
Abstract
The ternary-based Fe-24Mn-3Cr alloy has superior mechanical properties based on an attractive combination of high strength and ductility, with long-term environmental stability in highly corrosive environments compared to conventional ferritic steel alloys. This study reports that the environmental instability caused by the rapid electrochemical corrosion kinetics on the surface of conventional high Mn-bearing ferrous alloys could be overcome by a combination of high Mn–low Cr-balanced Fe and their synergistic interactions. In contrast to Cr-free Mn-bearing alloys, the high Mn–low Cr-bearing alloy showed comparatively lower corrosion kinetic parameters, without a continuously increasing trend, and higher polarization resistance according to electrochemical polarization and impedance spectroscopy measurements. Moreover, the rate of degradation caused by erosion–corrosion synergistic interaction under erosion–corrosion dynamic flow conditions was the lowest in the high Mn–low Cr-bearing alloy. These surface-inhibiting characteristics of the alloy were attributed primarily to the formation of a bilayer scale structure consisting of inner α-Fe2−xCrxO3/outer FexMn3−xO4 on the surface.
Similar content being viewed by others


Introduction
In comparison with rare, noble, and expensive elements, Fe and Mn are the 4th and 12th most abundant elements in the earth’s crust, respectively, and a ferrous alloy containing high quantities of Mn was developed under the name of Hadfield steel in the late nineteenth century1. With the advent of high Mn-bearing ferrous alloys with a face-centered cubic (FCC) structure at room temperature, they have attracted considerable attention owing to their superior mechanical properties of strength ductility compared to the conventional ferritic steel alloys2. High tensile strength (UTS) of more than ~900 MPa can be achieved even under exceptionally high ductility with a total elongation of >60%, owing to their high strain-hardening capability3. Moreover, the attractive combination of strength ductility can be implemented even in cryogenic environments4.
In spite of their superior mechanical performance, their susceptibility to corrosion and resulting surface deterioration features are recognized as a critical problem that has restricted their wider application5. In principle, their much faster kinetics of surface deterioration (i.e., rapid change in surface color to dark red) involves electrochemical corrosion and scale formation processes simultaneously, giving rise to a significantly reduced lifespan. This critical weakness is caused primarily by the rapid anodic dissolution of the active element (Mn) from the surface, which is an intrinsic characteristic of high Mn-bearing alloys from thermodynamic considerations (i.e., the standard reduction potential of Mn is much lower than that of Fe6). The alloy design concept proposed here as a radical solution is grounded on the reverse use of such inherent weaknesses by the synergistic interactions between more active and nobler elements in Fe-based alloys. Specifically, the combination of more active and nobler elements in the alloy can promote the preferential dissolution of more active elements on the surface by a sort of galvanic action among the elements, leaving the enrichment of nobler elements (Mnoble) on the surface. In the present study, for greater applicability of the alloy, Cr is a suitable element for Mnoble in both functional and economic aspects. Its content was determined within the range that does not deteriorate the superior mechanical properties. Kim et al.7 reported the effectiveness of Mn and Cr as alloying elements on the corrosion resistance of carbon steel. In their study, the higher corrosion resistance of 18 wt.% Mn steel with 5 wt.% Cr in a neutral solution was highlighted by the grain size in scale formed on the steel. Similarly, Park et al.8 reported the beneficial effects of Cr on the corrosion resistance of 18 wt.% Mn steel in a sweet environment. On the other hand, the marked decrease in the UTS of the high Mn steels with Cr contents higher than 4 wt.% was reported9. Therefore, the alloying of high Mn-bearing ferrous alloys needs to be considered, to ensure environmental sustainability in harsh and corrosive conditions without significant degradation of their mechanical properties.
Our work demonstrates that a combination of a more active element in larger quantities and nobler elements in smaller quantities (i.e., high Mn–low Cr couple in this investigation) in ferrous alloys enables the development of stronger, tougher, and more commercially available alloys with superior resistance to both static corrosion and flowing erosion–corrosion.
Results
Mechanical and H-permeation behaviors
As listed in Table 1, three types of high Mn-bearing ferrous alloy with FCC structure were classified according to the Mn (~13 and ~24 wt.%) and Cr (3–3.5 wt.%) contents, and are referred to as M_Mn, H_Mn, and H_Mn-L_Cr, respectively. Compared to H_Mn and H_Mn-L_Cr, the addition of a smaller amount of Mn to steel (M_Mn) can reduce the stain-hardening capability of austenite, resulting in lower mechanical strength and hardness. Hence, a higher C content (1.0–1.25 wt.%) was added, because it also acts as an austenite stabilizer. Three types of Mn-bearing ferrous alloys were considered as materials for transporting pipes operated at room- or even lower-temperature environments. Two types of commercial structural steel samples with a body-centered cubic (BCC)-based structure were used for comparison: thermo-mechanically controlled processed (TMCP) API X70 pipeline steel, and quenched and tempered 9% Ni steel, which are called “API X70” and “9Ni,” respectively.
Figure 1a shows the combination regime of the UTS and the total elongation of the samples used in the current study in comparison with several commercial steel alloys (low C-based mild steels10,11, medium C-based giga steels12, Ti-6Al-4V alloy13,14, 2205 duplex stainless steel13,15, 2024 Al alloy13,16, API-grade ferritic steels17,18, high Mn-bearing austenitic steel alloys3,19,20, 9% Ni steels21,22, and high entropy alloy13,23) reported elsewhere, evaluated at room temperature. In contrast to the reference materials showing a mutually exclusive relationship between strength and ductility, Mn-bearing ferrous alloys have a more desirable combination of high strength and ductility in terms of industrial applicability. The quantitative values of yield strength, UTS, and strain obtained from the engineering stress–strain curves were presented in Supplementary Fig. 1. Moreover, the hardness, which is one of the indices of the wear and erosion resistance of Mn-bearing ferrous alloys, was comparatively higher than those of low C-based ferritic steels, as shown in Fig. 1b.

a Chart for UTS-elongation of various ferrous alloys (mild steels (0.17C10/0.18C11), 1.5 G (0.2–0.3C), 1.8 G (0.3–0.4C), and 2.0 G (0.4–0.5C) post heat-treated steels12, Ti-6Al-4V alloy13,14, 2205 duplex stainless steel (0.03 C-22.42Cr-6.2Ni-2.94Mo)13,15, 2024 Al alloy (alloy 2024-T351 plate)13,16, API X70 steels (0.06 C–0.50(Ni + Cu + Cr)17/0.08 C–0.138Cr18), 13Mn-based steel (0.6C–13Mn)19, 18Mn-based steel (0.6C–18Mn)3, 25Mn-based steel (25Mn-0.4 C)20, 9Ni steels (0.05C–9Ni21/0.11C–9Ni12), and high entropy alloy (20Co-20Cr-20Fe-20Mn-20Ni)13,23). b Vickers hardness measurements of the tested samples (the error bars represent SDs of the mean values). c Electrochemical H-permeation behaviors of various ferrous alloys (samples in the current study and previously reported study (low C steel, 1.5 G (0.2–0.3C), 1.8 G (0.3–0.4C), and 2.0 G (0.4–0.5C) post heat-treated steels12)). d H-diffusivities of various ferrous alloys (samples in the current study and previously reported studies (low C steel (0.05 C24/0.05C-1.25Mn- ~0.1(Nb + Ti + V)- ~0.4(Cu + Ni)- ~0.1Cr- ~0.1Mo25), pressure vessel steel (0.16C-1 ~ 1.5Mn)26, TRIP steel (0.2C-1.45Mn)27, duplex stainless steel (0.04C-24.9Cr-5.61Ni-1.43Mo-0.11W-0.23Cu)28, austenitic stainless steel (0.017C-15.4Cr-15.3Mn-4.0Ni)29, and TWIP steel (0.81C-17.08Mn)30)) with respect to the crystal structure.
In addition to the superior mechanical performance of high Mn-bearing ferrous alloys, the remarkable inhibition of H-penetration in the alloys was validated by an electrochemical permeation experiment (Fig. 1c). The H-permeation flux through the 1 mm-thick alloy membrane was not measured in high Mn-bearing alloys compared to the other samples with a BCC-based structure within the given time. A chart constructed based on the H-diffusivity data obtained in this study and reported data24,25,26,27,28,29,30 (Fig. 1d) supports the H-impermeability through the high Mn-bearing alloys with considerably slower H-diffusion kinetics in a FCC structure. Assuming that the sample is composed of a perfect crystal lattice with no defects, a significantly longer break-through time (tbt = L2 (15.3 ∙ Diffusivity)−1, where L is the diffusion distance31) would be required for a 1 mm-thick sample with an FCC structure in the permeation experiment.
Electrochemical corrosion behaviors under static conditions
As presented in Fig. 2a, at the initial stage of immersion in a neutral solution, the open circuit potentials (OCPs) of three types of Mn-bearing alloy samples were comparatively lower than the other two samples tested. Among the Mn-bearing alloy samples, the OCPs of H_Mn-L_Cr were ennobled gradually with increasing immersion time to 7 days. Subsequently, the OCPs became stable with minor changes. Although the OCP evolution of M_Mn showed a similar trend to that of H_Mn-L_Cr, the OCPs of M_Mn were lower than those of H_Mn-L_Cr. On the other hand, no significant change was observed in the OCPs of H_Mn and much lower OCPs were maintained over the entire immersion period. In contrast, the OCPs of API X70 and 9Ni decreased markedly with increasing immersion time up to 3 days and became stable thereafter.

a OCP evolution. b Change in icorr and mpy, obtained by the LPR measurements. c Change in Rp, obtained by the EIS measurements. d Weight loss, measured after the immersion test.
After 28 days of immersion, the OCPs of 9Ni and H_Mn-L_Cr were the highest with similar values. Figure 2b presents the changes in the corrosion kinetic parameters (corrosion current density (icorr) and corrosion rate (mpy)) with an immersion time up to 70 days based on the curve fitting to linear polarization resistance (LPR) curves. The fitted curves and parameters were presented in Supplementary Fig. 2 and Table 1, respectively. Cr-free Mn-bearing alloy samples showed much higher parameters (icorr and mpy), with continuously increasing trends. In particular, a more remarkable increase in the parameters with the highest values was observed in M_Mn after ~35 days of immersion. Although a rapid increase in the parameters at the very early stages of immersion also appeared in H_Mn-L_Cr, they decreased markedly again within a few hours of immersion, showing a continuous decrease with a low downward slope. In API X70 and 9Ni, higher parameters were obtained in API X70, but both samples showed gradually increasing behavior with a very low upward slope. Considering general corrosion theory regarding the inverse relationship between icorr and polarization resistance (Rp)6, among the tested samples, the differences and changes in Rp (Fig. 2c) obtained from the Nyquist plots of electrochemical impedance spectroscopy (EIS) were in accordance with those in the LPR data. The fitted Nyquist plots and parameters were presented in Supplementary Fig. 3 and Table 2. As listed in Supplementary Table 2, H_Mn-L_Cr and 9Ni had the lowest double layer capacitance (Qdl), depending highly on the adsorption of dissolved metal ions at the surface32, giving rise to the highest levels of the charge transfer resistance (Rct). The reproducibility of the data obtained by the electrochemical measurements can be verified by the mean values and their SDs. The reliability of the electrochemical behaviors described above can also be supported by the weight-loss measurements after the immersion test of the four samples tested. As shown in Fig. 2d, the changes in weight loss exhibited a typical parabolic behavior. The mean weight losses of H_Mn and H_Mn-L_Cr obtained by ΔW(ti) − ΔW(ti−1)/ti − ti−1, where ΔW(ti) is the weight loss per unit area at immersion time ti, were the highest and lowest, respectively (refer to Supplementary Fig. 4 and Table 3).
Corrosion scale (adherent rust) on the surface
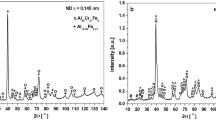
Figure 3 presents the surface and cross-section observations of the tested samples after the middle stage of immersion (28 days). The elemental compositions of each point (P1 ~ P6) marked on Fig. 3, analyzed by energy-dispersive spectroscopy (EDS) were listed in Supplementary Table 4. Higher Mn contents in the alloys resulted in the formation of scale with higher Mn concentrations. On the other hand, the addition of low Cr to high Mn-bearing alloys decreased the Mn concentration in the outer scale through the formation of inner scale enriched with Cr. It also appears that the Ni concentration in the scale on 9Ni was considerable. The additional data of the earlier (7 days) and later (56 days) stages of immersion were also found in Supplementary Figs. 5 and 6, respectively. With the M_Mn and H_Mn samples, coarse masses of oxides with high porosity that detached readily from the surface were distributed unevenly over the surface. The dark regions in the oxides shown in Fig. 3c,f indicate the pores filled with resin. The oxide scale formed on the two samples was composed primarily of MnxFe3−xO4 (i.e., partial substitution of Mn in Fe3O4), as characterized by X-ray diffraction (XRD, Fig. 4 with Supplementary Table 5). In contrast, a bilayer structure, consisting of the innermost layer of α-Fe2−xCrxO3 (i.e., Cr-substituted Fe2O3, characterized by XRD) with a brick-like structure and an outer layer of FexMn3−xO4, as densely formed on H_Mn-L_Cr. Further detailed data showing such a bilayer structure of the alloy are provided in Supplementary Fig. 7. Despite some cracks in the structure of α-Fe2−xCrxO3, as shown in Fig. 3h and Supplementary Fig. 7, the layer was seldom detached even after ultrasonic cleaning for more than 30 min (in a preliminary test), suggesting much stronger adhesion to the matrix. On the other hand, API X70 was not covered completely with the oxide scale composed mostly of typical Fe3O4 and γ-FeOOH (Figs. 3j and 4), suggesting that such oxide scale could be detached easily. 9Ni was also assumed to have a bilayer structure consisting of the inner layer of Ni2FeO4 and the outer layer of NiO/Ni2O3, characterized by X-ray photoelectron spectroscopy (XPS, see Supplementary Fig. 8 with Table 6) and XRD. One of the characteristic features in 9Ni was some localized pit-like damage on the surface (Fig. 3m).

a, b Surface and cross-section images of M_Mn, respectively. c Magnified view of b. d, e Surface and cross-sectional images of H_Mn, respectively. f Magnified view of e. g, h Surface and cross-sectional images of H_Mn-L_Cr, respectively. i Magnified view of h. j, k Surface and cross-sectional images of API X70, respectively. l Magnified view of k. m, n Surface and cross-sectional images of 9Ni, respectively. o Magnified view of n.

a–e M_Mn, H_Mn, H_Mn-L_Cr, API X70, and 9Ni, respectively.
Erosion–corrosion behaviors
The erosion–corrosion test was performed with the samples used in the aqueous corrosion tests under static conditions, except for M_Mn, which had the worst corrosion resistance. The four individual components consisting of total erosion–corrosion rate (VT): pure corrosion (C0), erosion-enhanced corrosion (CE), pure erosion (E0), and corrosion-enhanced erosion (EC), and the contributions of the synergistic effects (CE and EC) to VT were quantified and presented in Fig. 5. In contrast to the order of icorr measured under static conditions, the total corrosion rate (CT) of H_Mn was lower than that of 9Ni. The parameters for determining CT were presented in Supplementary Table 7. The VT of the samples increased in the order of H_Mn-L_Cr ~ 9Ni, H_Mn, and API X70. This trend among the samples was maintained even when the erosion–corrosion condition became more severe (i.e., increase in flow rate and erodent (SiO2) size), but the individual components and resulting VT of the tested samples were increased (Supplementary Fig. 9). Overall, C0 and E0 make a little contribution (only a few %) to VT, indicating that the synergistic effects account for most of the VT. Unlike static corrosion, API X70 was more vulnerable to erosion–corrosion than H_Mn. The other noticeable feature was that the contribution of EC to VT was comparatively higher in the Mn-bearing alloy samples. In contrast, the contribution of CE to VT was higher in the other two samples. The reproducibility of the data in Fig. 5 can be verified by the mean values and their SDs of the four individual components of the tested samples, as shown in Supplementary Table 8.

a Total erosion–corrosion rates. b, c Contribution of CE and EC to total erosion–corrosion rates, respectively.
Erosion–corrosion morphology
Figure 6 presents the surface and cross-section morphologies of the tested samples after the erosion–corrosion experiment. In the case of H_Mn, corrosion scale (FexMn3−xO4) mostly detached with only small amounts remaining. Some small crater-like areas, 7 ~ 10 μm in depth, produced by particle collisions were often observed in the matrix (Fig. 6a). In contrast, most of the corrosion scale (α-Fe2−xCrxO3), which had been damaged partially, remained on the H_Mn-L_Cr surface (Fig. 6e), suggesting that direct damage to the matrix by particle collision was mitigated partly by the corrosion scale. The depth of plastically damaged spot areas was in the range of 9 ~ 13 μm. In contrast, very little corrosion scale remained and much more severely damaged wear scars with an average depth of 23 μm were observed abundantly over the surface of API X70 (Fig. 6i), as expected from its highest VT. In the case of 9Ni, however, most of the surface was covered with a thin (Fe,Ni)-enriched oxide layer (magnified view can be found in Supplementary Fig. 10), except for the comparatively massive and deeper craters with an average depth of 52 μm, which were occasionally observed. Such damage shown in 9Ni appeared to be a more severe form of localized pit-like corrosion, usually appearing under static corrosion conditions (Fig. 3m). These damage characteristics among the samples were similar, even under more severe erosion–corrosion conditions.

a–d H_Mn. e–h H_Mn-L_Cr. i–l API X70. m–p 9Ni. a, e, i, m Surface morphologies with low magnification. b, f, j, n Magnified view of damaged areas in the surfaces. c, g, k, o Cross-sectional view of damaged areas. d, h, l, p Simplified cross-section schematics showing their damaged characteristics.
Discussion
Compared to several commercial alloys, high Mn-bearing ferrous alloys show superior mechanical properties with a more attractive combination of UTS and ductility, owing to their inherent high strain-hardening capability33. In principle, during deformation of the alloys, austenite (γ) deforms via three mechanistic processes: (i) dislocation glide, (ii) martensite transformation, and (iii) mechanical twinning34. In particular, the twin boundaries serving as planar obstacles can reduce the mean free path of the dislocation glide35 and refine the grain structure, causing the dynamic Hall–Petch effect36, which describes the beneficial effects of twinning on the strain hardening33 of the alloys. This deformation mechanism, ensuring the extraordinary superior hybrid performance, can be manifested even in cryogenic environments37, with no marked ductile/brittle transition temperature4. Although a similar level of low-temperature toughness can also be found in austenitic stainless steels38, which is a much more expensive alloy, their UTS and tensile toughness fall behind those of high Mn-bearing ferrous alloys. Hence, these high Mn-bearing alloys can be regarded as promising and more commercially available materials used in harsh mechanical and cryogenic environments.
On the other hand, the stronger metallic materials are generally more prone to embrittlement caused primarily by H-penetration and accumulation at metallurgical defects in the matrix39. Nevertheless, the H-impermeability of Mn-bearing alloys (Fig. 1c), which was maybe attributed to the considerably slower diffusion kinetics of H through their matrix with an FCC structure, suggests that the alloys are mostly free from the danger of H-embrittlement.
One of the noticeable features in the electrochemical characterization was that the Cr-bearing high Mn alloy and 9Ni showed comparatively slower corrosion kinetics during long-term polarization measurements in an aqueous environment. In addition, the two samples (H_Mn-L_Cr and 9Ni) had comparatively lower Qdl values obtained by the EIS, suggesting a higher tendency to adsorb dissolved metal ions32,40 and form scale at the surface. This leads to a higher Rct at the solution/metal interface (i.e., a representative index for the resistance of an electrochemical reaction at the metal surface) for the two samples. In contrast, the Cr-free Mn-bearing alloy samples had much higher corrosion kinetic parameters, showing continuously increasing trends with prolonged immersion. The higher anodic current densities (i.e., ianodic, anodic dissolution rates) of the Mn-bearing alloy samples at the very early stages of immersion, determined quantitatively by potentiodynamic polarization measurements (see Supplementary Fig. 11), can also be understood in this regard. The presence of Mn element with a much lower standard reduction potential (−1.19 VSHE) than Fe (−0.45 VSHE)6 in the ferrous alloys increases the thermodynamic driving force for corrosion. This facilitates anodic dissolution on the surfaces of Mn-bearing ferrous alloys at the early stages of immersion due to the higher stability of Mn2+ than Mn-based oxide/hydroxide under acidic and neutral conditions (refer to the Pourbaix diagram of Mn41). Hence, it cannot be expected to have corrosion scale with a protective nature, as evidenced morphologically by the formation of irregular and porous scale (MnxFe3−xO4) with poor adhesion to the matrix of the Cr-free Mn-bearing alloy samples. Under controversy over the role of Mn addition on the characteristics of rust formation (refinement of the grain size7, decrease in electronegativity42, increase in thermodynamic stability43, and generation of H+ 32), this study showed that the partial substitution of Fe with Mn with a small portion (5.5 ~ 8 at.%) in Fe3O4 produced little improvement of corrosion resistance during the early to long-term immersion periods in aqueous environments. The highest susceptibility to aqueous corrosion of M_Mn with a relatively lower Mn content was caused mainly by the detrimental impact of carbide precipitated along the grain boundaries in the matrix (Supplementary Fig. 12) due to its higher C content. The morphological evidence of the preferential dissolution at the interface between matrix and carbide, characterized as M7C3 (M: mostly Cr), during the early stage of corrosion, was shown in Supplementary Fig. 13.
In a ternary system consisting of high Mn–low Cr-balanced Fe, however, the driving force for Mn and Fe dissolution from the surface is expected to be higher, owing to the much higher standard reduction potential of Cr6 than Mn and Fe. Such preferential and rapid dissolution of Mn and Fe causes Cr-enrichment on the surface, leading to the (Fe,Cr)-enriched oxide characterized as α-Fe2−xCrxO3 by XRD (Fig. 4). Considering that the partial substitution of Fe by Cr in Fe2O3 results in lattice contraction44 and thereby a higher band gap energy (i.e., lower electron mobility), the corrosion inhibiting nature of α-Fe2−xCrxO3 formed on a Cr-bearing high Mn alloy may be closely associated with the refinement of the particle size in its structure. Although there were cracks in the inhibiting oxide, as observed in Fig. 3g, h and Supplementary Fig. 7, and the outward migration of dissolved cations may continue through the cracks, it results in the additional formation of FexMn3−xO4 on the inhibiting oxide, giving a bilayer structure consisting of α-Fe2−xCrxO3/FexMn3−xO4. This type of structure may suppress the transport of corrosive species and dissolved metallic cations during prolonged immersion. Based on this discussion, Fig. 7 presents a brief sketch of the corrosion and scale-forming processes of high Mn–low Cr-bearing ferrous alloys proposed in this study.

a–c Initial, middle, and later stages in aqueous environments, respectively.
In API X70, Fe3O4 formed on the surface is a metallic conductor with a band gap of 0.1 eV45 that cannot provide an efficient barrier against further corrosion. On the other hand, the addition of 9 wt.% Ni, which is a nobler element than Fe and thus more expensive than conventional steel alloys, is generally expected to have superior corrosion resistance. Although a significant decrease in the OCP and increase in icorr were observed at the early stages of immersion presumably due to microgalvanic corrosion46 in the matrix consisting of tempered martensite (α’) and dispersed austenite (γ) enriched with Ni, the corrosion kinetics was similar to that of H_Mn-L_Cr after prolonged immersion. Based on a previous investigation47, which reported the type of rust formed on Ni-bearing steel (Ni/Fe ratio < 0.5 : NixFe3−xO4 (X < 1) = 0.5 : NiFe3O4 and >0.5 : Ni-based compounds), and the analytical study (EDS/XRD/XPS) shown here, the inner and outer rusts were regarded reasonably as NixFe3−xO4 and NiO/Ni2O3, respectively. Similar to the case of Cr-substituted Fe3O4, a smaller lattice parameter and crystalline size48, higher band gap of more than 5 eV49, and higher Rp of Ni-substituted Fe3O4, providing the corrosion inhibiting effects were expected. Nevertheless, the localized pit-like attacks observed occasionally on the outer surface of 9Ni could be taken seriously under a combination of erosion and corrosion environment, which will be discussed below.
Under the erosion–corrosion dynamic flow system, two processes are typically involved: surface plastic deformation produced by the collision of erodent and flow-accelerated electrochemical corrosion. The two synergistic parameters, EC and CE, were adopted in this study, because these processes are mutually influenced. Based on the chemo-mechanical effect, the change in flowing stress (ΔσA) of the surface layer produced by strain hardening before and after corrosion process can be described simply as ΔσA = σafter − σbefore (σafter < σbefore) = α·ln (iÃ/ith), where α, iÃ, and ith are the parametric constant, average anodic current density over the sample surface, and threshold current density to degrade the surface strength at the equilibrium state, respectively50. This corrosion-induced surface hardness degradation, which was proposed previously51,52, can be a probable mechanism of EC. Hence, the higher EC/VT of Mn-bearing alloy samples could have resulted from their higher ià and subsequent ΔHv (= \(H_{v_c}\)−\(H_{V_a}\), \(H_{v_c}\) and \(H_{V_a}\) are the surface hardness measured under the corrosion and air condition, respectively) at the early stage (within 1 day) of erosion–corrosion. A longer erosion–corrosion duration would have resulted in a smaller EC/VT in H_Mn-L_Cr, which will be clarified more in further studies. Despite the higher EC/VT of H_Mn-L_Cr, the VT was similar to that of 9Ni, suggesting that surface softening by the corrosion process of 9Ni is less severe, but it has a higher CE/VT. The parameter, CE, can be understood by conventional electrochemical theory, indicating that the formation of a plastically deformed area on the metal surface leads to an increase in local anodic current density53. Although the plastically deformed areas in H_Mn-L_Cr appear broader, much of the deformation was confined to the damage to the corrosion product (α-Fe2−xCrxO3), not direct damage to the underlying matrix, indicating a cushion effect by α-Fe2−xCrxO3. Rather, local damage in the form of a coarse crater with a depth of more than 50 µm was more noticeable in 9Ni. Instead of softening over a large surface of 9Ni, induced by the uniform corrosion process (relating to iÃ), such local damage may lead to much higher local anodic current densities (iÅ), which would be a probable mechanistic reason for the higher CE/VT of 9Ni. These results suggest that EC is dependent greater on ià associated with uniform corrosion than on ià induced by localized damage, but vice versa in the case of CE.
In conclusion, this study highlighted ternary-based austenitic alloys (Fe1−x−yMnxCry with y << x << 1 and lower C contents) with superior resistances to static corrosion and flowing erosion–corrosion in an aqueous environment, in combination with attractive mechanical properties and inhibiting nature of H-penetration in the alloy. A combination of more active Mn–nobler Cr with larger–smaller quantities, respectively, with balanced Fe enables the alloys to have markedly enhanced corrosion and erosion–corrosion resistances by forming of a bilayer scale structure with inner α-Fe2−xCrxO3 and outer FexMn3−xO4. This work provides deeper insights into the combination of more active–noble elements in ferrous alloys, developed possibly through existing commercial processes in industry, and can also be widely applicable to the design of other alloys.
Methods
Sample information
The three types of high Mn-bearing ferrous alloys were produced by hot rolling and subsequent controlled cooling within the temperature range of 800–500 °C at an average rate faster than 10 °C s–1, which is in reference to ASTM A1106/A1106M54. For a steel plate equivalent to X70 grade according to the API specification (API X70), a TMCP involving the controlled rolling and on-line accelerated cooling process using a water spray in the temperature range 800–500 °C was applied. The 9Ni steel plate, which had been hot rolled and reheated to over the A3 temperature, was quenched in water and tempered within 500–600 °C.
Mechanical test and H-permeation measurements
The uniaxial tensile tests of the polished samples were performed in a tensile machine with an initial strain rate of 10−3 s−1 at room temperature, and the UTS and total elongation were obtained. To ensure repeatability, the tests were conducted three times. Vickers hardness testing with a 1 kgf load was also conducted on the sample surfaces after grinding to 2000 grit paper. At least ten indentations were measured on each sample for reproducibility.
The H-permeation experiment of the tested samples was conducted using a Devanathan–Stachurski cell. This involves the diffusion of atomic H generated on the H-charging side of a sample membrane polarized galvanostatically at −1 mA cm−2 in a 3.5% NaCl + 0.3% NH4SCN solution. The H-permeation current density was measured on the other side of the membrane electrodeposited with Pd under potentiostatic polarization with an anodic potential of 270 mVSCE. A more detailed experimental method can be found elsewhere39,55. The experiment was carried out three times to ensure reproducibility and only representative data of the samples were presented.
Electrochemical corrosion characterization and weight-loss measurement
The electrochemical corrosion experiments were conducted with a conventional three-electrode cell system in a 3.5% NaCl solution for 70 days using a potentiostat (Gamry Reference 600). A Pt grid and a saturated calomel electrode (SCE) were used as the counter and reference electrode, respectively. Before the experiments, the samples serving as the working electrode (WE) were ground to 1200 grit sand paper and cleaned ultrasonically in ethanol. For the LPR measurements, the WE (exposed area of 1 cm2), which had been immersed in the solution for 1 h to 70 days, was polarized from −20 to 20 mV vs. OCP at a scan rate of 0.2 mV s−1. To ensure reproducibility, the LPR measurements were conducted three times. The icorr as an index of the corrosion kinetics were quantified by fitting the experimental data to the Wagner–Traud equation56 (given in Eq. (1)) and the changes in icorr and mpy (derived from icorr by the unit conversion formula57) with immersion periods were plotted. The mpy was calculated from icorr by assuming the dissolution of the major alloying elements (Mn, Cr, Ni) as well as Fe from the samples.
where i, icorr, βa, βc, E, and Ecorr are the measured total current density (A cm−2), corrosion current density (A cm−2), anodic Tafel slope (V decade−1), cathodic Tafel slope (V decade−1), measured potential (V), and corrosion potential (V), respectively.
The potentiodynamic anodic polarization tests were also conducted by sweeping the potential from the OCP to 1.2 V vs. OCP at a scan rate of 0.2 mV s–1 for 1 and 28 days. Based on the polarization curves, the anodic current densities termed ianodic at a potential of −0.4 VSCE of the tested samples were compared. The changes in Rp with the immersion periods were additionally obtained by EIS, carried out over the frequency range from 100 kHz to 10 mHz, with an amplitude of ±10 mV AC potential vs. the OCP. The resulting Nyquist plots were fitted to an equivalent circuit model of the metal/solution interface (i.e., a Randle circuit consisting of the solution resistance (Rs) in series with the parallel combination of the double layer capacitance (Qdl) and charge transfer resistance at the solution/metal interface (Rct); see Supplementary Fig. 14). Under the simple Randle circuit model without considering the Warburg element and other resistances, the Rct was considered equal to Rp.
Before the weight-loss measurements, the samples, 35 mm in width, 35 mm in length, and 5 mm in thickness, were ground to 800 grit sand paper and cleaned with ethanol. They were then weighed (W) in an analytical balance to a precision of 0.0001 g. After immersion in a 3.5% NaCl solution for 7–56 days, the samples were cleaned ultrasonically in distilled water for 3 min, to remove the corrosion scale from the surface. The samples were then dried in air and re-weighed. The weight loss was quantified by the change in the measured weights (Winitial − Wfinal) per initial surface area. To ensure reproducibility, three independent weight-loss measurements were conducted.
Analysis of corrosion scale
The surface and cross-section of the samples after immersion in the solution were observed by field-emission scanning electron microscopy (FE-SEM). Before observing the cross-section, the cold-mounted samples were ground to 2400 grit sand paper and then micro-polished with a 1 µm diamond suspension. The elemental compositions of the rust formed on the samples were analyzed by EDS. XRD (Bruker type diffractometer using a Cu anode) and XPS (VG Scientific Escalab 250 using monochromatic Al Kα radiation (1486.7 eV)) were also employed to analyze the rust. The analyses were conducted directly from the surface of the corroded samples. For XPS, it was operated at a voltage, emission current, and spot size of 15 kV, 10 mA, and 500 μm in diameter, respectively. A constant analyzer energy mode with a 200 and 50 eV pass energy for the survey and high-resolution spectra was employed. The spectra were analyzed using a spectral data processor v 3.0, enabling the deconvolution of the curve by minimizing the χ2-value. The C 1s peak of adventitious carbon at 284.8 eV was used as a reference for charge correction58. The spectrum background was subtracted using the Shirley59 correction method.
Erosion–corrosion characterization
Erosion–corrosion experiments (electrochemical and weight-loss measurements) were conducted with an erosion–corrosion test cell equipped with a conventional rotating device, which is illustrated schematically in Supplementary Fig. 15. Before the experiment, all sides of the rectangular-shaped samples, 100 mm in length, 25 mm in width, and 5 mm in thickness, were ground to 1200 grit sand paper and cleaned ultrasonically in ethanol. The sample was mounted in a sample holder and exposed to a 3.5% NaCl solution containing SiO2 particles (3 wt.%) as an erodent with an average diameter of 75 μm for 1 day. The rotation speed was 570 r.p.m. (i.e., apparent flowing velocities V = ωr = 3 m s−1, where ω and r are the rotating angle speed and radius of the sample, respectively). For an additional study conducted under more severe conditions, the size of the SiO2 particle and rotating angle speed were increased to 250 μm and 1055 r.p.m. (i.e., V = 5.5 m s−1), respectively. The LPR test was conducted immediately after the erosion–corrosion experiments to obtain the total corrosion rate (CT), which was derived from a combination of Faraday’s law60 and Stern–Geary equation61, as shown below62:
where M, B, n, and F are the atomic weight of the metal, Stern–Geary constant, number of electrons involved, and Faraday constant, respectively.
CT can be separated into two components: C0, which was obtained by weight-loss measurement under static conditions, and CE (i.e., CT = C0 + CE), which can be determined by CT − C0. In the same context, the total erosion rate (ET) consisted of two components: E0, which was obtained by weight-loss measurements after the erosion–corrosion experiment in a de-aerated 0.1 M NaOH solution, and EC (i.e., ET = E0 + EC). Therefore, EC can be determined by ET (=VT − CT) − E0. These analytical designs separating the individual components in the total erosion–corrosion rate can also be referenced to a previous investigation63,64 and ASTM G119–0465. The erosion–corrosion measurements were conducted three times to ensure reproducibility.
The surface and cross-section morphologies of the tested samples after the erosion–corrosion experiments were observed by FE-SEM. The elemental compositions in some rusts remaining on the surfaces of the samples were analyzed by EDS.
Data availability
The data that support the plots and other findings of the current study are available from the corresponding author on reasonable request.
References
Podany, P., Duchek, M. & Rund, M. Properties of experimental low-carbon high-manganese twip steel. Int. J. Mech. Pord. Eng. 5, 69–72 (2017).
Cooman, B. C. D., Estrin, Y. & Kim, S. K. Twinning-induced plasticity (TWIP) steels. Acta Mater. 142, 283–362 (2018).
Vladimir, T., Andrey, B. & Rustam, K. Improving mechanical properties of 18%Mn TWIP steels by cold rolling and annealing. Metals 9, 776 (2019).
Nam, Y. H., Park, J. S., Baek, U. B., Suh, J. Y. & Nahm, S. H. Low-temperature tensile and impact properties of hydrogen-charged high-manganese steel. Int. J. Hydrog. Energy 8, 7000–7013 (2019).
Zhang, Y. S. & Zhu, X. M. Electrochemical polarization and passive film analysis of austenitic Fe-Mn-Al Steels in aqueous solutions. Corros. Sci. 41, 1817–1833 (1999).
Jones, D. A. Principles and Prevention of Corrosion (Prentice Hall, 1996).
Kim, B. M., Kim, S. J. & Kim, H. S. Effects of alloying elements (Cr, Mn) on corrosion properties of the high-strength steel in 3.5% NaCl solution. Adv. Mater. Sci. Eng. 2018, 1–13 (2018).
Park, J. H., Seo, H. S., Kim, K. Y. & Kim, S. J. The effect of Cr on the electrochemical corrosion of high Mn steel in a sweet environment. J. Electrochem. Soc. 163, 791–797 (2016).
ASTM Staff. Annual Book of ASTM Standards 1979 (ASTM, 1979).
Krolo, P., Grandić, D. & Smolčić, Ž. Experimental and numerical study of mild steel behavior under cyclic loading with variable strain ranges. Adv. Mater. Sci. Eng. 2016, 1–13 (2016).
Nasiri, Z. & Mirzadeh, H. Spheroidization heat treatment and intercritical annealing of low carbon steel. J. Min. Met. Sect. B Met. 55, 405–411 (2019).
Hwang, E. H., Seong, H. G. & Kim, S. J. Effect of carbon contents on corrosion and hydrogen diffusion behaviors of ultra-strong steels for automotive applications. Korean J. Met. Mater. 56, 570–579 (2018).
Luo, H. et al. A strong and ductile medium-entropy alloy resists hydrogen embrittlement and corrosion. Nat. Commun. 11, 1–8 (2020).
Yuan, B., Yu, H., Li, C. & Sun, D. Effect of hydrogen on fracture behavior of Ti-6Al-4V alloy by in-situ tensile test. Int. J. Hydrog. Energy 35, 1829–1838 (2010).
Hoyos, J. J. et al. In situ synchrotron radiation measurements during axial strain in hydrogen cathodically charged duplex stainless steel SAF 2205. Mater. Res. 21, e20170686 (2018).
Zeides, F. & Roman, I. Study of hydrogen embrittlement in aluminium alloy 2024 in the longitudinal direction. Mater. Sci. Eng. A 125, 21–30 (1990).
Natividad, C., Garcı́a, R., López, V. H., Falcón, L. A. & Salazar, M. in Characterization of Metals and Alloys 3–11 (Springer, 2017).
Lee, S. W., Im, I. H. & Hwang, B. Effect of microstructure on the strain aging properties of API X70 pipeline steels. Korean J. Mater. Res. 28, 702–708 (2018).
Sasaki, T. et al. Physical and mechanical properties of high manganese non-magnetic steel and its application to various products for commercial use. Trans. Iron. Steel Inst. Jpn 22, 1010 (1982).
Sung, H. K., Jeong, D. H., Park, T. D., Lee, J. S. & Kim, S. S. S-N fatigue behavior of Fe25Mn steel and its weld at 298 and 110K. Met. Mater. Int. 22, 755–763 (2016).
Yoon, Y. K., Kim, J. H. & Shim, K. T. Mechanical characteristics of 9% Ni steel welded joint for LNG storage tank at cryogenic. Int. J. Mod. Phys. 6, 355–360 (2012).
Nakada, N., Syarif, J., Tsuchiyama, T. & Takaki, S. Improvement of strength–ductility balance by copper addition in 9%Ni steels. Mater. Sci. Eng. A 374, 137–144 (2004).
Luo, H., Li, Z. & Raabe, D. Hydrogen enhances strength and ductility of an equiatomic high-entropy alloy. Sci. Rep. 7, 9892 (2017).
Fallahmohammadi, E. et al. Hydrogen diffusion into three metallurgical microstructures of a C-Mn X65 and low alloy F22 sour service steel pipelines. Int. J. Hydrog. Energy 39, 13300–13313 (2014).
Park, G. T., Koh, S. U., Jung, H. G. & Kim, K. Y. Effect of microstructure on the hydrogen trapping efficiency and hydrogen induced cracking of linepipe steel. Corros. Sci. 50, 1865–1871 (2008).
Park, J. S., Lee, J. W., Hwang, J. K. & Kim, S. J. Effects of alloying elements (C, Mo) on hydrogen assisted cracking behaviors of A516-65 steels in sour environments. Materials 13, 1–17 (2020).
Sojka, J., Váňová, P., Vodárek, V. & Sozańska, M. Diffusion of hydrogen in the TRIP 800 steel. Procedia Mater. Sci. 12, 66–71 (2016).
Olden, V., Saai, A., Jemblie, L. & Joghnsen, R. FE simulation of hydrogen diffusion in duplex stainless steel. Int. J. Hydrog. Energy 39, 1156–1163 (2014).
Kim, K. S., Kang, J. H. & Kim, S. J. Nitrogen effect on hydrogen diffusivity and hydrogen embrittlement behavior in austenitic stainless steels. Scr. Mater. 184, 70–73 (2020).
Kwon, Y. J., Seo, H. J., Kim, J. N. & Lee, C. S. Effect of grain boundary engineering on hydrogen embrittlement in Fe-Mn-C TWIP steel at various strain rates. Corros. Sci. 142, 213–221 (2018).
British Standard European Norm. ISO 17081. Method of Measurement of Hydrogen Permeation and Determination of Hydrogen Uptake and Transport in Metals by an Electrochemical Technique (BS EN Standards, 2014).
Singh, A. K., Shukla, S. K. & Quraishi, M. A. Corrosion behavior of mild steel in sulphuric acid solution in presence of ceftazidime. Int. J. Electrochem. Sci. 6, 5802–5814 (2011).
Bayraktar, E., Khalid, F. A. & Levaillant, C. Deformation and fracture behavior of high manganese austenitic steel. J. Mater. Process. Technol. 147, 145–154 (2004).
Adler, P. H., Olson, G. B. & Owen, W. S. Strain hardening of hadfield manganese steel. Met. Tans. A 17, 1725–1737 (1986).
Bouaziz, O., Allain, S., Scott, C. P., Cugy, P. & Barbier, D. High manganese austenitic twinning induced plasticity steels: a review of the microstructure properties relationships. Curr. Opin. Solid Stat. Mater. Sci. 15, 141–168 (2011).
Otto, F. et al. The influences of temperature and microstructure on the tensile properties of a CoCrFeMnNi high-entropy alloy. Acta Mater. 61, 5743–5755 (2013).
Tang, L. et al. Synergistic deformation pathways in a TWIP steel at cryogenic temperatures: In situ neutron diffraction. Acta Mater. 200, 943–958 (2020).
Zheng, C. & Yu, W. Effect of low-temperature on mechanical behavior for an AISI austenitic stainless steel. Mater. Sci. Eng. A 710, 359–365 (2018).
Park, J. S., Seong, H. G. & Kim, S. J. Effect of Heat treatment conditions on corrosion and hydrogen diffusion behaviors of ultra-strong steel used for automotive applications. Corros. Sci. Technol. 18, 267–276 (2019).
Kim, M. J. & Kim, J. G. Effect of manganese on the corrosion behavior of low carbon steel in 10 wt.% sulfuric acid. Int. J. Electrochem. Sci. 10, 6872–6885 (2015).
Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions (Pergamon, 1966).
Hao, L., Zhang, S., Dong, J. & Ke, W. Atmospheric corrosion resistance of MnCuP weathering steel in simulated environments. Corros. Sci. 53, 4187–4192 (2011).
Kamakshi, T., Sundari, G. S., Erothu, H. & Singh, R. S. Effect of nickel dopant on structural, morphological and optical characteristics of Fe3O4 nanoparticles. Rasayan J. Chem. 12, 531–536 (2019).
Paul Scherrer Institute. Report No. 02-04 (Paul Scherrer Institute, 2002).
Andreu, C. et al. Vacancy coalescence during oxidation of iron nanoparticles. J. Am. Chem. Soc. 129, 10358–10360 (2007).
Qin, M., Xu, W., Yang, L. & Li, Y. Corrosion behavior of 9% Ni steel for LNG strorage tanks in 3.5% NaCl solution. Int. J. Electrochem. Sci. 13, 6537–6549 (2018).
Electric Power Research Institute. Report No. EPRI/NP/3789 (Electric Power Research Institute, 1985).
Riaz, S., Bashir, M. & Naseem, S. Iron oxide nanoparticles prepared by modified Co-precipitation method. IEEE Trans. Mag. 50, 1–4 (2014).
Pouretedal, H. R. & Momenzadeh, F. Synthesis, characterization and study of photocatalytic activity of nanocomposites of oxides and sulfides of Ni(II) and Ni(III). Bulg. Chem. Commun. 47, 59–65 (2015).
Lu, B. T., Lu, J. F. & Luo, J. L. Erosion-corrosion of carbon steel in simulated tailing slurries. Corros. Sci. 53, 1000–1008 (2011).
Lu, B. T. & Luo, J. L. Synergism of electrochemical and mechanical factors in erosion-corrosion. J. Phys. Chem. B 110, 4217–4231 (2006).
Gutman, E. M. Mechanochemistry of Materials. (Cambridge International Science, 1998).
Kim, S. J. Effect of the elastic tensile load on the electrochemical corrosion behavior and diffusible hydrogen content of ferritic steel in acidic environment. Int. J. Hydrog. Energy 42, 19367–19375 (2017).
ASTM A1106/A1106M. Standard Specification for Pressure Vessel Plate, Alloy Steel, Austenitic High Manganese for Cryogenic Application (ASTM International, 2017).
Kim, S. J. & Kim, K. Y. Electrochemical hydrogen permeation measurement through high-strength steel under uniaxial tensile stress in plastic range. Scr. Mater. 66, 1069–1072 (2012).
Wagner, C. & Traud, W. Additive hypothesis for oxidation-reduction reactions. Z. Electrochem. 44, 391–454 (1938).
Metha, Y., Trivedi, S., Chandra, K. & Mishra, P. S. Effect of silicon on the corrosion behavior of powder-processed phosphoric irons. J. Miner. Mater. Charact. Eng. 9, 855–865 (2010).
Schön, G. High resolution auger electron spectroscopy of metallic copper. J. Electron Spectrosc. 1, 377–387 (1972).
Shirley, D. A. High-resolution X-ray photoemission spectrum of the valence bands of gold. Phys. Rev. B 5, 4709–4714 (1972).
Faraday, M. On electrochemical decompositions. Philos. Trans. 123, 675–710 (1833).
Stern, M. & Geary, A. L. Electrochemical polarization I. A theoretical analysis of the shape of polarization curves. J. Electrochem. Soc. 104, 56–63 (1957).
Liu, J., Wang, J. & Hu, W. Erosion-corrosion behavior of X65 carbon steel in oilfield formation water. Int. J. Electrochem. Sci. 14, 262–278 (2019).
Zeng, L., Zhang, G. A. & Guo, X. P. Erosion-corrosion at different locations of X65 carbon steel elbow. Corros. Sci. 85, 318–330 (2014).
Guo, H. X., Lu, B. T. & Luo, J. L. Interaction of mechanical and electrochemical factors in erosion-corrosion of carbon steel. Electrochim. Acta 51, 315–323 (2005).
ASTM G 119-04. Standard Guide for Determining Synergism Between Wear and Corrosion (ASTM International, 2016).
Acknowledgements
This research was supported in part by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (number 2019R1C1C1005007). In addition, this work was partly funded and conducted under the Competency Development Program for Industry Specialists of the Korean Ministry of Trade, Industry and Energy (MOTIE), operated by the Korea Institute for Advancement of Technology (KIAT) (number P0002019, HRD Program for High Value-Added Metallic Materials Expert).
Author information
Authors and Affiliations
Contributions
S.J.K. directed the research project. S.G.L. and J.K.C. designed and produced the samples. J.S.P., J.W.L., and J.K.H. analyzed the data. S.O.K. and Y.J.J. performed the experiments and analyses. They are co-first authors. S.O.K., Y.J.J., and S.J.K. wrote the paper. All authors discussed and commented on the results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jeong, Y.J., Kim, S.O., Park, J.S. et al. Strong and ductile Fe-24Mn-3Cr alloy resistant against erosion-corrosion. npj Mater Degrad 5, 47 (2021). https://doi.org/10.1038/s41529-021-00195-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-021-00195-0
