
Abstract
Experimental evolution experiments in which bacterial populations are repeatedly exposed to an antimicrobial treatment, and examination of the genotype and phenotype of the resulting evolved bacteria, can help shed light on mechanisms behind reduced susceptibility. In this review we present an overview of why it is important to include biofilms in experimental evolution, which approaches are available to study experimental evolution in biofilms and what experimental evolution has taught us about tolerance and resistance in biofilms. Finally, we present an emerging consensus view on biofilm antimicrobial susceptibility supported by data obtained during experimental evolution studies.
Similar content being viewed by others

Introduction
Experimental evolution (Box 1) is the study of evolutionary processes occurring in populations in response to conditions imposed and controlled by the experimenter1. While the first microbial experimental evolution studies date back to the 1880's2, experimental evolution was introduced to bacteriology in the 1950's by Francis J. Ryan3 and became well-known due the long-term evolution experiment (LTEE) that was started by Richard Lenski in the 1980's and has been running for >75000 generations4,5. The LTEE and many other experimental evolution experiments are carried out in unstructured environments, i.e., in liquid culture with shaking, with most bacteria in a planktonic state. However, already in early evolution experiments in structured environments, marked differences in terms of evolution of fitness (Box 1) and within-population variability were observed compared to what is typically observed in planktonic cultures6,7.
Biofilms are structured microenvironments—why does that matter during evolution?
Biofilms are structured microbial communities that are either attached to a surface or occur as suspended or embedded aggregates8. Various gradients (oxygen, nutrients, antimicrobial agents, …) are present in biofilms, resulting in the development of spatially structured niches with distinct environmental conditions9 and these microenvironments co-determine the outcome of biofilm-related infections, as they directly impact on bacterial growth and metabolism, as well as on the effect of antimicrobial treatment10,11,12,13.
Experimental evolution in general14 and specific aspects of experimental evolution in biofilms15 were recently reviewed; we refer readers to these reviews for more details. A brief summary of why biofilm populations become more diverse during evolution is presented below.
Due to their heterogeneity, biofilms contain multiple ecological niches, not all of which are used by existing genotypes; these unused niches present opportunities for novel genotypes16. Moreover, novel genotypes can create additional niches by altering the surrounding environment (‘niche construction’)7,17. Due to the spatial heterogeneity, biofilm populations can be considered as collections of independently evolving subpopulations and this population fragmentation reduces the effective population size. As the relative contribution of genetic drift (Box 1) towards diversity is higher in smaller subpopulations, spatial heterogeneity ultimately leads to more diversity16,18. Population fragmentation also allows fixation (Box 1) of beneficial mutations with a relatively small effect in particular subpopulations. Indeed, beneficial mutations that have a large effect are less frequent than beneficial mutations that have a small effect and the former are unlikely to appear in all subpopulations; as a consequence different beneficial mutations with a small effect are expected to occur and be maintained in different spatially separated subpopulations, leading to more diversity within the population as a whole19. Recent experimental work and modeling showed that in a spatially structured environment the spread of a beneficial mutation is amplified and that beneficial mutations are less likely to be lost20. The reason for this is that in structured environments selection can increase the frequency of a beneficial mutation in a certain subpopulation faster than the migration of this mutation to other subpopulations; as a consequence, the mutant harboring this beneficial mutation is likely to be able to migrate to novel subpopulations repeatedly, which ultimately reduces the likelihood of loss of this mutation due to genetic drift20. The competition between mutants harboring different beneficial mutations (clonal interference, Box 1) increases fixation times (i.e., it will take longer before a particular mutation outcompetes all others) and clonal interference is more frequent in spatially structured environments (as beneficial mutations show a slow, ‘wave-like’ spread throughout the population)21. As a consequence multiple beneficial mutations can co-occur in biofilms, again with a higher diversity as result22,23. The recent observation that in vitro evolution of Pseudomonas aeruginosa in conditions that are most similar to those encountered in the lung of cystic fibrosis (CF) patients (i.e., in a synthetic CF medium [SCFM]) leads to lower parallelism (i.e., more diversity) than evolution in a minimal medium, confirms the importance of the presence of spatially separated subpopulations for generating diversity24,25. In contrast to the minimal medium, SCFM contains mucin, which allows the creation of spatially structured subpopulations with smaller effective population sizes, making it less likely to find the same beneficial mutations in replicate populations25.
Finally, in homogeneous populations exposed to an antimicrobial agent, all mutations required for full resistance need to be acquired at the same time in order to avoid eradication by the uniform high concentrations of the antibiotic. However, penetration of antimicrobial agents into the biofilm can be hindered, leading to concentration gradients9,26,27,28,29 that can create ‘sanctuaries’, i.e., parts of the biofilm in which concentrations of antimicrobial agents are lower and that can act as ‘stepping stones’ allowing populations to acquire mutations one by one30. Other important aspects to consider are the increased mutation rates often observed in biofilm cells, as well as the increased rate of horizontal gene transfer (HGT) in bacterial biofilms (discussed in more detail in Box 2). It should be noted that as most experimental evolution studies are carried out with single species (see below), HGT is usually not a factor driving evolutionary changes in these studies.
Recent insights into development of reduced antimicrobial susceptibility from experimental evolution with planktonic populations
Antimicrobial resistance
Antimicrobial resistance is quantified by the minimal inhibitory concentration (MIC, Box 1)31. The ability of resistant organisms to grow at concentrations above the MIC for susceptible organisms is linked to the presence of one or more resistance mechanisms32 and evolutionary trajectories towards a resistant phenotype can be complex33. Experimental evolution in which cultures are serially passaged (in the presence of a constant or gradually increasing concentration of an antibiotic), combined with whole-genome sequencing (WGS) can be used to identify resistance of and trajectories towards resistance34. For example, serial passaging of Escherichia coli in the presence of carbapenems allowed identification of several previously unknown carbapenem resistance mechanisms, including mutations in mrdA (coding for PBP2) and ftsI (coding for PBP3), both targets of carbapenems, as well as mutations in acrB (coding for the inner membrane associated part of the AcrAB-TolC efflux pump)35. Experimentally evolving E. coli in the presence of chloramphenicol induced mutations in the DNA binding region of marR, which can upregulate the AcrAB-TolC efflux pump, as well as mutations in acrB and acrR (interruption of acrR leads to upregulation of acrAB)36. In Streptococcus pneumoniae, experimental evolution in the presence of increasing concentrations of moxifloxacin and levofloxacin led to the identification of novel mutations in gyrB, that in combination with mutations in gyrA and parC lead to high-level fluoroquinolone resistance37. In an experimental evolution study with P. aeruginosa, both expected (e.g., mutations leading to AmpC overproduction after evolution in the presence of ceftazidime, mutations in oprD leading to inactivation of the porin after evolution in the presence of meropenem) and novel (e.g., gain-of-function mutations leading to the structural modification of AmpC after evolution in the presence of ceftazidime, novel mutations in gyrA after evolution in the presence of ciprofloxacin) resistance mechanisms were identified38. There is a growing body of evidence that metabolic adaptations and reduced antimicrobial susceptibility go hand in hand39,40, and several experimental evolution studies with planktonic E. coli populations have recently confirmed this. When E. coli is grown in a minimal medium with glucose (supporting rapid growth with respiration or fermentation) or acetate (supporting slower growth with respiration only), resistance develops much faster on glucose, confirming that environmental conditions constrain the rate of resistance development41. Most changes observed involve metabolic processes that are not directly affected by the antibiotic treatment. For example, cultures evolved in the presence of glucose and chloramphenicol consume more glucose, secrete more acetate and show reduced oxygen uptake compared to wild type E. coli and E. coli adapted in the presence of glucose only, indicating a metabolic switch from respiration to fermentation. This switch is linked to overexpression of the AcrAB efflux pump (required for chloramphenicol resistance) and membrane proteome remodeling, due to competition for space between efflux pump and proteins involved in oxidative phosphorylation41. Further evidence for the role of metabolic changes in the development of antimicrobial resistance comes from experimental evolution of planktonic E. coli using both a conventional experimental evolution protocol and a ‘metabolic evolution protocol’ designed to ensure equivalent selection dynamics for all conditions, by exposing bacteria to antibiotics at different temperatures (i.e., at increasingly heightened metabolic states)42. Evolution under the conventional settings leads to more slowly growing populations with an increased MIC; mutations frequently found in these populations are in genes linked to known resistance mechanisms. However, a subset of clones acquires mutations in other genes, including genes related to central metabolism (TCA cycle, electron transport). Populations obtained at the end of the ‘metabolic evolution’ experiment exhibit increased survival in killing assays compared to the ancestral wild type strain, without reduction in exponential growth rate or increase in lag time (ruling out tolerance [Box 1] due to slow growth). Engineering mutants in six metabolic genes further confirmed the relevance of these mutations as in all mutants the MIC to at least one antibiotic was increased. The mechanism by which these mutations provide resistance vary, but for at least one of them (sucA, encoding the TCA cycle enzyme 2-oxoglutarate decarboxylase) the mutation provides resistance by lowering basal respiration and thereby preventing antibiotic-mediated induction of TCA cycle activity, a mechanism previously observed in different organisms43,44,45. 39% of coding sequence mutations identified in these evolution experiments can also be found in sequenced E. coli genomes; moreover, several mutations in metabolic genes are abundantly present in these genomes and some are statistically enriched in clinical E. coli isolates, suggesting they are relevant in vivo42.
Antimicrobial tolerance and persistence
Reduced susceptibility to antibiotics is not only due to resistance, as also tolerance and persistence (Box 1) play important roles31,32,46,47,48. Tolerant cells survive exposure to antibiotics without carrying conventional resistance mechanisms and will resume growth after removal of the antibiotic32. The factors that lead to tolerance can be genetic (e.g., mutations leading to increased lag time49,50) or environmental (e.g., production of a protective biofilm matrix51,52, slow growth due to microenvironmental conditions53,54). Tolerance and persistence can both be quantified by the minimum duration for killing (MDK, Box 1); in addition, persistence is typically characterized by the presence of a biphasic killing curve31,32. Cyclic exposure of planktonic E. coli cultures to ampicillin led to an increase of the MDK and this increase was due to an extended single-cell lag time; no changes in MIC were observed, ruling out resistance49. When planktonic populations of various ESKAPE pathogens were cycled between exposure to aminoglycosides and regrowth, a 37 to 213-fold increase in number of persister cells was observed upon treatment of evolved clones compared to the start culture, again without an increase in MIC55. WGS of evolved high-persistence clones showed that this phenotype could be attributed to a single mutation in either oppB, gadC or nuoN, genes not previously implicated in persistence56.
Tolerance and persistence as ‘stepping stones’ towards resistance
Experimental evolution has shown that the development of tolerance and persistence can be ‘stepping stones’ towards the development of resistance. When planktonic E. coli cultures were evolved in the presence of ampicillin, mutations in the promotor region of ampC, encoding a β-lactamase, increased the MIC after 7–17 cycles, while delayed growth was already observed after 3–4 cycles, i.e., development of tolerance preceded that of resistance57. WGS of the first resistant clones showed that all carried additional mutations of which some had previously been identified as increasing tolerance by increasing the lag time49; additional sequencing revealed that the same tolerance mutations had been present prior to the appearance of the ampC resistance mutations. As mutations in several genes can lead to tolerance, the target size for mutations leading to tolerance is larger than that for resistance (ampC being the only target); as a consequence tolerance mutations occur more frequently and can be detected earlier. Starting evolution experiments from wild type strains and from strains that had already developed tolerance demonstrated that resistance mutations established faster in tolerant clones The survival advantage conferred by resistance mutations upon exposure to high ampicillin concentrations is comparable to that of the tolerance mutations (as ampC resistance mutations only result in partial resistance). As a result, tolerance mutations start dominating the population after a few cycles and the presence of these mutations reduces the probability of loss of resistance mutations during antibiotic treatment57. Similar observations were made for P. aeruginosa: upon sequential exposure, P. aeruginosa rapidly adapts to high concentrations of tobramycin with a stepwise increase in survival rate and after 7-8 cycles all evolved lineages had reached MICs substantially higher than the ancestral strain58. WGS showed that alleles occurred and reached fixation in a specific order, with mutations in genes involved in respiration and energy metabolism (leading to tolerance) typically preceding the acquisition of resistance mutations, and periodically exposing P. aeruginosa wild type, and mutants with various levels of tolerance, to tobramycin confirmed that the rates of resistance acquisition were similar in all groups but that tolerant lineages were more likely to survive the initial selection. This suggests that bacterial populations with high tolerance have a better chance to develop resistance than populations with low or no tolerance58. Finally, several studies have pointed towards a link between persistence and the likelihood of developing resistance, e.g., in Mycobacterium tuberculosis59, Pseudomonas spp.60 and E. coli61.
Tools to study experimental evolution in biofilms
For a detailed overview of available biofilm methods, we refer to recent reviews62,63,64,65. Importantly, while the general set-up in most evolution experiments is similar (with repeating cycles of growth, treatment and transfer to a new environment) (Fig. 1a), the model used can profoundly impact the outcome of the experiment and not every in vitro model will mimic evolution in vivo, e.g., many models use surfaces and growth media that are poorly reflective of the in vivo conditions15.

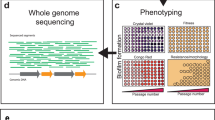
a Schematic overview of the general set-up of experimental evolution experiments involving antimicrobial treatment of biofilms. b P. aeruginosa readily forms aggregates in SCFM2, making this a suitable growth medium to study evolution in a relevant microenvironment. c Based on whole-genome sequence analysis (mutations occurring in P. aeruginosa PAO1 after repeated exposure to furanone C-30 shown as example89), frequency of mutations can be calculated and effect of mutations on protein function (fusA1 shown as example89) can be estimated. d Phenotypic characterization typically starts with determining antimicrobial susceptibility (illustrated here with disk diffusion) and the number of CFU (number of CFU in three replicate B. cenocepacia populations after repeated cycles of exposure to tobramycin are shown as example70). Experimental evolution in biofilms frequently leads to the occurrence of small colony variants (SCV) (P. aeruginosa AA2 shown as example, picture courtesy of Dr. A. Sass). Finally, changes in metabolism occur during evolution and can be measured using for example microcalorimetry; metabolic activity after treatment of WT P. aeruginosa PAO1 (left) or the same strain evolved in the presence of tobramycin (right) is shown as example.
Static and dynamic model systems
In static systems, biofilms will be grown and treated, and subsequently cells will be collected to initiate a new cycle. In dynamic systems a biofilm is continuously grown and treated, without being disrupted.
Biofilms formed on plastic or glass beads are frequently used to study evolution17,66,67,68,69,70,71,72. The bead model was originally developed for the selection for daily adherence to, and dispersal from, a bead by Burkholderia cenocepacia22. In this set-up, a bead is incubated together with bacteria that will attach to the bead to form a biofilm. The bead that contains the biofilm is then transferred to another recipient tube containing a new empty bead and fresh medium, which will allow colonization of the new bead, without disturbing the biofilm. Variants of this model that are more tailored towards studying the responses to antimicrobial treatment have also been developed70,73. Despite the fact that it only allows studying surface-attached biofilms, the ease of use and the compatibility with different organisms and growth media make this an attractive model.
Colony biofilms can be formed on membrane filters that are inoculated with the test organism and are subsequently placed on a suitable growth medium74,75. Nutrients will diffuse through the membrane, and the bacteria will form a biofilm on the filter membrane. During the experiment the filter can be moved to another agar plate and this way biofilms can easily be exposed to antimicrobial agents. At the end of a cycle, the bacterial cells are detached from the membrane and the resulting suspension can be used to inoculate a new filter membrane.
The Calgary biofilm device consists of a 96-well plate and a lid with pegs, which are each submerged in a well and support biofilm formation; this device was originally developed to determine the minimal biofilm eradication concentration76. During experimental evolution, the lid with pegs can easily be transferred to a new 96-well plate and biofilms can be dispersed from the pegs via sonication. The resulting cell suspensions can then be used to start biofilm formation on pegs on a new lid77. A conceptually-similar system (FlexiPeg) was recently developed and used to study competition and fitness in biofilms78,79.
In dynamic model systems biofilms are grown on a surface while nutrient and waste products are continuously added and removed, respectively. While technically more demanding, an advantage of these systems is that the biofilm does not have to be dispersed in between different treatment cycles. Examples include acrylic flow cells with a glass surface80, Sartorius bioreactors81,82,83 and various microfluidic devices84,85,86.
While many studies make use of standard growth media (e.g., LB broth), it is possible to more closely mimic the in vivo environment by using validated in vivo-like media. This includes various artificial CF sputum media87 in which suspended bacterial biofilm aggregates rapidly form (Fig. 1b) and which were used to study development of ciprofloxacin resistance88 as well as resistance to the combination tobramycin/furanone C-3089 in P. aeruginosa biofilms.
Selective pressure during experimental evolution
The choice of the appropriate selective pressure is an important decision in evolution experiments and can profoundly affect the outcome of the experiment.
When studying mechanisms of adaptation, the antibiotic concentration should be sufficiently high to have an effect on the bacteria, but cannot be too high in order to allow survival of a sufficient number of bacteria to initiate a next cycle of the experiment. There is no information on which concentration range constitutes the mutant selection window (MSW, Box 1) for biofilms and various aspects of biofilm biology likely affect this window90,91. While it has been predicted that biofilm growth leads to shifts and distortions of the MSW91, recent work with E. coli showed that minimal selective concentration values (for five different antibiotics) did not differ between planktonic cultures and biofilms79. Due to the uncertainty concerning the biofilm MSW, antibiotic concentrations are often selected based on the MIC70 or on the minimal biofilm inhibitory concentration74. An alternative strategy is using antimicrobial concentrations that are achievable in vivo, e.g., in sputum from CF patients after inhalation therapy75. Selection strength can profoundly influence evolutionary trajectories, e.g., sublethal concentrations of tigecycline select for P. aeruginosa mutants with lower tigecycline MICs and higher MICs to other antibiotics than mutants selected under lethal concentrations92. In general, in vitro evolution in the presence of a mild selective pressure leads to a more diverse population, while exposure to a high selective pressure eliminates bacteria with intermediate susceptibility, and will only result in the detection of those mutations that have the strongest effect93. The concentration of an antibiotic at the site of infection depends on the mode of administration, and while high concentrations may be achievable with inhalation therapy or topical application, the antibiotic concentration at the infection site will often be substantially lower when antibiotics are systemically administered94,95. In addition, biofilms can be considered as independent pharmacological microcompartments96,97 and diffusion limitations often lead to formation of gradients of antibiotic concentrations in a biofilm26,27,28,29,98.
Evolutionary trajectories will differ between antibiotics belonging to different classes, e.g., when P. aeruginosa was evolved in the presence of sublethal tobramycin or tigecycline concentrations, mutants were selected at sublethal concentrations of tigecycline only92. While these trajectories will depend on the mode of action of the antimicrobial agents, different classes of bactericidal antibiotics also have common aspects, including that they mostly inhibit biosynthesis of macromolecules (DNA, proteins, peptidoglycan) and induce changes in metabolism that promote the formation of reactive oxygen species99,100. In addition, the activity of some antibiotics strongly depends on microbial metabolism while other antibiotics only weakly depend on metabolism for their killing activity101; tolerance will quickly develop towards the former group of antibiotics, but not the latter102.
Finally, the treatment regime can have an influence on the evolutionary trajectory that is followed. The concentration of the antimicrobial agent can be kept constant during the course of the evolution experiment70,75 or bacteria can be exposed to gradually increasing antimicrobial concentrations82,85, and exposure can be continuous69,74,75,82,85 or intermittent70,77,103,104. Regrowth of the biofilm after each treatment cycle ensures that biofilms with similar cell densities are studied throughout the experiment, and the regrowth phase can mimic the decrease of antibiotic concentration in between two treatments. In addition, continuous exposure may impose growth-dependent selection which can be avoided by separating treatments by rounds of antibiotic-free growth42.
Studying evolution in multispecies communities and in vivo
Experimental evolution of antimicrobial susceptibility has not yet extensively been studied in more complex settings although several studies demonstrate that evolution experiments with polymicrobial biofilms are feasible; examples include a dual species biofilm (Acinetobacter sp. + Pseudomonas putida) evolved on benzyl alcohol105, evolution of P. aeruginosa in the presence of Staphylococcus aureus106 or members of the CF microbiome107, and a 34-species model bacterial community repeatedly exposed to streptomycin108. Examples of in vivo studies include serial propagation of S. pneumoniae by repeated murine nasal colonization109 and adaptation of Shewanella oneidensis to life in the intestines of larval zebrafish110. Recently a Caenorhabditis elegans infection model was used to show that repeated exposure of B. cenocepacia to anti-virulence compound FR900098, an inhibitor of the non-mevalonate pathway, did not lead to changes in susceptibility to this compound111.
Interstrain variability and selection of isolates
When selecting isolates for experimental evolution studies and when analyzing the results, interstrain variability should be taken into account. For example, based on in vitro biofilm morphology and transcriptional profiles, clinical P. aeruginosa isolates can be grouped in different clusters and strains in different clusters share only a restricted core biofilm transcriptional profile; these differences appear shaped by the genetic background of the individual strains rather than the maturation status of the biofilm112. Also tolerance is to a large extent determined by the individual strain background and this strain-dependent tolerance is also antibiotic-dependent, with cross-tolerance of clinical P. aeruginosa isolates observed for ciprofloxacin and tobramycin, but not colistin113. This interstrain variability may have a profound impact on evolutionary trajectories during experimental evolution and will likely complicate the elucidation of the contribution of specific tolerance and resistance mechanisms to reduced susceptibility. At the same time it highlights the versatility of bacterial pathogens to come up with parallel solutions.
What has experimental evolution in the presence of antibiotics taught us about tolerance and resistance in microbial biofilms?
Changes in antimicrobial susceptibility are not only observed when populations are evolved in the presence of an antimicrobial agent, but have also been observed in some experimental evolution studies in which biofilms are evolved in the absence of antibiotics (e.g., in E. coli114 and P. aeruginosa74,115). These changes are likely the result of higher mutation rates in biofilms (Box 2) and, combined with a range of other mechanisms involved in reduced susceptibility12,39, the resulting diversity helps survival of the population (‘insurance hypothesis’)15,116,117,118. In the next section we however focus on experimental evolution studies investigating changes in biofilm antimicrobial susceptibility occurring during exposure to antibiotics.
P. aeruginosa
An non-exhaustive overview of genes mutated in P. aeruginosa biofilms during experimental evolution in the presence of antibiotics is shown in Table 1.
In P. aeruginosa PAO1 colony biofilms formed on polycarbonate membranes, exposure to subinhibitory concentrations of ciprofloxacin rapidly induced reduced susceptibility to this antibiotic74. After 7 passages, the size of the resistant subpopulations was significantly larger in biofilms than in planktonic populations and the mean MIC of ciprofloxacin towards selected colonies derived from ciprofloxacin-evolved biofilms increased significantly during the experimental evolution; the latter was not observed for colonies derived from planktonic cultures (although the clones with the highest MIC values were derived from planktonic cultures)74. Both the number of mutations and the mutational spectrum differed between evolved populations: a significantly higher number of nonsynonymous mutations was observed in the ciprofloxacin-evolved populations, transitions were more frequent in planktonic populations, and transversion and indels were more frequent in biofilms (the latter potentially linked to higher activity of insertion sequences under oxygen-limited conditions119,120). Mutations in mexR (regulator of efflux pump MexAB-OprM), nfxB (MexCD-OprJ) and mexS (MexEF-OprN) were frequent in biofilms evolved in the presence of ciprofloxacin, while mutations in nalC and nalD (regulators of MexAB-OprM) as well as in gyrA and gyrB were frequently found in ciprofloxacin-evolved planktonic populations. In addition, low-frequency mutations in genes related to metabolism were found in several biofilms evolved in the presence of ciprofloxacin; mutated genes include PA1252 (malate dehydrogenase), nuoJ and PA1054 (NADH dehydrogenase)74. Additional mutations linked to metabolism identified in ciprofloxacin-exposed biofilms include mutations in genes related to the TCA cycle (e.g., sdhA) and polyamine and arginine metabolism and transport (e.g., argS), as well as in genes encoding various sigma factors (including rpoN and rpoS)121. The latter mutations might help explain the prolonged lag phase and increased doubling times observed in ciprofloxacin-resistant clones recovered from evolved biofilms. Overall these data suggest that biofilm-grown P. aeruginosa cells exposed to subinhibitory ciprofloxacin concentrations more frequently carry mutations leading to low-level resistance, which could in turn accelerate the stepwise development of ciprofloxacin resistance in vivo74. Interestingly, under the same experimental conditions, lack of the major P. aeruginosa catalase KatA increased the fraction of the ciprofloxacin-resistant population in biofilms and more mutations were observed in evolved ΔkatA biofilms75, again highlighting the role oxidative stress can play in generating diversity in biofilms122. Nevertheless, the observation that ciprofloxacin-resistant mutants also appear after evolving biofilms under anaerobic conditions demonstrates oxidative stress is not the only mechanism75.
Using a bead based model, P. aeruginosa PA14 biofilms were evolved in the absence or presence of increasing tobramycin concentrations. In the biofilm evolved in the presence of tobramycin, MIC values increased 16-fold and at the end of the experiment all tobramycin-exposed biofilms had acquired mutations in fusA1 (encoding elongation factor G)73. While fusA1 mutations also occurred in tobramycin-exposed planktonic populations, they dominated in all final biofilm populations while in planktonically evolved populations their frequencies were more variable. Investigating evolved mutant clones revealed that fusA1 mutations alone lead to 2 to 4-fold increase of tobramycin MIC and at least a 6-fold increase in the tobramycin concentration in which biofilms survived. P. aeruginosa biofilm populations also frequently acquired mutations in the orfKHLN genes (encoding O antigen biosynthesis enzymes) and mutants with mutations in orfN and fusA1 mutations were more resistant than mutants with mutations in fusA1 alone.
Finally, experimental evolution in P. aeruginosa biofilms and planktonic cultures was recently used to identify mechanism of resistance towards the engineered cationic antimicrobial peptide WLBU2123. WGS revealed that surviving populations had minimum two mutations among three key functional categories, i.e., LPS modification (pmrB), O-antigen biosynthesis (orfN) and biofilm formation (wspF and morA). While pmrB and orfN are known to be involved in resistance to cationic peptides, the occurrence of mutations in genes of the wsp pathway (selected both in biofilms and planktonic cultures) was more unexpected. Resistant clones with wsp mutations showed more aggregation, suggesting that increased aggregate and/or biofilm formation itself could contribute to WLBU2 resistance123.
Acinetobacter baumannii
An non-exhaustive overview of genes mutated in A. baumannii biofilms during experimental evolution in the presence of antibiotics is shown in Table 1.
Using a flow model in which A. baumannii biofilms are formed in plastic tubes attached to a peristaltic pump, the effect of exposure to ciprofloxacin (0.5 x MIC) and tetracycline (0.25 x MIC) was investigated124. Cells dispersed from biofilms exposed to antibiotics had a higher MIC with 93% of isolates from ciprofloxacin-treated biofilms showed increased resistance towards ciprofloxacin and 53% isolates from tetracycline-treated biofilms showed increased resistance towards tetracycline; 80% of isolates from ciprofloxacin-treated biofilms also showed increased resistance to tetracycline but cross-resistance was not observed in isolates from tetracycline-treated biofilms. Mutations selected in cells from ciprofloxacin-treated biofilms could often directly be linked to resistance, e.g., mutations in smpB (the deletion of which leads to increased resistance to fluoroquinolones, possibly due to a preventive effect on chromosome fragmentation) and in adeS (leading to overexpression of the AdeABC efflux system)124. Mutations in two genes belonging to the K-locus (production of capsular polysaccharide) were found in samples exposed to either antibiotic and these mutations were often linked with antibiotic resistance phenotypes. Several genes were commonly mutated in isolates from tetracycline-treated biofilms; these mutations often positively correlated with increased biofilm formation rather than increased resistance to tetracycline and include a large 8706 bp deletion in a region encoding proteins involved in regulating c-di-GMP levels124.
The above-mentioned bead model has also been used to study evolution of A. baumannii biofilms in the presence of ciprofloxacin69 or tobramycin73. Comparison of planktonic cultures and biofilms exposed to increasing concentration of ciprofloxacin showed that high-level resistance quickly developed in planktonic cultures (~160-fold increase in MIC) while mutants with low levels of resistance (~6-fold increase in MIC) occurred in biofilms69. Mutations disrupting the repressors adeL (regulator of the AdeFGH efflux pump) or adeN (regulator of the AdeIJK efflux pump) dominate in biofilm and planktonic clones, respectively, suggesting the presence of lifestyle-specific efflux systems, as previously identified in other organisms125. Interestingly, mutations in adeS (regulator of the AdeABC efflux pump) appeared in exposed biofilms, but were subsequently outcompeted by adeL mutations, something not observed in another study with A. baumannii124. While a couple of mutations quickly reached fixation in planktonic populations (including a single high frequency mutation in gyrA in genetic backgrounds containing an adeN mutation) more diversity was maintained in biofilms. A. baumannii biofilms propagated under tobramycin selection demonstrated an 8 to 32-fold increase in MIC and also in this species mutations in fusA1 occurred in all replicate populations exposed to tobramycin73. In contrast to P. aeruginosa biofilms, tobramycin-treated A. baumannii biofilms quickly accumulated mutations in ptsP (encoding phosphoenolpyruvate phosphotransferase), and fusA1 and ptsP mutations reached similar frequencies in treated biofilm and planktonic populations. Evolved mutant clones with only a mutation in fusA1 showed a 4-fold increase in MIC, while fusA1 ptsP double mutants showed an 8-fold increase. In contrast to fusA1 (which is an essential gene), ptsP mutations are likely loss-of-function mutations as they are indels that lead to a frameshift. In addition, six mutations in cyoAB (coding for two subunits of cytochrome bo3 ubiquinol oxidase involved in the electron transport chain) only occurred in biofilms; these mutations were however outcompeted by the fusA1 ptsP genotype at higher tobramycin concentrations73.
E. coli and Salmonella
E. coli biofilms grown in flow cells in the presence of rifampicin or kanamycin were used to address the question how growth in a biofilm can protect resistant cells from being outcompeted by fitter non-resistant cells in the absence of antibiotics80. Because of physical constraints and biofilm heterogeneity, it can reasonably be assumed that individual cells only have to compete with a subset of other cells15, while in unstructured planktonic populations cells would experience global competition in which they have to compete against all other cells126. The inoculum already contained low levels of kanamycin and rifampicin‐resistant mutants and during biofilm formation in the absence of antibiotics, their number increased ~45-fold. Treatment with rifampicin led to fixation of rifampicin resistance (i.e., the entire population became resistant), while kanamycin treatment resulted in a population with 52% resistant cells. When the treatment was stopped, the fraction of resistant cells did not change, but when biofilm cells were transferred to planktonic cultures, kanamycin (but not rifampicin) resistance gradually returned to the original low levels80. This study shows that resistance in biofilms can be the result of de novo mutations, but can also be due to selection of pre-existing mutants that are less fit outside the biofilm environment. E. coli biofilms that are grown on silicone disks and are intermittently exposed to high (5 x MIC) and very high (80 x MIC) concentrations of amikacin experience a strong drop in surviving cell number after the first treatment, but the number of surviving cells quickly increases (to ~100% survival for exposure to 5 x MIC and ~1% survival for exposure to 80 x MIC)104. In planktonic cultures, the decrease after the first treatment is more pronounced and only ~0.1% of the cells ultimately survive exposure to 5 x MIC (no survivors are observed after three cycles with exposure to 80 x MIC). This increased survival in biofilms is associated with a rapid MIC increase in treated biofilms, while the MIC increase in planktonic cultures is much lower. Mutations in sbmA (coding for an inner membrane peptide transporter previously associated to increased E. coli resistance to aminoglycosides) were found in all treated biofilm populations and two out of three treated planktonic populations, but not in non-treated controls; five out of six evolved biofilm populations had multiple sbmA mutations, suggesting clonal interference. Mutations in fusA were selected in several intermediate biofilm populations and at the end of the experiment in one biofilm population; no fusA mutations were selected in planktonic cultures. fusA and sbmA can coexist in biofilm populations but fusA mutations appear sooner (or the latest at the same time) than sbmA mutations104. In the absence of antibiotics fusA mutants have a lower fitness than sbmA mutants, suggesting the former were counter-selected in the periods between treatment in planktonic cultures while they were maintained in biofilms. Loss-of-function mutations in the sbmA gene lead to a moderate increase of the MIC (from 16 to 24 µg/ml), while fusA mutations lead to MIC values of 48 µg/ml. Highest MIC values (128 µg/ml) were observed in clones that harbored a mutation in fusA combined with a loss-of-function mutation in sbmA and a mutation in fre (coding for a NAD(P)H flavin reductase); or harbored a mutation in fusA combined with a mutation in yfgZ (encoding a protein involved in repair during oxidative stress and Fe-S cluster synthesis). In general, in planktonic cultures clones were selected that had mutations in a diverse set of genes and MICs of these clones were typically lower than for clones evolved under biofilm conditions104. Interestingly, clones recovered from treated biofilms had higher survival rates upon treatment when grown in biofilms as compared to when grown in planktonic cultures, and the majority of evolved biofilm populations contained mutations in fimH, coding for the FimH tip-adhesin of type 1 fimbriae; the fimH mutants show enhanced biofilm formation and reduced amikacin susceptibility. Together these data suggest that the biofilm environment as such contributes to higher survival upon exposure to amikacin, by increasing the occurrence of new genetic resistance mutations, even in the absence of mutations that lead to increased tolerance104.
Experimental evolution of Salmonella Typhimurium biofilms grown on glass beads and planktonic cultures, in the presence and absence of azithromycin, cefotaxime and ciprofloxacin showed that biofilms and planktonic cultures develop resistance to these antibiotics in the same time frame72. However, the phenotype of evolved mutants differs between different conditions; e.g., in contrast to planktonic populations exposed to cefotaxime (which become mainly resistant to cefotaxime), biofilms evolved in the presence of cefotaxime show resistance to a wide range of antibiotics72,127. The same genes were often mutated in evolved planktonic and biofilm populations, e.g., mutations in acrB and ramR (after exposure to azithromycin), envZ (cefotaxime) and gyrA (ciprofloxacin); although the exact mutation sometimes differed (e.g., in ramR: term194Tyr in planktonic cultures vs. Thr18Pro in biofilms; in gyrA: Ser83Tyr in planktonic cultures vs. Ser83Phe in biofilms)72,127. These mutations suggest efflux (azithromycin), reduced membrane permeability (cefotaxime) and target modification (ciprofloxacin) are the most important mechanisms involved in the observed reduced susceptibility, although many other mutations were identified, and WGS clearly showed that different mutants followed different paths of adaptation.
How does experimental evolution of biofilm susceptibility compare to evolution of susceptibility in vivo?
From the LTEE and many other studies we have learned that overall there is a high degree of parallelism in diversification and that evolution appears to be reproducible between replicate lineages and between different experiments carried out in different labs, suggesting the observed evolutionary changes are not random artefacts15. Additional proof for this comes from a direct comparison of mutations in experimentally evolved P. aeruginosa isolates and in clinical isolates, including those from chronic respiratory tract infections in CF. Overall these comparisons confirm that the changes observed in vitro are relevant for evolution of susceptibility in vivo. For example, selection of different ciprofloxacin resistance mechanisms is lifestyle-dependent74 which is in line with the high prevalence of mutations in ciprofloxacin target genes in isolates from acute infections (e.g., urinary tract infections), which are less common in isolates recovered from chronic infections128. Likewise, mutations in P. aeruginosa genes fusA1 and ptsP occur in high frequency during in vitro evolution and identical mutations have been observed in clinical isolates73,89. P. aeruginosa adaptation to chronic infection not only occurs in CF; e.g., also in isolates recovered from chronic obstructive pulmonary disease patients mutations occur in genes that are frequently identified in experimental evolution studies (including mexA, mexB, oprM and oprF)129.
Indirect evidence comes from the comparison of phenotypes of isolates evolved in vitro with those involved during chronic infection54. For example, P. aeruginosa isolates recovered from younger CF patients typically display low resistance and low tolerance to antibiotics, and the frequency of drug-tolerant isolates increased with increasing age; increased frequencies of resistant isolates were only observed in older patients58. In these older patients two subpopulations were present, one consisting of highly resistant isolates and one consisting of hyper-tolerant isolates that retained low-level resistance, suggesting that also in vivo tolerance can be a ‘stepping stone’ towards resistance development58. Finally, the recent finding that biofilms are also present in at least some acute respiratory tract infections and that the main difference between acute and chronic infection may not be the association with the planktonic and biofilm lifestyle, respectively, but rather be related to differences in metabolism130 is in line with observations from in vitro experimental evolution studies as mutations in genes related to metabolism are frequently identified during experimental evolution73,74,121.
While these similarities between evolution in vitro and in vivo strongly suggest that genetic changes identified in vitro are relevant for what happens in vivo, experimental validation of the link between these genetic changes (in metabolism-related genes and others) on the one hand, and reduced antimicrobial susceptibility on the other, remains necessary.
Changes in biofilm formation during experimental evolution
Changes in biofilm forming capacity during experimental evolution can also affect biofilm susceptibility. In the presence of daptomycin, Enterococcus faecalis biofilms grown in a bioreactor quickly develop resistance to the antibiotic, but at the same time biofilm formation increased in daptomycin-resistant strains81. WGS identified combinations of mutations that ultimately lead to an increase in biofilm formation and while this increase in biofilm formation is not a prerequisite for increased resistance, it was observed in the majority of the resistant lineages81. Increases in biofilm formation were also observed during experimental evolution of A. baumannii biofilms (both in the bead model69 and in a flow system124), with isolates from untreated and ciprofloxacin-treated biofilms showing increased biofilm formation capability compared to start cultures in both studies. Moreover, in the flow system, many isolates from tetracycline-treated biofilms showed an additional increase in biofilm formation124. While some mutations linked to increased biofilm formation occurred in treated and untreated samples (e.g., mutations in ABUW_0885 coding for biofilm-associated protein Bap), others (e.g., mutations in ABUW_2055, encoding a fimbrial adhesin) only occurred in untreated biofilms124. As already outlined above, fimH mutations were found in the majority of E. coli biofilm populations treated with amikacin as well as in the untreated controls; fimH mutants showed increased biofilm forming capacity and increased survival upon exposure to high concentrations of amikacin104. In studies with Salmonella Typhimurium biofilms grown on glass beads, a clear trade-off between antimicrobial resistance and biofilm formation was observed72,127. Over the course of the experiment, biofilm forming capacity (as measured by crystal violet staining) increased in colonies recovered from untreated glass beads and this was associated with a missense mutation in cytR (which is known to increase biofilm formation) that occurred in multiple untreated lineages72. However, colonies recovered from biofilms evolved in the presence of antibiotics (especially azithromycin and cefotaxime) showed reduced biofilm formation compared to unexposed biofilms and none of them contained mutations in cytR72. Exposure to subinhibitory concentrations of cefotaxime selects for mutations in the C-terminal catalytic/ATP-binding domain of EnvZ which result in lower levels of the porin OmpF and reduced permeability. However, EnvZ also regulates curli production and reduced curli production and biofilm formation was observed in envZ mutants, suggesting a trade-off between biofilm susceptibility and biofilm formation. Overall, these data suggest that the association between changes in biofilm formation and antimicrobial susceptibility during experimental evolution is complex and probably species, model and antibiotic-dependent.
Looking at biofilm antimicrobial susceptibility through the lens of experimental evolution—a consensus view emerges
Although the studies discussed above used different model systems, antibiotics, species and strains, some common patterns emerge.
While decreased susceptibility during experimental evolution develops both in planktonic and biofilm populations, the mechanisms involved and the trajectories towards this reduced susceptibility are not identical. Mutations in genes that code for targets of antibiotics are frequently encountered in planktonic populations evolved in the presence of antibiotics (e.g., mutations in gyrA following evolution in the presence of ciprofloxacin), while evolved biofilm populations also contain a wide range of mutations in genes involved in efflux and metabolism69,74,121. When subinhibitory concentrations of antibiotics are used, growth in well-mixed planktonic cultures selected for high-level resistance, while growth in spatially structured biofilms favored mutants with lower levels of resistance69,74,121. However, this is not always the case when stepwise increasing or lethal concentrations of antibiotic are used during evolution69,73,88,104. While species- and/or antibiotic-dependent effects cannot yet be ruled out, this suggest that the treatment regime itself plays an important role in determining final MIC levels in planktonic and biofilm populations.
Evolved biofilm populations maintain a higher diversity than corresponding planktonic populations, in which successful mutations reach fixation quickly, and the biofilm environment may protect against negative selection of less fit resistant mutants that would be quickly outcompeted in planktonic cultures121. However, a recent study indicated that fitness costs for resistance in surface-associated E. coli biofilms did not differ from those in planktonic cultures79. In addition, another recent study has shown that the specific environment co-determines fitness and resistance levels associated with specific mutations131. Clearly more work is needed to gain deeper insight in parameters affecting fitness in different (structured) environments. In addition, mutations that lead to increased biofilm formation can increase the size of the tolerant population that survives antimicrobial exposure, in which resistance can subsequently develop104.
While mutations in some genes are found across organisms (e.g., mutations in fusA have been observed in P. aeruginosa, A. baumannii and E. coli), different organisms will also accumulate mutations in different genes although the resulting phenotype could be similar (Table 1). An example of such a parallel strategy are mutations in P. aeruginosa orfKHLN and A. baumannii cyoAB: while these genes are involved in very different cellular process (O antigen biosynthesis and electron transport, respectively) mutations in either result in reduced permeability for aminoglycosides and may lead to reduced aminoglycoside susceptibility73. Likewise, mutations in many different metabolic genes or sigma factors might lead to reduced growth, and ‘tolerance by lag’. This suggests that the fundamental mechanisms behind reduced biofilm susceptibility could be similar for different classes of antibiotics and in different organisms, even when it is not possible to identify mutations, mutated genes, or differences in metabolism or gene expression shared between different organisms. As such, data from experimental evolution are in line with the conclusion of a recent study that could not find evidence for a common genetic or biochemical basis for antimicrobial tolerance in biofilms but concluded that many genes, proteins, and metabolic pathways collectively determine the physiological state and susceptibility of bacterial cells in a biofilm132.
We believe experimental evolution has and will continue to help to elucidate the interplay of resistance, tolerance and persistence that is behind the reduced antimicrobial susceptibility of biofilms and determines the outcome of antimicrobial treatment. However, identifying the complex patterns of mutations, changes in gene expression and metabolism in different organisms as well as polymicrobial communities will require an interdisciplinary and holistic approach and will greatly benefit from the use of relevant model systems.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Kawecki, T. J. et al. Experimental evolution. Trends Ecol. Evol. 27, 547–560 (2012).
Haas, J. W. The Reverend Dr William Henry Dallinger, F.R.S. (1839–1909). Notes Rec. R. Soc. Lond. 54, 53–65 (2000).
Atwood, K. C., Schneider, L. K. & Ryan, F. J. Periodic selection in Escherichia coli. Proc. Natl. Acad. Sci. USA 37, 146–155 (1951).
Lenski, R. E. Convergence and divergence in a long-term experiment with bacteria. Am. Nat. 190, S57–S68 (2017).
Lenski, R. E. Experimental evolution and the dynamics of adaptation and genome evolution in microbial populations. ISME J. 11, 2181–2194 (2017).
Korona, R., Nakatsu, C. H., Forney, L. J. & Lenski, R. E. Evidence for multiple adaptive peaks from populations of bacteria evolving in a structured habitat. Proc. Natl. Acad. Sci. USA 91, 9037–9041 (1994).
Rainey, P. B. & Travisano, M. Adaptive radiation in a heterogeneous environment. Nature 394, 69–72 (1998).
Sauer, K. et al. The biofilm life cycle: expanding the conceptual model of biofilm formation. Nat. Rev. Microbiol. 20, 608–620 (2022).
Stewart, P. S. & Franklin, M. J. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 6, 199–210 (2008).
Sonderholm, M. et al. Pseudomonas aeruginosa aggregate formation in an alginate bead model system exhibits in vivo-like characteristics. Appl. Environ. Microbiol. 83, e00113–17 (2017).
Bjarnsholt, T. et al. The importance of understanding the infectious microenvironment. Lancet Infect. Dis. 22, e88–e92 (2022).
Ciofu, O., Moser, C., Jensen, P. O. & Hoiby, N. Tolerance and resistance of microbial biofilms. Nat. Rev. Microbiol. 20, 621–635 (2022).
Livingston, J., Spero, M. A., Lonergan, Z. R. & Newman, D. K. Visualization of mRNA expression in Pseudomonas aeruginosa aggregates reveals spatial patterns of fermentative and denitrifying metabolism. Appl. Environ. Microbiol. 88, e0043922 (2022).
Van den Bergh, B., Swings, T., Fauvart, M. & Michiels, J. Experimental design, population Dynamics, and diversity in microbial experimental evolution. Microbiol. Mol. Biol. Rev. 82, e00008–18 (2018).
Steenackers, H. P., Parijs, I., Dubey, A., Foster, K. R. & Vanderleyden, J. Experimental evolution in biofilm populations. FEMS Microbiol. Rev. 40, 373–397 (2016).
Habets, M. G., Rozen, D. E., Hoekstra, R. F. & de Visser, J. A. The effect of population structure on the adaptive radiation of microbial populations evolving in spatially structured environments. Ecol. Lett. 9, 1041–1048 (2006).
Poltak, S. R. & Cooper, V. S. Ecological succession in long-term experimentally evolved biofilms produces synergistic communities. ISME J. 5, 369–378 (2011).
Armbruster, C. R. et al. Adaptation and genomic erosion in fragmented Pseudomonas aeruginosa populations in the sinuses of people with cystic fibrosis. Cell Rep. 37, 109829 (2021).
Rozen, D. E., Habets, M. G., Handel, A. & de Visser, J. A. Heterogeneous adaptive trajectories of small populations on complex fitness landscapes. PLoS One 3, e1715 (2008).
Chakraborty, P. P., Nemzer, L. R. & Kassen, R. Experimental evidence that metapopulation structure can accelerate adaptive evolution. bioRxiv https://doi.org/10.1101/2021.07.13.452242 (2021).
Martens, E. A. & Hallatschek, O. Interfering waves of adaptation promote spatial mixing. Genetics 189, 1045–1060 (2011).
Traverse, C. C., Mayo-Smith, L. M., Poltak, S. R. & Cooper, V. S. Tangled bank of experimentally evolved Burkholderia biofilms reflects selection during chronic infections. Proc. Natl. Acad. Sci. USA 110, E250–E259 (2013).
Harris, K. B., Flynn, K. M. & Cooper, V. S. Polygenic adaptation and clonal interference enable sustained diversity in experimental pseudomonas aeruginosa populations. Mol. Biol. evolution 38, 5359–5375 (2021).
Schick, A. & Kassen, R. Rapid diversification of Pseudomonas aeruginosa in cystic fibrosis lung-like conditions. Proc. Natl. Acad. Sci. USA 115, 10714–10719 (2018).
Schick, A., Shewaramani, S. & Kassen, R. Genomics of diversification of Pseudomonas aeruginosa in cystic fibrosis lung-like conditions. Genome Biol. Evol. 14, evac074 (2022).
Stewart, P. S. Diffusion in biofilms. J. Bacteriol. 185, 1485–1491 (2003).
Tseng, B. S. et al. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ. Microbiol. 15, 2865–2878 (2013).
Davies, S. K. et al. Visualizing antimicrobials in bacterial biofilms: three-dimensional biochemical imaging using TOF-SIMS. mSphere 2, e00211–17 (2017).
Stewart, P. S. et al. Conceptual model of biofilm antibiotic tolerance that integrates phenomena of diffusion, metabolism, gene expression, and physiology. J. Bacteriol. 201, e00307–19 (2019).
Baquero, F., Negri, M. C., Morosini, M. I. & Blazquez, J. Antibiotic-selective environments. Clin. Infect. Dis. 27, S5–S11 (1998).
Brauner, A., Fridman, O., Gefen, O. & Balaban, N. Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14, 320–330 (2016).
Balaban, N. Q. et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 17, 441–448 (2019).
Baquero, F. et al. Evolutionary pathways and trajectories in antibiotic resistance. Clin. Microbiol. Rev. 34, e0005019 (2021).
Jansen, G., Barbosa, C. & Schulenburg, H. Experimental evolution as an efficient tool to dissect adaptive paths to antibiotic resistance. Drug Resist. Update. 16, 96–107 (2013).
Adler, M., Anjum, M., Andersson, D. I. & Sandegren, L. Combinations of mutations in envZ, ftsI, mrdA, acrB and acrR can cause high-level carbapenem resistance in Escherichia coli. J. Antimicrobial Chemother. 71, 1188–1198 (2016).
Langevin, A. M., El Meouche, I. & Dunlop, M. J. Mapping the role of AcrAB-TolC efflux pumps in the evolution of antibiotic resistance reveals near-MIC treatments facilitate resistance acquisition. mSphere 5, e01056–20 (2020).
Zhang, G., Wang, C., Sui, Z. & Feng, J. Insights into the evolutionary trajectories of fluoroquinolone resistance in Streptococcus pneumoniae. J. Antimicrobial Chemother. 70, 2499–2506 (2015).
Cabot, G. et al. Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation rates. Antimicrob. Agents Chemother. 60, 1767–1778 (2016).
Crabbe, A., Jensen, P. O., Bjarnsholt, T. & Coenye, T. Antimicrobial tolerance and metabolic adaptations in microbial biofilms. Trends Microbiol. 27, 850–863 (2019).
Stokes, J. M., Lopatkin, A. J., Lobritz, M. A. & Collins, J. J. Bacterial metabolism and antibiotic efficacy. Cell Metab. 30, 251–259 (2019).
Zampieri, M. et al. Metabolic constraints on the evolution of antibiotic resistance. Mol. Syst. Biol. 13, 917 (2017).
Lopatkin, A. J. et al. Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science 371, eaba0862 (2021).
Van Acker, H. et al. Biofilm-grown Burkholderia cepacia complex cells survive antibiotic treatment by avoiding production of reactive oxygen species. PLoS one 8, e58943 (2013).
Van Acker, H. & Coenye, T. The role of reactive oxygen species in antibiotic-mediated killing of bacteria. Trends Microbiol. 25, 456–466 (2017).
Meylan, S., Andrews, I. W. & Collins, J. J. Targeting antibiotic tolerance, pathogen by pathogen. Cell 172, 1228–1238 (2018).
Kester, J. C. & Fortune, S. M. Persisters and beyond: mechanisms of phenotypic drug resistance and drug tolerance in bacteria. Crit. Rev. Biochem. Mol. Biol. 49, 91–101 (2014).
Windels, E. M., Van den Bergh, B. & Michiels, J. Bacteria under antibiotic attack: different strategies for evolutionary adaptation. PLoS Pathog. 16, e1008431 (2020).
Verstraete, L., Van den Bergh, B., Verstraeten, N. & Michiels, J. Ecology and evolution of antibiotic persistence. Trends Microbiol. 30, 466–479 (2022).
Fridman, O., Goldberg, A., Ronin, I., Shoresh, N. & Balaban, N. Q. Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 513, 418–421 (2014).
Dengler Haunreiter, V. et al. In-host evolution of Staphylococcus epidermidis in a pacemaker-associated endocarditis resulting in increased antibiotic tolerance. Nat. Commun. 10, 1149 (2019).
Jennings, L. K. et al. Pseudomonas aeruginosa aggregates in cystic fibrosis sputum produce exopolysaccharides that likely impede current therapies. Cell Rep. 34, 108782 (2021).
Chakraborty, P., Bajeli, S., Kaushal, D., Radotra, B. D. & Kumar, A. Biofilm formation in the lung contributes to virulence and drug tolerance of Mycobacterium tuberculosis. Nat. Commun. 12, 1606 (2021).
Sonderholm, M. et al. The consequences of being in an infectious biofilm: microenvironmental conditions governing antibiotic tolerance. Int. J. Mol. Sci. 18, 2688 (2017).
Rossi, E. et al. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat. Rev. Microbiol. 19, 331–342 (2021).
Michiels, J. E., Van den Bergh, B., Verstraeten, N., Fauvart, M. & Michiels, J. In vitro emergence of high persistence upon periodic aminoglycoside challenge in the ESKAPE pathogens. Antimicrob. Agents Chemother. 60, 4630–4637 (2016).
Van den Bergh, B. et al. Frequency of antibiotic application drives rapid evolutionary adaptation of Escherichia coli persistence. Nat. Microbiol. 1, 16020 (2016).
Levin-Reisman, I. et al. Antibiotic tolerance facilitates the evolution of resistance. Science 355, 826–830 (2017).
Santi, I., Manfredi, P., Maffei, E., Egli, A. & Jenal, U. Evolution of antibiotic tolerance shapes resistance development in chronic Pseudomonas aeruginosa infections. mBio 12, e03482–20 (2021).
Sebastian, J. et al. De Novo emergence of genetically resistant mutants of Mycobacterium tuberculosis from the persistence phase cells formed against antituberculosis drugs in vitro. Antimicrob. Agents Chemother. 61, e01343–16 (2017).
Vogwill, T., Comfort, A. C., Furio, V. & MacLean, R. C. Persistence and resistance as complementary bacterial adaptations to antibiotics. J. Evolut. Biol. 29, 1223–1233 (2016).
Windels, E. M. et al. Bacterial persistence promotes the evolution of antibiotic resistance by increasing survival and mutation rates. ISME J. 13, 1239–1251 (2019).
Coenye, T. & Nelis, H. J. In vitro and in vivo model systems to study microbial biofilm formation. J. Microbiol. Methods 83, 89–105 (2010).
Azeredo, J. et al. Critical review on biofilm methods. Crit. Rev. Microbiol. 43, 313–351 (2017).
Mai-Prochnow, H. K. N. V. B. X. A. Clinically relevant in vitro biofilm models: a need to mimic and recapitulate the host environment. Biofilm 4, 100069 (2022).
Lebeaux, D., Chauhan, A., Rendueles, O. & Beloin, C. From in vitro to in vivo models of bacterial biofilm-related infections. Pathogens. 2, 288–356 (2013).
Cooper, V. S., Staples, R. K., Traverse, C. C. & Ellis, C. N. Parallel evolution of small colony variants in Burkholderia cenocepacia biofilms. Genomics 104, 447–452 (2014).
O’Rourke, D., FitzGerald, C. E., Traverse, C. C. & Cooper, V. S. There and back again: consequences of biofilm specialization under selection for dispersal. Front. Genet. 6, 18 (2015).
Martin, M., Holscher, T., Dragos, A., Cooper, V. S. & Kovacs, A. T. Laboratory evolution of microbial interactions in bacterial biofilms. J. Bacteriol. 198, 2564–2571 (2016).
Santos-Lopez, A., Marshall, C. W., Scribner, M. R., Snyder, D. J. & Cooper, V. S. Evolutionary pathways to antibiotic resistance are dependent upon environmental structure and bacterial lifestyle. eLife 8, e47612 (2019).
Sass, A. et al. Various evolutionary trajectories lead to loss of the tobramycin-potentiating activity of the Quorum-sensing inhibitor baicalin hydrate in Burkholderia cenocepacia biofilms. Antimicrob. Agents Chemother. 63, e02092–18 (2019).
Oakley, J. L. et al. Phenotypic and genotypic adaptations in Pseudomonas aeruginosa biofilms following long-term exposure to an alginate oligomer therapy. mSphere 6, e01216–20 (2021).
Trampari, E. et al. Exposure of Salmonella biofilms to antibiotic concentrations rapidly selects resistance with collateral tradeoffs. NPJ biofilms microbiomes 7, 3 (2021).
Scribner, M. R., Santos-Lopez, A., Marshall, C. W., Deitrick, C. & Cooper, V. S. Parallel evolution of tobramycin resistance across species and environments. mBio 11, e00932–20 (2020).
Ahmed, M. N., Porse, A., Sommer, M. O. A., Hoiby, N. & Ciofu, O. Evolution of antibiotic resistance in biofilm and planktonic Pseudomonas aeruginosa populations exposed to subinhibitory levels of ciprofloxacin. Antimicrob. Agents Chemother. 62, e00320–18 (2018).
Ahmed, M. N. et al. Lack of the major multifunctional catalase KatA in Pseudomonas aeruginosa accelerates evolution of antibiotic resistance in ciprofloxacin-treated biofilms. Antimicrob. Agents Chemother. 63, e00766–19 (2019).
Ceri, H. et al. The Calgary biofilm device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol. 37, 1771–1776 (1999).
Maiden, M. M. & Waters, C. M. Triclosan depletes the membrane potential in Pseudomonas aeruginosa biofilms inhibiting aminoglycoside induced adaptive resistance. PLoS Pathog. 16, e1008529 (2020).
Zaborskyte, G., Wistrand-Yuen, E., Hjort, K., Andersson, D. I. & Sandegren, L. Modular 3D-printed Peg biofilm device for flexible setup of surface-related biofilm studies. Front. Cell. Infect. Microbiol. 11, 802303 (2021).
Hjort, K., Fermer, E., Tang, P. C. & Andersson, D. I. Antibiotic minimal selective concentrations and fitness costs during biofilm and planktonic growth. mBio 13, e0144722 (2022).
France, M. T., Cornea, A., Kehlet-Delgado, H. & Forney, L. J. Spatial structure facilitates the accumulation and persistence of antibiotic-resistant mutants in biofilms. Evolut. Appl. 12, 498–507 (2019).
Miller, C. et al. Adaptation of Enterococcus faecalis to daptomycin reveals an ordered progression to resistance. Antimicrob. Agents Chemother. 57, 5373–5383 (2013).
Hammerstrom, T. G., Beabout, K., Clements, T. P., Saxer, G. & Shamoo, Y. Acinetobacter baumannii repeatedly evolves a hypermutator phenotype in response to tigecycline that effectively surveys evolutionary trajectories to resistance. PLoS One 10, e0140489 (2015).
Mehta, H. H., Prater, A. G. & Shamoo, Y. Using experimental evolution to identify druggable targets that could inhibit the evolution of antimicrobial resistance. J. Antibiotics 71, 279–286 (2018).
Zhang, Q. et al. Acceleration of emergence of bacterial antibiotic resistance in connected microenvironments. Science 333, 1764–1767 (2011).
Zoheir, A. E., Spath, G. P., Niemeyer, C. M. & Rabe, K. S. Microfluidic evolution-on-a-chip reveals new mutations that cause antibiotic resistance. Small 17, e2007166 (2021).
Tang, P. C. et al. A microfluidic chip for studies of the dynamics of antibiotic resistance selection in bacterial biofilms. Front. Cell. Infect. Microbiol. 12, 896149 (2022).
Palmer, K. L., Aye, L. M. & Whiteley, M. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 189, 8079–8087 (2007).
Wong, A., Rodrigue, N. & Kassen, R. Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PLoS Genet. 8, e1002928 (2012).
Bove, M., Bao, X., Sass, A., Crabbe, A. & Coenye, T. The Quorum-sensing inhibitor furanone C-30 rapidly loses its tobramycin-potentiating activity against Pseudomonas aeruginosa biofilms during experimental evolution. Antimicrob. Agents Chemother. 65, e0041321 (2021).
Drlica, K. & Zhao, X. Mutant selection window hypothesis updated. Clin. Infect. Dis. 44, 681–688 (2007).
Trubenova, B., Roizman, D., Moter, A., Rolff, J. & Regoes, R. R. Population genetics, biofilm recalcitrance, and antibiotic resistance evolution. Trends Microbiol. 30, 841–852 (2022).
Sanz-Garcia, F., Sanchez, M. B., Hernando-Amado, S. & Martinez, J. L. Evolutionary landscapes of Pseudomonas aeruginosa towards ribosome-targeting antibiotic resistance depend on selection strength. Int. J. antimicrobial agents 55, 105965 (2020).
Barrick, J. E. & Lenski, R. E. Genome dynamics during experimental evolution. Nat. Rev. Genet. 14, 827–839 (2013).
Bos, A. C., Passe, K. M., Mouton, J. W., Janssens, H. M. & Tiddens, H. A. The fate of inhaled antibiotics after deposition in cystic fibrosis: How to get drug to the bug? J. Cyst. Fibros. 16, 13–23 (2017).
van Os, W. & Zeitlinger, M. Predicting antimicrobial activity at the target site: pharmacokinetic/pharmacodynamic indices versus time-kill approaches. Antibiotics 10, 1485 (2021).
Cao, B. et al. Antibiotic penetration and bacterial killing in a Pseudomonas aeruginosa biofilm model. J. Antimicrobial Chemother. 70, 2057–2063 (2015).
Christophersen, L. et al. In vivo demonstration of Pseudomonas aeruginosa biofilms as independent pharmacological microcompartments. J. Cyst. Fibros. 19, 996–1003 (2020).
Messiaen, A. S., Forier, K., Nelis, H., Braeckmans, K. & Coenye, T. Transport of nanoparticles and tobramycin-loaded liposomes in Burkholderia cepacia complex biofilms. PLoS One 8, e79220 (2013).
Kohanski, M. A., Dwyer, D. J., Hayete, B., Lawrence, C. A. & Collins, J. J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810 (2007).
Dwyer, D. J., Kohanski, M. A. & Collins, J. J. Role of reactive oxygen species in antibiotic action and resistance. Curr. Opin. Microbiol. 12, 482–489 (2009).
Zheng, E. J., Stokes, J. M. & Collins, J. J. Eradicating bacterial persisters with combinations of strongly and weakly metabolism-dependent antibiotics. Cell Chem. Biol. 27, 1544–1552.e1543 (2020).
Zheng, E. J. et al. Modulating the evolutionary trajectory of tolerance using antibiotics with different metabolic dependencies. Nat. Commun. 13, 2525 (2022).
Davies, E. V., James, C. E., Brockhurst, M. A. & Winstanley, C. Evolutionary diversification of Pseudomonas aeruginosa in an artificial sputum model. BMC Microbiol. 17, 3 (2017).
Usui, M., Yoshii, Y., Thiriet-Rupert, S., Ghigo, J.-M. & Beloin, C. Intermittent antibiotic treatment of bacterial biofilms favors the rapid evolution of resistance. bioRxiv https://doi.org/10.1101/2022.05.03.490405 (2022).
Hansen, S. K., Rainey, P. B., Haagensen, J. A. & Molin, S. Evolution of species interactions in a biofilm community. Nature 445, 533–536 (2007).
Tognon, M. et al. Co-evolution with Staphylococcus aureus leads to lipopolysaccharide alterations in Pseudomonas aeruginosa. ISME J. 11, 2233–2243 (2017).
Vandeplassche, E. et al. In vitro evolution of Pseudomonas aeruginosa AA2 biofilms in the presence of cystic fibrosis lung microbiome members. Sci. Rep. 9, 12859 (2019).
Cairns, J., Jokela, R., Becks, L., Mustonen, V. & Hiltunen, T. Repeatable ecological dynamics govern the response of experimental communities to antibiotic pulse perturbation. Nat. Ecol. Evol. 4, 1385–1394 (2020).
Cooper, V. S. et al. Experimental evolution in vivo to identify selective pressures during pneumococcal colonization. mSystems 5, e00352–20 (2020).
Lebov, J. F., Schlomann, B. H., Robinson, C. D. & Bohannan, B. J. M. Phenotypic parallelism during experimental adaptation of a free-living bacterium to the zebrafish gut. mBio 11, e01519–20 (2020).
Bove, M. & Coenye, T. The anti-virulence activity of the non-mevalonate pathway inhibitor FR900098 towards Burkholderia cenocepacia is maintained during experimental evolution. Microbiology 168, 001170 (2022).
Thoming, J. G. et al. Parallel evolutionary paths to produce more than one Pseudomonas aeruginosa biofilm phenotype. NPJ Biofilms Microbiomes 6, 2 (2020).
Thoming, J. G. & Haussler, S. Pseudomonas aeruginosa is more tolerant under biofilm than under planktonic growth conditions: a multi-isolate survey. Front. Cell. Infect. Microbiol. 12, 851784 (2022).
Tyerman, J. G., Ponciano, J. M., Joyce, P., Forney, L. J. & Harmon, L. J. The evolution of antibiotic susceptibility and resistance during the formation of Escherichia coli biofilms in the absence of antibiotics. BMC Evolut. Biol. 13, 22 (2013).
Azimi, S. et al. Allelic polymorphism shapes community function in evolving Pseudomonas aeruginosa populations. ISME J. 14, 1929–1942 (2020).
Allegrucci, M. & Sauer, K. Formation of Streptococcus pneumoniae non-phase-variable colony variants is due to increased mutation frequency present under biofilm growth conditions. J. Bacteriol. 190, 6330–6339 (2008).
Boles, B. R., Thoendel, M. & Singh, P. K. Self-generated diversity produces “insurance effects” in biofilm communities. Proc. Natl. Acad. Sci. USA 101, 16630–16635 (2004).
Ryder, V. J., Chopra, I. & O’Neill, A. J. Increased mutability of Staphylococci in biofilms as a consequence of oxidative stress. PLoS One 7, e47695 (2012).
Shewaramani, S. et al. Anaerobically grown Escherichia coli has an enhanced mutation rate and distinct mutational spectra. PLoS Genet. 13, e1006570 (2017).
Maharjan, R. P. & Ferenci, T. A shifting mutational landscape in 6 nutritional states: Stress-induced mutagenesis as a series of distinct stress input-mutation output relationships. PLoS Biol. 15, e2001477 (2017).
Ahmed, M. N. et al. The evolutionary trajectories of P. aeruginosa in biofilm and planktonic growth modes exposed to ciprofloxacin: beyond selection of antibiotic resistance. NPJ biofilms microbiomes 6, 28 (2020).
Boles, B. R. & Singh, P. K. Endogenous oxidative stress produces diversity and adaptability in biofilm communities. Proc. Natl. Acad. Sci. USA 105, 12503–12508 (2008).
Santos-Lopez, A. et al. Evolved resistance to a novel cationic peptide antibiotic requires high mutation supply. Evol. Med. Public Health 10, 266–276 (2022).
Penesyan, A., Nagy, S. S., Kjelleberg, S., Gillings, M. R. & Paulsen, I. T. Rapid microevolution of biofilm cells in response to antibiotics. NPJ biofilms microbiomes 5, 34 (2019).
Van Acker, H., Van Dijck, P. & Coenye, T. Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol. 22, 326–333 (2014).
Hibbing, M. E., Fuqua, C., Parsek, M. R. & Peterson, S. B. Bacterial competition: surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 8, 15–25 (2010).
Trampari, E. et al. Cefotaxime exposure selects mutations within the CA-domain of envZ which promote antibiotic resistance but repress biofilm formation in Salmonella. Microbiol. Spectr. 10, e0214521 (2022).
Wong, A. & Kassen, R. Parallel evolution and local differentiation in quinolone resistance in Pseudomonas aeruginosa. Microbiol. (Read., Engl.) 157, 937–944 (2011).
Eklof, J. et al. Persistence and genetic adaptation of Pseudomonas aeruginosa in patients with chronic obstructive pulmonary disease. Clin. Microbiol. Infect. 28, 990–995 (2022).
Kolpen, M. et al. Bacterial biofilms predominate in both acute and chronic human lung infections. Thorax 77, 1015–1022 (2022).
Laborda, P., Martinez, J. L. & Hernando-Amado, S. Evolution of Habitat-Dependent Antibiotic Resistance in Pseudomonas aeruginosa. Microbiol. Spectrum 10, e0024722 (2022).
Stewart, P. S. et al. Search for a Shared Genetic or Biochemical Basis for Biofilm Tolerance to Antibiotics across Bacterial Species. Antimicrob. Agents Chemother. 66, e0002122 (2022).
Schroeder, J. W., Yeesin, P., Simmons, L. A. & Wang, J. D. Sources of spontaneous mutagenesis in bacteria. Crit. Rev. Biochem. Mol. Biol. 53, 29–48 (2018).
Oliver, A., Canton, R., Campo, P., Baquero, F. & Blazquez, J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288, 1251–1254 (2000).
Mena, A. et al. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J. Bacteriol. 190, 7910–7917 (2008).
Oliver, A. & Mena, A. Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin. Microbiol. Infect. 16, 798–808 (2010).
Driffield, K., Miller, K., Bostock, J. M., O’Neill, A. J. & Chopra, I. Increased mutability of Pseudomonas aeruginosa in biofilms. J. Antimicrobial Chemother. 61, 1053–1056 (2008).
Perez-Osorio, A. C., Williamson, K. S. & Franklin, M. J. Heterogeneous rpoS and rhlR mRNA levels and 16S rRNA/rDNA (rRNA gene) ratios within Pseudomonas aeruginosa biofilms, sampled by laser capture microdissection. J. Bacteriol. 192, 2991–3000 (2010).
Brown, M. R., Allison, D. G. & Gilbert, P. Resistance of bacterial biofilms to antibiotics: a growth-rate related effect? J. Antimicrobial Chemother. 22, 777–780 (1988).
Molin, S. & Tolker-Nielsen, T. Gene transfer occurs with enhanced efficiency in biofilms and induces enhanced stabilisation of the biofilm structure. Curr. Opin. Biotechnol. 14, 255–261 (2003).
Madsen, J. S., Burmolle, M., Hansen, L. H. & Sorensen, S. J. The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol. Med. Microbiol. 65, 183–195 (2012).
Krol, J. E. et al. Increased transfer of a multidrug resistance plasmid in Escherichia coli biofilms at the air-liquid interface. Appl. Environ. Microbiol. 77, 5079–5088 (2011).
Krol, J. E. et al. Invasion of E. coli biofilms by antibiotic resistance. Plasmids. Plasmid 70, 110–119 (2013).
Bender, N., Hennes, M. & Maier, B. Mobility of extracellular DNA within gonococcal colonies. Biofilm 4, 100078 (2022).
Merod, R. T. & Wuertz, S. Extracellular polymeric substance architecture influences natural genetic transformation of Acinetobacter baylyi in biofilms. Appl. Environ. Microbiol. 80, 7752–7757 (2014).
Acknowledgements
T.C. and T.B. acknowledge support from the Lundbeck Foundation. T.C. acknowledges support from FWO-Vlaanderen. M.B. was supported by the Special Research Fund of Ghent University.
Author information
Authors and Affiliations
Contributions
T.C. conceived the review. All authors contributed to the writing and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Coenye, T., Bové, M. & Bjarnsholt, T. Biofilm antimicrobial susceptibility through an experimental evolutionary lens. npj Biofilms Microbiomes 8, 82 (2022). https://doi.org/10.1038/s41522-022-00346-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-022-00346-4
This article is cited by
-
Synergistic use of anti-inflammatory ketorolac and gentamicin to target staphylococcal biofilms
Journal of Translational Medicine (2024)
-
Antibiofilm and antivirulence activities of laminarin-gold nanoparticles in standard and host-mimicking media
Applied Microbiology and Biotechnology (2024)
-
Microfluidics for adaptation of microorganisms to stress: design and application
Applied Microbiology and Biotechnology (2024)
-
Intermittent antibiotic treatment of bacterial biofilms favors the rapid evolution of resistance
Communications Biology (2023)
-
Biofilms preserve the transmissibility of a multi-drug resistance plasmid
npj Biofilms and Microbiomes (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
