
Abstract
Over the past few decades, molecular dynamics simulations and first-principles calculations have become two major approaches to predict the lattice thermal conductivity (κL), which are however limited by insufficient accuracy and high computational cost, respectively. To overcome such inherent disadvantages, machine learning (ML) has been successfully used to accurately predict κL in a high-throughput style. In this review, we give some introductions of recent ML works on the direct and indirect prediction of κL, where the derivations and applications of data-driven models are discussed in details. A brief summary of current works and future perspectives are given in the end.
Similar content being viewed by others


Introduction
The lattice thermal conductivity (κL) is a key design parameter for various technological applications. For example, heat sinks in the electronic devices require higher κL to dissipate the excessive thermal energy1, while reducing κL is an effective approach to improve the efficiency of thermoelectric (TE) conversion2. It is thus quite necessary to discover/design particular systems with desired κL. On the theoretical side, the most reliable approach for predicting κL is the solution of phonon Boltzmann transport equation (BTE) within the framework of density functional theory (DFT)3,4. However, the required calculations of the interatomic force constants (IFCs) are time-consuming, especially for those with large unit cell and low symmetry. As an alternative, the classic molecular dynamics (MD) simulations can be utilized to predict the κL of systems with complex crystal structure5. Nevertheless, the accuracy of MD significantly depends on the choice of interatomic potentials, which also limits its wide application. In a word, there remains some challenges or difficulties to accurately predict the κL, especially in a high-throughput way.
As an important technique of artificial intelligence, machine learning (ML) can efficiently determine the underlying connectivity among enormous data at extremely low cost6,7,8,9. During the past few decades, many efforts have been devoted to evaluate the κL of various systems, both theoretically and experimentally10,11,12,13,14. Based on these available data, ML can establish a mapping between the target property (κL) and the input features (such as the atomic mass, the phonon frequency, and the volume of unit cell8,15). Compared with first-principles calculations and MD simulations, the data-driven ML models enable high-throughput evaluation of κL, which exhibit strong predictive power for systems both inside and beyond the training set15,16. In addition to such direct prediction of κL, ML has been successfully used to build accurate interatomic potentials for MD simulations. Generally speaking, the machine learning potential (MLP) employs regression algorithm to determine the ab-initio potential energy surface (PES), and the atomic configurations are usually adopted as input features17,18. Recently, the MLPs have been utilized to accurately predict the κL of systems with complex crystal structures and chemical compositions, such as the alloys19, the heterostructures20, and the molten salts21. On the other hand, as the derivative of total energy with respect to the atomic displacement, IFCs can be obtained from the Taylor expansion of the PES4,22. The MLPs determine the accurate PES and thus can derive the IFCs at almost negligible computational cost, which enable accelerated solution of phonon BTE for the evaluation of κL22,23. Collectively speaking, ML can overcome the inherent disadvantages of MD simulations and first-principles calculations to accurately and readily predict κL.
The remainder of this review is organized as follows. In the section of “Direct prediction”, we give a brief introduction of the dataset construction, the feature selection, and the training algorithms, which are then combined to obtain the high-throughput models for predicting κL. In the section of “Indirect approach”, we focus on the construction of MLPs, and highlight their first-principles level accuracy as well as advantages over general approaches in predicting κL. The review is concluded with a summary of current works and future perspectives.
Direct prediction
Dataset construction and related features
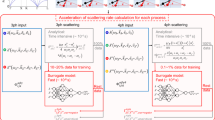
As a data-driven technique, ML requires a dataset that contains the κL for a substantial number of systems to derive reliable prediction model. In general, the κL can be collected from first-principles calculations, MD simulations, and experimental measurements. As an addition, one can also obtain the κL from some materials databases. For example, thousands of entries in the Automatic FLOW (AFLOW) database have included the κL values calculated by the so-called Automatic Gibbs Library (AGL) method12,24. Here, the GIBBS quasiharmonic Debye model25 is employed to evaluate the Debye temperature and the Grüneisen parameter based on the computationally feasible adiabatic bulk modulus, which are then inserted into the well-known Slack model for the determination of κL26. We should emphasize that it would be better to collect all the κL obtained using the same approach, for example, either first-principles or MD. However, if there is not enough data available for ML, one can also consider both of them, as long as the interatomic potentials adopted in MD are well-tested and the results exhibit sufficient accuracy. Figure 1 is a schematic illustration of ML for the high-throughput prediction of κL, where the dataset is usually divided into two subsets including the training and testing sets. To avoid random selection of training data, the principal component analysis (PCA) can be used to identify systems that possess distinct features in the dataset. For example, Tranås et al. demonstrated that a model based on a semi-random pool of half-Heusler (HH) compounds (i.e. assumed “bad luck” in the training set) was unable to correctly predict the small κL values of those systems in the testing set from the rest27. As an alternative, they used active sample selection based on PCA, where three compounds with extremely low κL were included in the training process. Such an approach can significantly improve the model performance, in particular the ability to identify the low κL compounds in the testing set.

Three components are usually involved in machine learning: dataset construction, input features, and training algorithms.
As mentioned in the introduction, ML is implemented to establish a mapping between the κL and some related input features, which usually contains the information about: (1) the structural properties, such as the lattice constant, the volume of unit cell, the number of atoms, and the bond length; (2) the elemental properties of the constituent atoms, including the atomic number, the atomic mass, and the Pauli electronegativity; (3) the phonon properties, such as the phonon frequency, the group velocity, the heat capacity, and the Grüneisen parameter. To be compatible with most ML algorithms, systems with different size needs to be represented by feature vectors with a fixed length. Such a problem can be solved by adopting statistical values of the elemental properties, such as the maximum, the minimum, the composition-weighted (CW) value, and the standard deviation28. Obviously, it is quite important to screen out features that are closely related to the target property. For instance, Juneja et al. revealed high Pearson correlation between κL and several fundamental properties, including the maximum phonon frequency, the average atomic mass, the volume of the unit cell, and the integrated Grüneisen parameter up to 3 THz15. Using these input features, they developed a Gaussian process regression-based ML model by training the κL of 120 dynamically stable and nonmetallic compounds. To keep only those features that are highly related to κL, Chen et al. performed the so-called recursive feature elimination (RFE) on the initial feature vector, which significantly reduces the dimensionality from 63 to 2929. It should be also noted that highly intercorrelated features could increase the computational cost and may affect the predictive power. It is thus quite necessary to check the feature relevancy and redundancy before any training process.
Machine learning algorithms
With the rapid development of artificial intelligence, various ML algorithms have been proposed, such as the Bayesian Optimization (BO)30, the eXtreme Gradient Boosting (XGBoost)31, the Neural Network (NN)32, the Kernel Ridge Regression (KRR)33, the Least Absolute Shrinkage and Selection Operator (LASSO)34, the Sure Independence Screening and Sparsifying Operator (SISSO)35, the Generalized Linear Regression (GLR)36, the Random Forest (RF)37, the Gaussian Process Regression (GPR)38, and etc. In this section, we give a brief introduction of the NN, SISSO, and RF, which are widely used to construct high-throughput ML models for the prediction of κL.
In the learning process, NN algorithm first feeds the input layer with feature data, which is then manipulated by several hidden layers, and the output layer finally generates the target value. Each neuron is connected with all the neurons from the previous layer, and it deals with the data according to a specific activation function. When data is transferred between neurons, its value will be multiplied by a weight parameter. To generate the best model, one should optimize the hyperparameters (such as the activation function, the number of neurons and the hidden layers) and minimize the loss function. It has been demonstrated that NN can effectively handle non-linear and complex problems, whereas the obtained model is usually treated as a black box.
The SISSO algorithm relies on two key steps: the feature-space construction and the descriptor identification. Specifically, the input features are first combined by iteratively using the algebraic operators \(\left\{{I, + , - , \times ,/,\exp ,\log ,\left| - \right|,\sqrt {} ,^{- 1},^2,^3} \right\}\), which could construct a huge feature space. The sure independence screening (SIS) then scores each new feature with a metric (correlation magnitude), and selects the subspace that contains the descriptors highly related with the training data. The sparsifying operators (SO) is finally utilized to find the optimal n-dimensional descriptor. Compared with many other ML algorithms, the SISSO can identify descriptors that are explicit and analytic functions of key inputs, which is very beneficial for understanding the inherent physical mechanisms.
As an ensemble learning algorithm, RF combines multiple decision trees (DT)39 to avoid overfitting. Each DT in the “forest” is individually trained by randomly selecting a subset of features. Here, the training data is divided into two or more categories at the root node based on the feature values, and each subsequent node will receive a data subgroup and then output the separations to the next nodes until all the generated groups are homogeneous. The output of a final node (called a “leaf”) gives the mean value of the corresponding separated samples. In a word, the trained DT model obtains the predicted value by determining the interval of input features. Collectively, the RF model can rank the importance of each feature according to the order of nodes, and output the average value of predicted results by all the DT ensemble models.
High-throughput prediction models
As an effective high-throughput method, ML has been widely used to predict κL of various systems in recent years15,16,27,28,29,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57. For example, Wang et al. developed a XGBoost model and its predictive power was checked by 549 compounds in the testing set, as shown in Fig. 2a44. Among 75 input features, the average atomization enthalpy (ΔHatomic) and the density (ρ) were found to be most relevant with the κL. Note that the training set contains 4937 κL calculated by the above-mentioned AGL method12, which were collected from the AFLOW database24. The model was then employed on all the entries in the Inorganic Crystallographic Structure Database (ICSD)58, and it was found that compounds containing halogen elements or heavy atoms exhibit low κL (see Fig. 2b). Among them, potential TE materials (such as BiTe2Tl and Cl2CsI) were screened out and the prediction accuracy was validated by first-principles calculations. Besides, the NN algorithm has been successfully applied to predict the κL of random multilayer (RML) and gradient multilayer (GML) structures composed of two types of conceptual atoms with different mass values, as shown in Fig. 2c45. To construct the training set, the κL of 1600 RMLs and their corresponding 1600 GMLs were calculated by MD simulations. In contrast to generally used crystal and elemental properties, the input features include several key parameters for quantifying the disorder in layer thicknesses of RMLs, as listed in Table 1. Figure 2d shows the predicted κL for 200 multilayer structures beyond the training set, which are in good agreement with the MD results. Unlike most ML models which appears as black boxes, Liu et al. employed the SISSO method to establish a physically intuitive descriptor for predicting the κL of HH compounds46. They found that the first term \(D_1 = \frac{{\bar m \times \chi _{{{\mathrm{B}}}} \times \left| {\chi _{{{\mathrm{A}}}} - \chi _{{{\mathrm{B}}}}} \right|}}{{e^{2a}}}\) dominates the three-dimensional (3D) descriptor, where the κL decreases with the lattice constant a but increases with the electronegativity difference |χA − χB| between atoms at site A and B. This is consistent with the general belief that systems with larger unit cell usually have smaller κL, and stronger chemical bonding would lead to higher κL. Beyond the initial 86 training data, the strong predictive power of the descriptor was confirmed by 75 HH compounds and 15 full-Heusler (FH) systems.

a The XGBoost model-predicted log-scaled κL versus the calculated values for the testing set. The top and right histograms show the corresponding data distributions. b Dependence of the predicted κL on specific elements for compounds in the ICSD, and the values are shown by colors along with the ΔHatomic and ρ. a and b are reproduced with permission from ref. 44. c Schematics of the multilayer structures and the MD simulation setup. d Comparisons of the real and predicted κL for 100 randomly generated RMLs and their corresponding 100 GMLs in the testing set. c and d are reproduced with permission from ref. 45.
As typical input features for many ML models, accurate structural parameters are usually obtained by first-principles calculations40,41,54,56 if experimental results are not available. Alternatively, Jaafreh et al. utilized the crystal features of a series prototype structures to establish a RF-based model, which can be applied to related systems without the use of any DFT-relaxed structural parameters16. It should be noted that the crystal features are generated by using the area of each face of the Wigner-Seitz cell (see Fig. 3a) and the characteristics of neighboring atoms. The training set contains 2146 κL of 119 compounds at a series of temperatures from 100 to 1000 K. As shown in Fig. 3b, the RF-based model exhibits strong predictive power for 4 systems in the testing set. To go further, the model was used to predict the room temperature κL for 32,116 compounds in the ICSD, where 273 have ultralow values and 4 are even <0.1 Wm−1K−1 suggesting very promising applications in the field of energy harvesting.

a The Wigner-Seitz cell used to construct the feature space. b Comparisons of the calculated and predicted κL of 4 compounds in the testing set. a and b are reproduced with permission from ref. 16. c Schematic of transfer learning based on CGCNN for the prediction of κL. Here, the crystal structure is converted to a graph, where the nodes represent atoms and the edges connect the neighboring nodes. Reproduced with permission from ref. 51.
In recent years, the Convolutional Neural Network (CNN) algorithm has been adopted to predict the κL of porous graphene47, hybrid carbon-boron nitride honeycombs50, aperiodic superlattices57, and etc. The input layer of CNN is fed with particular arrays, which can be obtained by extracting characteristics of an image representing the system, instead of selecting features from various physical properties. In particular, the Crystal Graph Convolutional Neural Network (CGCNN) algorithm enables the prediction of target properties by a graph representing the connection of atoms in the crystal59. As a major advance, Zhu et al. employed CGCNN to predict the κL of all known inorganic crystals directly from their atomic structures51. As shown in Fig. 3c, they established a model based on the dataset60 containing 2668 calculated lattice thermal conductivities (named κC in their work), where the crystal structure was converted to a graph with the nodes and the edges respectively representing atoms and connections between neighboring atoms. The CNN was initialized by feature vectors that characterize each node and edge. It should be noted that these κC were firstly calculated by a semi-empirical model, which inevitably exhibit insufficient accuracy51,61. To address such a problem, they collected 132 experimentally measured lattice thermal conductivities (κexp). Due to the small size of the training set, the established model exhibits a large mean absolute error (MAE) of 0.51 (for log-scaled κexp). As correlated datasets share similar domain knowledge, they developed a transfer learning scheme (see Fig. 3c) where all layers from the model trained by κC was transferred to initialize a second CGCNN with reduced MAE of ~0.27.
It should be noted that ML models can be further optimized via active learning, which is very useful for the inverse design of systems with desired κL. For example, it is very time-consuming to identify the optimal distribution of holes that can minimize the κL of two-dimensional (2D) materials since the design space expands dramatically with increasing hole density. Taking porous graphene as a prototypical class of examples, Wan et al. adopted a CNN-based inverse design approach to determine the structure with the lowest κL, which only needs to simulate ~103 systems by MD out of the total 106 possible candidates47. By performing MD simulations, Chowdhury et al. obtained the κL of 300 randomly generated Si/Ge RMLs which is used as the initial dataset57. They iteratively identified RMLs with locally enhanced phonon transport and included them as additional training data. Using the CNN model, RMLs with unexpectedly higher κL are discovered, which can be attributed to the presence of closely spaced interfaces.
Summarizing this section, considerable progress has been made in the high-throughput prediction of κL by leveraging various data-driven models. To have a fast understanding, Table 1 provides the training and testing sets, the input features, and the adopted algorithms of representative ML works in recent 5 years. With the increasing growth of big data and accelerated development of artificial intelligence, it is expected that ML would become a major scientific paradigm for accurately predicting κL, and more ML models or descriptors could be emerged to give physical insights into different mechanisms to manipulate κL, such as phonon coherence, weak coupling of phonons, and high-order phonon anharmonicity62.
Indirect approach
Due to the lack of available training data, the above-mentioned ML models cannot be directly applicable to various 2D materials, nanowires, alloys, ternary salts, and etc. As an alternative, ML can be also utilized to construct accurate interatomic potentials or force constants so that efficient evaluation of κL become feasible using MD simulations or even first-principles calculations.
It is known that the atomic-scale simulations need to determine the PES that provides the potential energy as a function of atomic positions. In principle, the most accurate PES can be obtained by quantum mechanics calculations, which is however very time-consuming and even prohibitive for large systems. Based on the physical knowledge of the interatomic bonding, many specific analytic expressions have been proposed, known as the empirical potentials63. However, the PES is a multidimensional real-valued function, which cannot be completely fitted by these specific functional forms64. The empirical interatomic potentials thus usually exhibit insufficient accuracy and the involved parameters should be carefully optimized for different systems. Taking the bulk silicon as an example, the κL calculated by using the original Stillinger-Weber potential (~244 Wm−1K−1 at room temperature) is much higher than the experimentally measured result (~148 Wm−1K−1)65. It is thus quite necessary to develop alternative potentials for accurate prediction of κL, and MLP is one of good choices.
Training data and input features
In principle, ML can be used to fit the correlation between atomic configurations and physical properties of given systems. Compared with empirical potentials, MLPs determine the PES in a data-driven manner to describe the interatomic interactions. To ensure that the MLPs exhibit first-principles level accuracy, the dataset is usually constructed by performing ab-initio molecular dynamics (AIMD) simulations at a series of temperatures, where the energies, the forces, and the stresses of different atomic configurations are then recorded66,67,68. It should be noted that the atomic configurations are sampled from AIMD trajectories and uncorrelated with each other.
Unlike many ML models for high-throughput prediction of κL, establishing MLPs requires to input features that can represent the local environment around each atom, usually within a specific cutoff radius. The adopted features must be invariant to Euclidean transformations and permutation of chemically equivalent atom69. A simple example is the atomic Cartesian coordinates which cannot be used for training. The reason is that when the system is rotated or the chemically equivalent atoms are exchanged, a new list of Cartesian coordinates is generated, which however corresponds to the same atomic configuration. For the evaluation of κL, the widely used features are the moment tensor70, the atom-centered symmetry functions (ACSFs)71,72, and the smooth overlap of atomic positions (SOAP)18. Taking the ACSFs as an example, the \(G_i^2\) and \(G_i^4\) in the following expressions respectively describe the radial and angular environment of atom i,
and
Here Rij is the distance between atom i and j, θijk is the angle centered at atom i, and fc is the smooth cutoff function. ηs and Rs define the width and the center of the Gaussians, respectively. In Eq. (2), the angular resolution and distribution can be determined by ζ and ηa, and λ has the values of +1 and −1. It should be emphasized that the construction of appropriate features is a very challenging task, and we refer the interested reader to a review article73 that summarizes recent work on the efficient representations of atomic and molecular structures.
Machine learning potentials
Table 2 summarizes several important MLPs used for the evaluation of κL, which includes the Moment Tensor Potentials (MTPs)70, the Neural Network Potentials (NNPs)71,74, and the Gaussian Approximation Potentials (GAPs)75. In particular, the MTPs exhibit an excellent balance between accuracy and computational efficiency76, which have been widely used to predict the κL of various systems, such as monolayers, alloys, and complex compounds68,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93. In principle, the purpose of training MTP is minimizing the difference between the predicted and DFT-calculated energies (E), forces (f), and stresses (σ) for K atomic configurations:70,94,95
Here, we, wf, and ws are respectively positive weights that express the importance of energies, forces, and stresses in the training process. In order to improve the quality of MTPs, active learning is usually implemented, where the atomic configuration will be included in the training set if its extrapolation grade (a feature correlated with the prediction error94) is above the threshold and below the allowed maximum. Figure 4 show the widely used active learning scheme for training a MTP, which usually contains six stages labeled as A to F.

By selecting extrapolative configurations from the MD trajectories, the MTPs are trained in a loop until simulations are finished without exceeding the allowed maximum of extrapolation grade. Reproduced with permission from ref. 94.
To validate the accuracy of the trained MLP, the energies, the forces, and the stresses of different atomic configurations are checked in the testing and predicting sets. For instance, Huang et al. proposed a single atom neural network potential (SANNP) for the amorphous silicon based on the training set containing 800 atomic configurations from AIMD simulations67. Figure 5a−c respectively show the predicted total energies, atomic energies, and atomic forces in the testing set, which agree well with those obtained from DFT calculations.

The intuitive linear correlations between the predicted a atomic energies, b total energies, c atomic forces and those calculated by DFT in the testing set for the amorphous silicon. Reproduced with permission from ref. 67.
Application examples
As mentioned in the introduction part, the MLPs with first-principles level accuracy can be implemented into the MD simulations or the phonon BTE to indirectly predict κL of given systems. Unlike conventional first-principle calculations or classic MD with empirical potentials, the evaluation of κL by employing MLPs simultaneously exhibits strong reliability and high efficiency, which have been demonstrated by various systems as also summarized in Table 220,21,22,23,66,67,68,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119. For example, Korotaev et al. used the active learning algorithm to develop the MTP for the CoSb3 skutterudite and accurately predict its κL at different temperatures68. Indeed, we see from Fig. 6a that the κL indirectly obtained from MTP almost coincide with the experimentally measured results. It should be emphasized that, compared with conventional first-principles calculations, the MTP can significantly accelerate the prediction process (the computational speed increased by more than four orders of magnitude).

a Comparison of the calculated and experimentally measured κL of CoSb3. The vertical lines show doubled standard deviation of the results calculated by Green-Kubo method. Reproduced with permission from ref. 68. b The lattice thermal conductivities along two different directions (κa and κc) and their average value (κp) of (Ti0.2Zr0.2Hf0.2Nb0.2Ta0.2)B2, which are plotted as a function of temperature. The inset shows the auto-correlation function. Reproduced with permission from ref. 107. c κph-v and d κ3ph+ph-v of silicon with respect to vacancy concentration at 300 K, predicted by the DFT, GAP, and empirical potentials. Reproduced with permission from ref. 116.
Due to the compositional complexity, predicting the κL of high-entropy materials is usually a challenging task. Recently, Dai et al. established a deep learning potential for the thermal insulting material (Ti0.2Zr0.2Hf0.2Nb0.2Ta0.2)B2120, which was then used in the MD simulations to calculate its average lattice thermal conductivity along two directions (κp), as shown in Fig. 6b107. At room temperature, the κp is predicted to be 4.0 Wm−1K−1, which is close to the experimentally measured value of ~4.8 Wm−1K−1.
In addition, the GAP of crystalline Si with vacancies was adopted by Babaei et al. to determine the lattice thermal conductivities contributed from phonon-vacancy scattering (κph-v)116. As can be found from Fig. 6c, the κph-v predicted from the GAP show good agreement with the DFT-calculated results at different vacancy concentrations, while those with empirical potentials exhibit much larger errors. Similar picture can be found in Fig. 6d, where the effects of three-phonon and phonon-vacancy scatterings are both included (κ3ph+ph-v). Note that the computational cost of the GAP is five orders of magnitude smaller than that of DFT calculations in their own work, indicating the high efficiency of such kind of MLP for the prediction of κL.
Last but not least, we note that several other MLPs were proposed recently, such as the spectral neighbor analysis potential, the bond order potential, the force constant potential, and the spatial density neural network force fields, which have been also demonstrated to accurately evaluate the κL of many systems at low computational cost19,121,122,123,124,125,126,127. With the deep understanding of interatomic interactions, it is reasonable to expect that more and more reliable and universal MLPs could be developed in the future.
Summary and perspective
To conclude, we hope this mini review could enable the interested reader to have a preliminary understanding of predicting κL via ML, either directly or indirectly. As high-throughput ML models for the direct prediction of κL, the input features usually contain several fundamental physical properties related to the investigated systems and the constituent elements, such as the lattice constants, the phonon frequency, the atomic mass, and so on. Such kind of data-driven models can be utilized for the rapid screening and inverse design of materials with desired κL, and their predictive power has been demonstrated by checking many systems both inside or beyond the training sets. In addition, the MLPs can be readily implemented into the MD simulations or the phonon BTE, which offer an indirect but quite efficient prediction of κL for particular systems, including crystal with defects, high-entropy compounds, amorphous structures, and so on. Compared with conventional DFT and MD approaches, the MLPs can significantly accelerate the evaluation of κL and simultaneously retain first-principles level accuracy.
Although considerable advances have been made in the direct prediction of κL via ML, there remain several challenges to be addressed in the future. For example, it is still difficult to construct large and reliable dataset required for training, which definitely affects the predictive power and the transferability of such data-driven method. In particular, many ML models are severely limited to some specific systems (e.g. HH compounds, zincblende and rocksalt structures), leaving a much larger materials space unexplored40,41,43,46,52,54. Although one can find the κL values for thousands of compounds in the AFLOW and the TE Design Lab repositories, they are usually calculated by using empirical models12,61 and may exhibit insufficient accuracy compared with those obtained from first-principles calculations, MD simulations, or experimental measurements. On the other hand, substantial advances have been made in the high-throughput discovery of 2D materials, while their thermal transport properties are less known so far128,129,130,131. Due to the limited experimental and theoretical data, it is rather difficult to derive a reliable ML model to predict the κL of various 2D materials. It is believed that the transfer learning can overcome the disadvantage of small data size by pretraining the correlated dataset. However, it still requires accurate first-principles calculations to obtain the scattering phase space of numerous systems53, which remains a tough task. To take better advantage of transfer learning, much efforts should be devoted to identify readily available physical properties that are highly correlated with κL.
In the case of developing efficient MLPs, it is usually necessary to calculate the energies, the forces, and the stresses of a substantial number of atomic configurations within the framework of DFT. Such a task is however very time-consuming for the systems with large unit cell and complex chemical composition, such as molten salts21, skutterudites68, high-entropy compounds107 and so on. Besides, the employed features usually indicate the atomic environment within a certain cutoff radius, and the established MLPs thus ignore the long-range interactions that could be very important for thermal transport properties in some cases. It is expected that the efficiency and accuracy of MLPs can be further improved by careful selection and optimization of input features and/or learning schemes.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
He, Z., Yan, Y. & Zhang, Z. Thermal management and temperature uniformity enhancement of electronic devices by micro heat sinks: a review. Energy 216, 119223 (2021).
Zhao, L.-D. et al. Ultralow thermal conductivity and high thermoelectric figure of merit in SnSe crystals. Nature 508, 373–377 (2014).
Broido, D. A., Malorny, M., Birner, G., Mingo, N. & Stewart, D. A. Intrinsic lattice thermal conductivity of semiconductors from first principles. Appl. Phys. Lett. 91, 231922 (2007).
Li, W., Carrete, J., Katcho, N. A. & Mingo, N. ShengBTE: A solver of the Boltzmann transport equation for phonons. Comput. Phys. Commun. 185, 1747–1758 (2014).
Fan, Z. et al. Force and heat current formulas for many-body potentials in molecular dynamics simulations with applications to thermal conductivity calculations. Phys. Rev. B 92, 094301 (2015).
Agrawal, A. & Choudhary, A. Perspective: Materials informatics and big data: realization of the “fourth paradigm” of science in materials science. APL Mater. 4, 053208 (2016).
Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O. & Walsh, A. Machine learning for molecular and materials science. Nature 559, 547–555 (2018).
Wang, T., Zhang, C., Snoussi, H. & Zhang, G. Machine learning approaches for thermoelectric materials research. Adv. Funct. Mater. 30, 1906041 (2020).
Ryu, B., Wang, L., Pu, H., Chan, M. K. Y. & Chen, J. Understanding, discovery, and synthesis of 2D materials enabled by machine learning. Chem. Soc. Rev. 51, 1899–1925 (2022).
Massot, M. et al. Critical behavior of CoO and NiO from specific heat, thermal conductivity, and thermal diffusivity measurements. Phys. Rev. B 77, 134438 (2008).
Toberer, E. S., Zevalkink, A., Crisosto, N. & Snyder, G. J. The Zintl compound Ca5Al2Sb6 for low-cost thermoelectric power generation. Adv. Funct. Mater. 20, 4375–4380 (2010).
Toher, C. et al. High-throughput computational screening of thermal conductivity, Debye temperature, and Grüneisen parameter using a quasiharmonic Debye model. Phys. Rev. B 90, 174107 (2014).
Lindsay, L., Broido, D. A. & Reinecke, T. L. Ab initio thermal transport in compound semiconductors. Phys. Rev. B 87, 165201 (2013).
Qian, X., Zhou, J. & Chen, G. Phonon-engineered extreme thermal conductivity materials. Nat. Mater. 20, 1188–1202 (2021).
Juneja, R., Yumnam, G., Satsangi, S. & Singh, A. K. Coupling the high-throughput property map to machine learning for predicting lattice thermal conductivity. Chem. Mater. 31, 5145–5151 (2019).
Jaafreh, R., Kang, Y. S. & Hamad, K. Lattice thermal conductivity: an accelerated discovery guided by machine learning. ACS Appl. Mater. Interfaces 13, 57204–57213 (2021).
Arabha, S., Aghbolagh, Z. S., Ghorbani, K., Hatam-Lee, S. M. & Rajabpour, A. Recent advances in lattice thermal conductivity calculation using machine-learning interatomic potentials. J. Appl. Phys. 130, 210903 (2021).
Caro, M. A. Optimizing many-body atomic descriptors for enhanced computational performance of machine learning based interatomic potentials. Phys. Rev. B 100, 024112 (2019).
Gu, X. & Zhao, C. Y. Thermal conductivity of single-layer MoS2(1−x)Se2x alloys from molecular dynamics simulations with a machine-learning-based interatomic potential. Comput. Mater. Sci. 165, 74–81 (2019).
Mortazavi, B. et al. Machine-learning interatomic potentials enable first-principles multiscale modeling of lattice thermal conductivity in graphene/borophene heterostructures. Mater. Horiz. 7, 2359–2367 (2020).
Rodriguez, A., Lam, S. & Hu, M. Thermodynamic and transport properties of LiF and FLiBe molten salts with deep learning potentials. ACS Appl. Mater. Interfaces 13, 55367–55379 (2021).
Wyant, S., Rohskopf, A. & Henry, A. Machine learned interatomic potentials for modeling interfacial heat transport in Ge/GaAs. Comput. Mater. Sci. 200, 110836 (2021).
Yang, H. et al. Dual adaptive sampling and machine learning interatomic potentials for modeling materials with chemical bond hierarchy. Phys. Rev. B 104, 094310 (2021).
Curtarolo, S. et al. AFLOWLIB.ORG: a distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 58, 227–235 (2012).
Blanco, M. A., Francisco, E. & Luaña, V. GIBBS: isothermal-isobaric thermodynamics of solids from energy curves using a quasi-harmonic Debye model. Comput. Phys. Commun. 158, 57–72 (2004).
Slack, G. A. Nonmetallic crystals with high thermal conductivity. J. Phys. Chem. Solids 34, 321–335 (1973).
Tranås, R., Løvvik, O. M., Tomic, O. & Berland, K. Lattice thermal conductivity of half-Heuslers with density functional theory and machine learning: Enhancing predictivity by active sampling with principal component analysis. Comput. Mater. Sci. 202, 110938 (2022).
Seko, A., Hayashi, H., Nakayama, K., Takahashi, A. & Tanaka, I. Representation of compounds for machine-learning prediction of physical properties. Phys. Rev. B 95, 144110 (2017).
Chen, L., Tran, H., Batra, R., Kim, C. & Ramprasad, R. Machine learning models for the lattice thermal conductivity prediction of inorganic materials. Comput. Mater. Sci. 170, 109155 (2019).
Frazier, P. I. In Recent Advances in Optimization and Modeling of Contemporary Problems (eds Ntaimo, L. & Gel, E.) 255−278 (INFORMS, 2018).
Chen, T. & Guestrin, C. In Proc. 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 785−794 (ACM, 2016).
LeCun, Y. et al. The Handbook of Brain Theory and Neural Networks 3361 (MIT press Cambridge, MA, USA 1995).
Orsenigo, C. & Vercellis, C. Kernel ridge regression for out-of-sample mapping in supervised manifold learning. Expert Syst. Appl. 39, 7757–7762 (2012).
Tibshirani, R. Regression shrinkage and selection via the lasso: a retrospective. J. R. Stat. Soc. Ser. B Stat. Methodol. 73, 273–282 (2011).
Ouyang, R. H., Curtarolo, S., Ahmetcik, E., Scheffler, M. & Ghiringhelli, L. M. SISSO: A compressed-sensing method for identifying the best low-dimensional descriptor in an immensity of offered candidates. Phys. Rev. Mater. 2, 083802 (2018).
Dias, S., Sutton, A. J., Ades, A. E. & Welton, N. J. Evidence synthesis for decision making 2: a generalized linear modeling framework for pairwise and network meta-analysis of randomized controlled. Trials Med. Decis. Mak. 33, 607–617 (2013).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Schulz, E., Speekenbrink, M. & Krause, A. A tutorial on Gaussian process regression: modelling, exploring, and exploiting functions. J. Math. Psychol. 85, 1–16 (2018).
Quinlan, J. R. Simplifying decision trees. Int. J. Hum. Comput. Stud. 51, 497–510 (1999).
Carrete, J., Li, W., Mingo, N., Wang, S. & Curtarolo, S. Finding unprecedentedly low-thermal-conductivity half-Heusler semiconductors via high-throughput materials modeling. Phys. Rev. X 4, 011019 (2014).
Seko, A. et al. Prediction of low-thermal-conductivity compounds with first-principles anharmonic lattice-dynamics calculations and Bayesian optimization. Phys. Rev. Lett. 115, 205901 (2015).
Wan, X. et al. Materials discovery and properties prediction in thermal transport via materials informatics: a mini review. Nano Lett. 19, 3387–3395 (2019).
Yang, L. et al. Investigation of mechanical and thermal properties of rare earth pyrochlore oxides by first-principles calculations. J. Am. Ceram. Soc. 102, 2830–2840 (2019).
Wang, X., Zeng, S., Wang, Z. & Ni, J. Identification of crystalline materials with ultra-low thermal conductivity based on machine learning study. J. Phys. Chem. C. 124, 8848–8495 (2020).
Chakraborty, P. et al. Quenching thermal transport in aperiodic superlattices: a molecular dynamics and machine learning study. ACS Appl. Mater. Interfaces 12, 8795–8804 (2020).
Liu, J. et al. A high-throughput descriptor for prediction of lattice thermal conductivity of half-Heusler compounds. J. Phys. D: Appl. Phys. 53, 315301 (2020).
Wan, J., Jiang, J.-W. & Park, H. S. Machine learning-based design of porous graphene with low thermal conductivity. Carbon 157, 262–269 (2020).
Juneja, R. & Singh, A. K. Guided patchwork kriging to develop highly transferable thermal conductivity prediction models. J. Phys. Mater. 3, 024006 (2020).
Juneja, R. & Singh, A. K. Unraveling the role of bonding chemistry in connecting electronic and thermal transport by machine learning. J. Mater. Chem. A 8, 8716–8721 (2020).
Du, Y., Ying, P. & Zhang, J. Prediction and optimization of the thermal transport in hybrid carbon-boron nitride honeycombs using machine learning. Carbon 184, 492–503 (2021).
Zhu, Y. et al. Charting lattice thermal conductivity for inorganic crystals and discovering rare earth chalcogenides for thermoelectrics. Energy Environ. Sci. 14, 3559–3566 (2021).
Loftis, C., Yuan, K., Zhao, Y., Hu, M. & Hu, J. Lattice thermal conductivity prediction using symbolic regression and machine learning. J. Phys. Chem. A 125, 435–450 (2021).
Ju, S. et al. Exploring diamondlike lattice thermal conductivity crystals via feature-based transfer learning. Phys. Rev. Mater. 5, 053801 (2021).
Miyazaki, H. et al. Machine learning based prediction of lattice thermal conductivity for half-Heusler compounds using atomic information. Sci. Rep. 11, 13410 (2021).
Hong, Y., Han, D., Hou, B., Wang, X. & Zhang, J. High-throughput computations of cross-plane thermal conductivity in multilayer stanene. Int. J. Heat. Mass Transf. 171, 121073 (2021).
Torres, P. et al. Descriptors of intrinsic hydrodynamic thermal transport: screening a phonon database in a machine learning approach. J. Phys: Condens. Matter 34, 135702 (2022).
Chowdhury, P. R. & Ruan, X. Unexpected thermal conductivity enhancement in aperiodic superlattices discovered using active machine learning. npj Comput. Mater. 8, 12 (2022).
Belsky, A., Hellenbrandt, M., Karen, V. L. & Luksch, P. New developments in the inorganic crystal structure database (ICSD): accessibility in support of materials research and design. Acta Crystallogr. Sect. B. Struct. Sci. 58, 364–369 (2002).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Gorai, P. et al. TE design lab: a virtual laboratory for thermoelectric material design. Comput. Mater. Sci. 112, 368–376 (2016).
Yan, J. et al. Material descriptors for predicting thermoelectric performance. Energy Environ. Sci. 8, 983–994 (2015).
Chen, J. et al. Emerging theory and phenomena in thermal conduction: a selective review. Sci. China-Phys. Mech. Astron. 65, 117002 (2022).
Ali et al. The structure of atomic and molecular clusters, optimised using classical potentials. Comput. Phys. Commun. 175, 451–464 (2006).
Behler, J. Perspective: Machine learning potentials for atomistic simulations. J. Chem. Phys. 145, 170901 (2016).
Lee, Y. & Hwang, G. S. Force-matching-based parameterization of the Stillinger-Weber potential for thermal conduction in silicon. Phys. Rev. B 85, 125204 (2012).
Barry, M. C., Wise, K. E., Kalidindi, S. R. & Kumar, S. Voxelized atomic structure potentials: predicting atomic forces with the accuracy of quantum mechanics using convolutional neural networks. J. Phys. Chem. Lett. 11, 9093–9099 (2020).
Huang, Y., Kang, J., Goddard, W. A. & Wang, L.-W. Density functional theory based neural network force fields from energy decompositions. Phys. Rev. B 99, 064103 (2019).
Korotaev, P., Novoselov, I., Yanilkin, A. & Shapeev, A. Accessing thermal conductivity of complex compounds by machine learning interatomic potentials. Phys. Rev. B 100, 144308 (2019).
Pozdnyakov, S. N. et al. Incompleteness of atomic structure representations. Phys. Rev. Lett. 125, 166001 (2020).
Shapeev, A. V. Moment tensor potentials: a class of systematically improvable interatomic potentials. Multiscale Model. Simul. 14, 1153 (2016).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 134, 074106 (2011).
Musil, F. et al. Physics-inspired structural representations for molecules and materials. Chem. Rev. 121, 9759–9815 (2021).
Behler, J. Representing potential energy surfaces by high-dimensional neural network potentials. J. Phys: Condens. Matter 26, 183001 (2014).
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, C. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010).
Zuo, Y. et al. Performance and cost assessment of machine learning interatomic potentials. J. Phys. Chem. A 124, 731–745 (2020).
Ghosal, S., Chowdhury, S. & Jana, D. Impressive thermoelectric figure of merit in two-dimensional tetragonal pnictogens: a combined first-principles and machine-learning approach. ACS Appl. Mater. Interfaces 13, 59092–59103 (2021).
Mortazavi, B., Novikov, I. S. & Shapeev, A. V. A machine-learning-based investigation on the mechanical/failure response and thermal conductivity of semiconducting BC2N monolayers. Carbon 188, 431–441 (2022).
Mortazavi, B., Zhuang, X., Rabczuk, T. & Shapeev, A. V. Outstanding thermal conductivity and mechanical properties in the direct gap semiconducting penta-NiN2 monolayer confirmed by first-principles. Phys. E 140, 115221 (2022).
Mohebpour, M. A. et al. Mechanical, optical, and thermoelectric properties of semiconducting ZnIn2X4 (X = S, Se, Te) monolayers. Phys. Rev. B 105, 134108 (2022).
Raeisi, M. et al. High thermal conductivity in semiconducting Janus and non-Janus diamanes. Carbon 167, 51–61 (2020).
Mortazavi, B. et al. Efficient machine-learning based interatomic potentials for exploring thermal conductivity in two-dimensional materials. J. Phys. Mater. 3, 02LT02 (2020).
Arabha, S. & Rajabpour, A. Thermo-mechanical properties of nitrogenated holey graphene (C2N): a comparison of machine-learning-based and classical interatomic potentials. Int. J. Heat. Mass Transf. 178, 121589 (2021).
Mortazavi, B. et al. A first-principles and machine-learning investigation on the electronic, photocatalytic, mechanical and heat conduction properties of nanoporous C5N monolayers. Nanoscale 14, 4324–4333 (2022).
Ghosal, S., Chowdhury, S. & Jana, D. Electronic and thermal transport in novel carbon-based bilayer with tetragonal rings: a combined study using first-principles and machine learning approach. Phys. Chem. Chem. Phys. 23, 14608–14616 (2021).
Wang, Q., Zeng, Z. & Chen, Y. Revisiting phonon transport in perovskite SrTiO3: anharmonic phonon renormalization and four-phonon scattering. Phys. Rev. B 104, 235205 (2021).
Korotaev, P. & Shapeev, A. Lattice dynamics of YbxCo4Sb12 skutterudite by machine-learning interatomic potentials: effect of filler concentration and disorder. Phys. Rev. B 102, 184305 (2020).
Marmolejo-Tejada, J. M. & Mosquera, M. A. Thermal properties of single-layer MoS2−WS2 alloys enabled by machine-learned interatomic potentials. Chem. Commun. 58, 6902–6905 (2022).
Liu, Z., Yang, X., Zhang, B. & Li, W. High thermal conductivity of Wurtzite boron arsenide predicted by including four-phonon scattering with machine learning potential. ACS Appl. Mater. Interfaces 13, 53409–53415 (2021).
Ouyang, Y. et al. Accurate description of high-order phonon anharmonicity and lattice thermal conductivity from molecular dynamics simulations with machine learning potential. Phys. Rev. B 105, 115202 (2022).
Liu, H., Qian, X., Bao, H., Zhao, C. Y. & Gu, X. High-temperature phonon transport properties of SnSe from machine-learning interatomic potential. J. Phys: Condens. Matter 33, 405401 (2021).
Ouyang, N., Wang, C. & Chen, Y. Temperature- and pressure-dependent phonon transport properties of SnS across phase transition from machine-learning interatomic potential. Int. J. Heat. Mass Transf. 192, 122859 (2022).
Zeng, Z. et al. Ultralow and glass-like lattice thermal conductivity in crystalline BaAg2Te2: strong fourth-order anharmonicity and crucial diffusive thermal transport. Mater. Today Phys. 21, 100487 (2021).
Novikov, I. S., Gubaev, K., Podryabinkin, E. V. & Shapeev, A. V. The MLIP package: moment tensor potentials with MPI and active learning. Mach. Learn. Sci. Technol. 2, 025002 (2021).
Mortazavi, B. et al. Accelerating first-principles estimation of thermal conductivity by machine-learning interatomic potentials: a MTP/ShengBTE solution. Comput. Phys. Commun. 258, 107583 (2021).
Choi, J. M. et al. Accelerated computation of lattice thermal conductivity using neural network interatomic potentials. Comput. Mater. Sci. 211, 111472 (2022).
Takeshita, Y., Shimamura, K., Fukushima, S., Koura, A. & Shimojo, F. Thermal conductivity calculation based on Green−Kubo formula using ANN potential for β-Ag2Se. J. Phys. Chem. Solids 163, 110580 (2022).
Watanabe, S. et al. High-dimensional neural network atomic potentials for examining energy materials: some recent simulations. J. Phys. Energy 3, 012003 (2021).
Li, R. et al. A deep neural network interatomic potential for studying thermal conductivity of β-Ga2O3. Appl. Phys. Lett. 117, 152102 (2020).
Mirhosseini, H., Tahmasbi, H., Kuchana, S. R., Ghasemi, A. & Kühne, T. D. An automated approach for developing neural network interatomic potentials with FLAME. Comput. Mater. Sci. 197, 110567 (2021).
Han, L. et al. Neural network potential for studying the thermal conductivity of Sn. Comput. Mater. Sci. 200, 110829 (2021).
Li, R., Lee, E. & Luo, T. A unified deep neural network potential capable of predicting thermal conductivity of silicon in different phases. Mater. Today Phys. 12, 100181 (2020).
Faraji, S., Allaei, S. M. V. & Amsler, M. Thermal conductivity of CaF2 at high pressure. Phys. Rev. B 103, 134301 (2021).
Mangold, C. et al. Transferability of neural network potentials for varying stoichiometry: phonons and thermal conductivity of MnxGey compounds. J. Appl. Phys. 127, 244901 (2020).
Fan, Z. et al. Neuroevolution machine learning potentials: combining high accuracy and low cost in atomistic simulations and application to heat transport. Phys. Rev. B 104, 104309 (2021).
Tahmasbi, H., Goedecker, S. & Ghasemi, S. A. Large-scale structure prediction of near-stoichiometric magnesium oxide based on a machine-learned interatomic potential: Crystalline phases and oxygen-vacancy ordering. Phys. Rev. Mater. 5, 083806 (2021).
Dai, F.-Z., Sun, Y., Wen, B., Xiang, H. & Zhou, Y. Temperature dependent thermal and elastic properties of high entropy (Ti0.2Zr0.2Hf0.2Nb0.2Ta0.2)B2: molecular dynamics simulation by deep learning potential. J. Mater. Sci. Tech. 72, 8–15 (2021).
Dai, F.-Z., Wen, B., Sun, Y., Xiang, H. & Zhou, Y. Theoretical prediction on thermal and mechanical properties of high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C by deep learning potential. J. Mater. Sci. Tech. 43, 168–174 (2020).
Pan, G., Ding, J., Du, Y., Lee, D.-J. & Lu, Y. A DFT accurate machine learning description of molten ZnCl2 and its mixtures: 2. Potential development and properties prediction of ZnCl2-NaCl-KCl ternary salt for CSP. Comput. Mater. Sci. 187, 110055 (2021).
Bosoni, E. et al. Atomistic simulations of thermal conductivity in GeTe nanowires. J. Phys. D: Appl. Phys. 53, 054001 (2020).
Sun, J. et al. Four-phonon scattering effect and two-channel thermal transport in two-dimensional paraelectric SnSe. ACS Appl. Mater. Interfaces 14, 11493–11499 (2022).
Pegolo, P., Baroni, S. & Grasselli, F. Temperature- and vacancy-concentration-dependence of heat transport in Li3ClO from multi-method numerical simulations. npj Comput. Mater. 8, 24 (2022).
Liu, Y.-B. et al. Machine learning interatomic potential developed for molecular simulations on thermal properties of β-Ga2O3. J. Chem. Phys. 153, 144501 (2020).
Verdi, C., Karsai, F., Liu, P., Jinnouchi, R. & Kresse, G. Thermal transport and phase transitions of zirconia by on-the-fly machine-learned interatomic potentials. npj Comput. Mater. 7, 156 (2021).
Zeng, Z. et al. Nonperturbative phonon scatterings and the two-channel thermal transport in Tl3VSe4. Phys. Rev. B 103, 224307 (2021).
Babaei, H., Guo, R., Hashemi, A. & Lee, S. Machine-learning-based interatomic potential for phonon transport in perfect crystalline Si and crystalline Si with vacancies. Phys. Rev. Mater. 3, 074603 (2019).
Zhang, Y., Shen, C., Long, T. & Zhang, H. Thermal conductivity of h-BN monolayers using machine learning interatomic potential. J. Phys: Condens. Matter 33, 105903 (2021).
Zhang, C. & Sun, Q. Gaussian approximation potential for studying the thermal conductivity of silicene. J. Appl. Phys. 126, 105103 (2019).
Qian, X., Peng, S., Li, X., Wei, Y. & Yang, R. Thermal conductivity modeling using machine learning potentials: application to crystalline and amorphous silicon. Mater. Today Phys. 10, 100140 (2019).
Chen, H., Xiang, H., Dai, F.-Z., Liu, J. & Zhou, Y. Porous high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)B2: a novel strategy towards making ultrahigh temperature ceramics thermal insulating. J. Mater. Sci. Tech. 35, 2404–2408 (2019).
Legrain, F. Vibrational properties of metastable polymorph structures by machine learning. J. Chem. Inf. Model. 58, 2460–2466 (2018).
Eriksson, F., Fransson, E. & Erhart, P. The Hiphive package for the extraction of high-order force constants by machine learning. Adv. Theory Simul. 2, 1800184 (2019).
Chan, H. et al. Machine learning a bond order potential model to study thermal transport in WSe2 nanostructures. Nanoscale 11, 10381–10392 (2019).
Zhang, Y., Lunghi, A. & Sanvito, S. Pushing the limits of atomistic simulations towards ultra-high temperature: a machine-learning force field for ZrB2. Acta Mater. 186, 467–474 (2020).
Rodriguez, A., Liu, Y. & Hu, M. Spatial density neural network force fields with first-principles level accuracy and application to thermal transport. Phys. Rev. B 102, 035203 (2020).
Plata, J. J., Posligua, V., Márquez, A. M., Sanz, J. F. & Grau-Crespo, R. Charting the Lattice thermal conductivities of I−III−VI2 chalcopyrite semiconductors. Chem. Mater. 34, 2833–2841 (2022).
Blancas, E. J. et al. Unraveling the role of chemical composition in the lattice thermal conductivity of oxychalcogenides as thermoelectric materials. J. Mater. Chem. A 10, 19941–19952 (2022).
Haastrup, S. et al. The Computational 2D Materials Database: high-throughput modeling and discovery of atomically thin crystals. 2D Matter 5, 042002 (2018).
Gjerding, M. N. et al. Recent progress of the Computational 2D Materials Database (C2DB). 2D Mater. 8, 044002 (2021).
Mounet, N. et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 13, 246–252 (2018).
Zhou, J. et al. 2DMatpedia, an open computational database of two-dimensional materials from top-down and bottom-up approaches. Sci. Data 6, 86 (2019).
Acknowledgements
We thank financial support from the National Natural Science Foundation of China (Grant No. 62074114).
Author information
Authors and Affiliations
Contributions
H.J.L. and Y.F. conceived and initiated the study. Y.F.L., M.K.L. and H.M.Y. discussed the results. Y.F.L. and H.J.L. wrote the manuscript. All authors reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, Y., Li, M., Yuan, H. et al. Predicting lattice thermal conductivity via machine learning: a mini review. npj Comput Mater 9, 4 (2023). https://doi.org/10.1038/s41524-023-00964-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-023-00964-2
This article is cited by
-
A Deep Neural Network Potential to Study the Thermal Conductivity of MnBi2Te4 and Bi2Te3/MnBi2Te4 Superlattice
Journal of Electronic Materials (2023)
