkin-deficient Mice. J. Biol. Chem. 279, 18614–18622 (2004).
Piccoli, C. et al. Mitochondrial respiratory dysfunction in familiar Parkinsonism associated with PINK1 mutation. Neurochem. Res. 33, 2565–2574 (2008).
Narendra, D., Tanaka, A., Suen, D. F. & Youle, R. J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 (2008).
Lin, Q. et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 26, 101254 (2019).
Wang, B. et al. AMPKa2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ. Res. 122, 712–729 (2018).
Kong, H. et al. Morphine induces dysfunction of PINK1/Parkin-mediated mitophagy in spinal cord neurons implying involvement in antinociceptive tolerance. J. Mol. Cell Biol. 11, 1056–1068 (2019).
Meade, R. M., Fairlie, D. P. & Mason, J. M. Alpha-synuclein structure and Parkinson’s disease - Lessons and emerging principles. Mol. Neurodegener. 14, 1–14 (2019).
Grünewald, A., Kumar, K. R. & Sue, C. M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 177, 73–93 (2019).
Liu, J. et al. Pink1 interacts with α-synuclein and abrogates α-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis. 8, e3056 (2017).
Di Maio, R. et al. α-synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 8, 1–15 (2016).
Ryan, S. D. et al. Isogenic Human iPSC Parkinson’s Model Shows Nitrosative Stress-Induced Dysfunction in MEF2-PGC1α Transcription. Cell 155, 1351–1364 (2013).
Cannon, J. R. et al. Expression of human E46K-mutated α-synuclein in BAC-transgenic rats replicates early-stage Parkinson’s disease features and enhances vulnerability to mitochondrial impairment. Exp. Neurol. 240, 44–56 (2013).
Ryan, T. et al. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat. Commun. 9, 1–17 (2018).
Taira, T. et al. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 5, 213–218 (2004).
Girotto, S. et al. Dopamine-derived quinones affect the structure of the redox sensor DJ-1 through modifications at Cys-106 and Cys-53. J. Biol. Chem. 287, 18738–18749 (2012).
Ryan, B. J., Hoek, S., Fon, E. A. & Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 40, 200–210 (2015).
Li, J. Q., Tan, L. & Yu, J. T. The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 9, 47 (2014).
Xiao, B. et al. Association of LRRK2 haplotype with age at onset in Parkinson disease. JAMA Neurol. 75, 127–128 (2018).
Nguyen, H. N. et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280 (2011).
Angeles, D. C. et al. Thiol peroxidases ameliorate LRRK2 mutant-induced mitochondrial and dopaminergic neuronal degeneration in Drosophila. Hum. Mol. Genet. 23, 3157–3165 (2014).
Angeles, D. C. et al. Antioxidants inhibit neuronal toxicity in Parkinson’s disease-linked LRRK2. Ann. Clin. Transl. Neurol. 3, 288–294 (2016).
Walter, J. et al. Neural Stem Cells of Parkinson’s Disease Patients Exhibit Aberrant Mitochondrial Morphology and Functionality. Stem Cell Rep. 12, 878–889 (2019).
Schwab, A. J. et al. Decreased Sirtuin Deacetylase Activity in LRRK2 G2019S iPSC-Derived Dopaminergic Neurons. Stem Cell Rep. 9, 1839–1852 (2017).
Wauters, F. et al. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 16, 203–222 (2020).
Migdalska-Richards, A. & Schapira, A. H. V. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 139, 77–90 (2016).
Gegg, M. E. & Schapira, A. H. V. Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol. Dis. 90, 43–50 (2016).
Li, H. et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 15, 113–130 (2019).
Nalls, M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993 (2014).
Do, C. B. et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for parkinson’s disease. PLoS Genet. 7, e1002141 (2011).
Billingsley, K. J. et al. Mitochondria function associated genes contribute to Parkinson’s Disease risk and later age at onset. NPJ Park. Dis. 5, 8 (2019).
Lesage, S. et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 98, 500–513 (2016).
Ivatt, R. M. et al. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc. Natl Acad. Sci. USA. 111, 8494–8499 (2014).
Schwartzentruber, A. et al. Oxidative switch drives mitophagy defects in dopaminergic parkin mutant patient neurons. Sci. Rep. 10, 15485 (2020).
Beyrath, J. et al. KH176 Safeguards Mitochondrial Diseased Cells from Redox Stress-Induced Cell Death by Interacting with the Thioredoxin System/Peroxiredoxin Enzyme Machinery. Sci. Rep. 8, 6577 (2018).
Sonn, S. K. et al. Peroxiredoxin 3 deficiency induces cardiac hypertrophy and dysfunction by impaired mitochondrial quality control. Redox Biol. 51, 102275 (2022).
Ma, S. et al. Peroxiredoxin 6 Is a Crucial Factor in the Initial Step of Mitochondrial Clearance and Is Upstream of the PINK1-Parkin Pathway. Antioxid. Redox Signal. 24, 486–501 (2016).
Xiao, L. et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 11, 297–311 (2017).
Frudd, K., Burgoyne, T. & Burgoyne, J. R. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 9, 95 (2018).
Radoshevich, L. et al. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 142, 590–600 (2010).
Towers, C. G. et al. Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev. Cell 56, 2029–2042.e5 (2021).
Tilley, B. C. et al. Effect of Creatine Monohydrate on Clinical Progression in Patients With Parkinson Disease. Jama 313, 584–593 (2015).
Shoulson, I. DATATOP: a decade of neuroprotective inquiry. Parkinson Study Group. Deprenyl And Tocopherol Antioxidative Therapy Of Parkinsonism. Ann. Neurol. 44, S160–S166 (1998).
Investigators, T. N. N.-P. A randomized clinical trial of coenzyme Q 10 and GPI-1485 in early Parkinson disease. Neurology 68, 20 LP–20 28 (2007).
Onphachanh, X. et al. Enhancement of high glucose-induced PINK1 expression by melatonin stimulates neuronal cell survival: Involvement of MT2/Akt/NF-κB pathway. J. Pineal Res. 63, 1–17 (2017).
Sun, B., Yang, S., Li, S. & Hang, C. Melatonin upregulates nuclear factor erythroid-2 related factor 2 (Nrf2) and mediates mitophagy to protect against early brain injury after subarachnoid hemorrhage. Med. Sci. Monit. 24, 6422–6430 (2018).
Zhang, H. M. & Zhang, Y. Melatonin: A well-documented antioxidant with conditional pro-oxidant actions. J. Pineal Res. 57, 131–146 (2014).
Filograna, R., Beltramini, M., Bubacco, L. & Bisaglia, M. Anti-Oxidants in Parkinson’s Disease Therapy: A Critical Point of View. Curr. Neuropharmacol. 14, 260–271 (2016).
Yoon, Y. M. et al. Pioglitazone protects mesenchymal stem cells against P-cresol-induced mitochondrial dysfunction via up-regulation of PINK-1. Int. J. Mol. Sci. 19, 2898 (2018).
Palacios-Ramírez, R. et al. Pioglitazone Modulates the Vascular Contractility in Hypertension by Interference with ET-1 Pathway. Sci. Rep. 9, 16461 (2019).
Ulusoy, G. K. et al. Effects of pioglitazone and retinoic acid in a rotenone model of Parkinson’s disease. Brain Res. Bull. 85, 380–384 (2011).
Zhu, Y., Pu, J., Chen, Y. & Zhang, B. Decreased risk of Parkinson’s disease in diabetic patients with thiazolidinediones therapy: An exploratory meta-analysis. PLoS One 14, e0224236 (2019).
Brakedal, B. et al. Glitazone use associated with reduced risk of Parkinson’s disease. Mov. Disord. 32, 1594–1599 (2017).
Lou, G. et al. Mitophagy and Neuroprotection. Trends Mol. Med. 26, 8–20 (2020).
Bové, J., Martínez-Vicente, M. & Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 12, 437–452 (2011).
Wang, Y. et al. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 9, 1–14 (2018).
Li, Q. et al. Rapamycin Enhances Mitophagy and Attenuates Apoptosis After Spinal Ischemia-Reperfusion Injury. Front. Neurosci. 12, 865 (2018).
Ko, J. H., Yoon, S. O., Lee, H. J. & Oh, J. Y. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFκB pathways in autophagy- and p62-dependent manners. Oncotarget 8, 40817–40831 (2017).
Dai, C. et al. Rapamycin Confers Neuroprotection against Colistin-Induced Oxidative Stress, Mitochondria Dysfunction, and Apoptosis through the Activation of Autophagy and mTOR/Akt/CREB Signaling Pathways. ACS Chem. Neurosci. 9, 824–837 (2018).
Chen, W. et al. Synergistic antitumor activity of rapamycin and EF24 via increasing ROS for the treatment of gastric cancer. Redox Biol. 10, 78–89 (2016).
Wang, C. et al. Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci. Trends 12, 605–612 (2018).
Chen, L. et al. Metformin mitigates gastrointestinal radiotoxicity and radiosensitises P53 mutation colorectal tumours via optimising autophagy. Br. J. Pharmacol. 177, 3991–4006 (2020).
Bhansali, S., Bhansali, A., Dutta, P., Walia, R. & Dhawan, V. Metformin upregulates mitophagy in patients with T2DM: A randomized placebo-controlled study. J. Cell. Mol. Med. 24, 2832–2846 (2020).
Algire, C. et al. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev. Res. 5, 536–543 (2012).
Kelly, B., Tannahill, G. M., Murphy, M. P. & O’Neill, L. A. J. Metformin inhibits the production of reactive oxygen species from NADH: Ubiquinone oxidoreductase to limit induction of interleukin-1β (IL-1β) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. J. Biol. Chem. 290, 20348–20359 (2015).
Shi, Q., Liu, S., Fonseca, V. A., Thethi, T. K. & Shi, L. Effect of metformin on neurodegenerative disease among elderly adult US veterans with type 2 diabetes mellitus. BMJ Open 9, e024954 (2019).
Qi, Y., Qiu, Q., Gu, X., Tian, Y. & Zhang, Y. ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Sci. Rep. 6, 24700 (2016).
Yang, X. et al. Spermidine inhibits neurodegeneration and delays aging via the PINK1-PDR1-dependent mitophagy pathway in C. elegans. Aging (Albany NY). 12, 16852–16866 (2020).
Veen, S. et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 578, 419–424 (2020).
Vrijsen, S. et al. ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc. Natl Acad. Sci. USA. 117, 31198–31207 (2020).
Anand, N. et al. Dysregulated iron metabolism in C. elegans catp-6/ATP13A2 mutant impairs mitochondrial function. Neurobiol. Dis. 139, 104786 (2020).
Chai, N. et al. Spermidine Prevents Heart Injury in Neonatal Rats Exposed to Intrauterine Hypoxia by Inhibiting Oxidative Stress and Mitochondrial Fragmentation. Oxid. Med. Cell. Longev. 2019, 5406468 (2019).
Wang, J. et al. Spermidine alleviates cardiac aging by improving mitochondrial biogenesis and function. Aging (Albany NY). 12, 650–671 (2020).
Li, R. & Chen, J. Salidroside Protects Dopaminergic Neurons by Enhancing PINK1/Parkin-Mediated Mitophagy. Oxid. Med. Cell. Longev. 2019, 9341018 (2019).
Zhong, Z. et al. Pharmacological activities, mechanisms of action, and safety of salidroside in the central nervous system. Drug Des. Devel. Ther. 12, 1479–1489 (2018).
Fang, E. F. et al. Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Sci. Rep. 7, 1–13 (2017).
Hu, B. et al. Tomatidine suppresses osteoclastogenesis and mitigates estrogen deficiency-induced bone mass loss by modulating TRAF6-mediated signaling. FASEB J. 33, 2574–2586 (2019).
Castrejón-Jiménez, N. S. et al. Ursolic and oleanolic acids induce mitophagy in a549 human lung cancer cells. Molecules 24, 3444 (2019).
Wang, J. et al. Small molecule natural compound agonist of SIRT3 as a therapeutic target for the treatment of intervertebral disc degeneration. Exp. Mol. Med. 50, 146 (2018).
Pillai, V. B. et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 6, 6656 (2015).
Tang, P. et al. Honokiol alleviates the degeneration of intervertebral disc via suppressing the activation of TXNIP-NLRP3 inflammasome signal pathway. Free Radic. Biol. Med. 120, 368–379 (2018).
Qiao, H. et al. Liraglutide repairs the infarcted heart: The role of the SIRT1/Parkin/mitophagy pathway. Mol. Med. Rep. 17, 3722–3734 (2018).
Zhu, H. et al. The neuroprotection of liraglutide against ischaemia-induced apoptosis through the activation of the PI3K/AKT and MAPK pathways. Sci. Rep. 6, 1–11 (2016).
Zhang, Y. et al. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca2+-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic. Biol. Med. 95, 278–292 (2016).
Liu, W. et al. Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 303, 42–50 (2015).
Masaldan, S., Callegari, S. & Dewson, G. Therapeutic targeting of mitophagy in Parkinson’s disease. Biochem. Soc. Trans. 50, 783–797 (2022).
Clark, E. H., de la Torre, A. V., Hoshikawa, T. & Briston, T. Targeting mitophagy in Parkinson’s disease. J. Biol. Chem. 296, 100209 (2021).
Kluge, A. F. et al. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg. Med. Chem. Lett. 28, 2655–2659 (2018).
Tsefou, E. et al. Investigation of USP30 inhibition to enhance Parkin-mediated mitophagy: tools and approaches. Biochem. J. 478, 4099–4118 (2021).
Phu, L. et al. Dynamic Regulation of Mitochondrial Import by the Ubiquitin System. Mol. Cell 77, 1107–1123.e10 (2020).
Jin, H. et al. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta - Mol. Basis Dis. 1842, 1282–1294 (2014).
Begley, C. G. et al. Drug repurposing: Misconceptions, challenges, and opportunities for academic researchers. Sci. Transl. Med. 13, eabd5524 (2021).
Pushpakom, S. et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58 (2019).
Xie, C., Markesbery, W. R. & Lovell, M. A. Survival of hippocampal and cortical neurons in a mixture of MEM+ and B27-supplemented neurobasal medium. Free Radic. Biol. Med. 28, 665–672 (2000).
Labinskyy, N. et al. Comparison of endothelial function, O2- and H2O2 production, and vascular oxidative stress resistance between the longest-living rodent, the naked mole rat, and mice. Am. J. Physiol. Heart Circ. Physiol. 291, H2698–H2704 (2006).
Panov, A. et al. Species- and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. Am. J. Physiol. Cell Physiol. 292, C708–C718 (2007).
Jo, J. et al. Lewy Body-like Inclusions in Human Midbrain Organoids Carrying Glucocerebrosidase and α-Synuclein Mutations. Ann. Neurol. 90, 490–505 (2021).
Iwashita, H. et al. Live Cell Imaging of Mitochondrial Autophagy with a Novel Fluorescent Small Molecule. ACS Chem. Biol. 12, 2546–2551 (2017).
Acknowledgements
The authors thank Singapore Ministry of Health’s National Medical Research Council (Open Fund Large Collaborative Grant (MOH-000207) and Singapore Translational Research (STaR) Investigator Award (NMRC/STaR/0030/2018) to TEK, CS-IRG-NIG and OF-YIRG to XB) for their support. Figures 1 and 3 were partially generated using templates from Servier Medical Art, which is licensed under a Creative Commons Attribution 3.0 Unported License.
Author information
Authors and Affiliations
Contributions
B.X. and E.-K.T. conceived the manuscript. All authors wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, B., Kuruvilla, J. & Tan, EK. Mitophagy and reactive oxygen species interplay in Parkinson’s disease. npj Parkinsons Dis. 8, 135 (2022). https://doi.org/10.1038/s41531-022-00402-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-022-00402-y
This article is cited by
-
Exploring the therapeutic potential of Sirt6-enriched adipose stem cell-derived exosomes in myocardial ischemia–reperfusion injury: unfolding new epigenetic frontiers
Clinical Epigenetics (2024)
-
GUCY2C signaling limits dopaminergic neuron vulnerability to toxic insults
npj Parkinson's Disease (2024)
-
PINK1 and Parkin regulate IP3R-mediated ER calcium release
Nature Communications (2023)
-
Zinc Overload Induces Damage to H9c2 Cardiomyocyte Through Mitochondrial Dysfunction and ROS-Mediated Mitophagy
Cardiovascular Toxicology (2023)
