
Abstract
The meticulous design of active sites and light absorbers holds the key to the development of high-performance photothermal catalysts for CO2 hydrogenation. Here, we report a nonmetallic plasmonic catalyst of Mo2N/MoO2-x nanosheets by integrating a localized surface plasmon resonance effect with two distinct types of active sites for CO2 hydrogenation. Leveraging the synergism of dual active sites, H2 and CO2 molecules can be simultaneously adsorbed and activated on N atom and O vacancy, respectively. Meanwhile, the plasmonic effect of this noble-metal-free catalyst signifies its promising ability to convert photon energy into localized heat. Consequently, Mo2N/MoO2-x nanosheets exhibit remarkable photothermal catalytic performance in reverse water-gas shift reaction. Under continuous full-spectrum light irradiation (3 W·cm−2) for a duration of 168 h, the nanosheets achieve a CO yield rate of 355 mmol·gcat−1·h−1 in a flow reactor with a selectivity exceeding 99%. This work offers valuable insights into the precise design of noble-metal-free active sites and the development of plasmonic catalysts for reducing carbon footprints.
Similar content being viewed by others


Introduction
The conversion of CO2 into high-value fuels and chemicals has emerged as a promising approach to address the global energy crisis and mitigate greenhouse gas emissions1,2,3. Among various CO2 utilization routes, the reverse water-gas shift (RWGS) reaction is a critical step to produce C1 feedstock for Fischer-Tropsch synthesis4. However, the inherent stability of CO2 necessitates substantial energy inputs for CO2 reduction in traditional thermal catalysis5,6. Photothermal catalysis offers a promising alternative to energy-intensive thermal catalysis by harnessing solar energy to actuate reactions7,8,9. This approach can drive catalytic reactions under mild conditions and reduce energy consumption effectively. The remarkable potential of photothermal catalysis in improving the efficiency of chemical processes and solar energy utilization will promote advancements in energy, sustainability and materials science10,11.
Photothermal catalysis toward practical applications requires the development of efficient catalysts by precisely designing and constructing catalytically active sites. Generally, the adsorption and activation of reactants on the active sites together with the light-harvesting process are crucial to photothermal catalysis. Noble-metal/oxide composite nanostructures have been commonly employed as effective constituents for photothermal CO2 hydrogenation12,13,14,15. In such systems, noble-metal nanoparticles with an localized surface plasmon resonance (LSPR) effect can enhance light utilization and generate hot carriers for chemical reactions10,13. Meanwhile, oxide components provide oxygen vacancies (Vo) to facilitate CO2 adsorption and activation16,17. For example, Au/TiO2 is one typical photocatalyst with LSPR effect18,19,20. Fan et al. proved that hot electrons generated by LSPR can promote the formation of oxygen vacancies in Au/TiO2 catalyst, facilitating the adsorption and activation of CO219. Sastre et al. demonstrated that plasmonic Au/TiO2 nanostructure could drive photothermal RWGS reaction with a CO generation rate of 429 mmol·gAu−1·h−1 (13.4 mmol·gcat−1·h−1) under 14.4 sun irradiation20. The photothermal catalysts composed of noble metals and oxides were usually synthesized via a multi-step process21. In addition to noble metal nanostructures, some semiconductor nanostructures also exhibit LSPR in visible-NIR regions. The adaptable surface of semiconductors can be engineered to provide plentiful active sites for photothermal reactions, such as the creation of oxygen vacancies in WO3 and MoO322,23,24. However, these catalysts still require the involvement of noble metals to activate H2 and boost their catalytic activity, which increases the intricacy of the catalyst synthesis process14,15,25. For instance, Mo-doped Pt/WOy nanostructures were formed using a multi-process method, which could achieve a CO production rate (3.1 mmol·gcat−1·h−1) at 140 °C benefiting from the incorporation of Pt nanoparticles14. To fulfil the requirements for application-oriented photothermal CO2 conversion, it is imperative to develop rational design strategies and feasible fabrication techniques for nonmetallic plasmonic catalysts along with high activity and durability under mild conditions.
Here, we report a Mo2N/MoO2-x nonmetallic plasmonic catalyst with two specific active sites for highly efficient photothermal CO2 hydrogenation. Implementing a facile one-step annealing treatment under ammonia, MoO3 precursor can be transformed into Mo2N with dispersed Vo-rich MoO2-x nanoclusters. The obtained Mo2N/MoO2-x catalyst shows excellent activity, selectivity and durability for photothermal RWGS reaction. In such a system, the optimal catalyst can produce CO with a yield rate as high as 355 mmol·gcat−1·h−1 (selectivity > 99.9%) for a continuous 168 h test in a flow reactor, indicating a great potential for practical application. Combining in-situ spectroscopic characterizations and density functional theory (DFT) calculations, we elucidate the in-depth mechanism of plasmonic Mo2N/MoO2-x catalyst for photothermal RWGS reaction. In the RWGS reaction, two types of active sites, i.e., N atoms and oxygen vacancies, work synergistically, enabling the adsorption and activation of H2 and CO2 molecules simultaneously. Owing to the synergism of dual active sites, the activation energy barrier can be reduced significantly. The strong LSPR effect in the catalyst also plays a vital role in promoting the RWGS reaction and boosting energy conversion efficiency by locally converting photon energy into thermal energy. The LSPR-induced photothermal catalysis utilizing nonmetallic plasmonic catalysts offers a viable approach to optimize localized heat management, catalytic property and cost performance toward practical applications7,21,26.
Results and discussion
Synthesis and characterization of Mo2N/MoO2-x catalyst
The Mo2N/MoO2-x nanosheets are synthesized by high-temperature ammoniation from MoO3 precursor (Fig. 1a). The resulting Mo2N/MoO2-x samples are designated as MNO-450, MNO-550 and MNO-650, respectively, corresponding to their annealing temperatures. Scanning electron microscopy (SEM), high-resolution transmission electron microscopy (HRTEM) and energy-dispersive X-ray spectroscopy (EDS) mapping analysis demonstrate the well-shaped nanosheet morphology, crystallographic structure and uniform element distribution of MNO-550 (Fig. 1b–d and Supplementary Fig. 1a). The lattice spacing of 0.24 nm can be assigned to the (111) plane of Mo2N, and that of 0.28 nm corresponds to the (101) plane of MoO2. Additionally, a considerable number of pore structures are discernible with an approximate diameter of ~3 nm, which is consistent to the result from Brunauer-Emmett-Teller (BET) measurements (Supplementary Fig. 1b, c). By varying the annealing temperature, we can tune the composition ratio of MoO2-x and Mo2N. X-ray diffraction (XRD) shows that the diffraction peaks for cubic Mo2N become more pronounced as the annealing temperature increases (Fig. 1e), indicating the positive correlation of Mo2N content with annealing temperature.

a The synthesis route of Mo2N/MoO2-x. b SEM image, c HRTEM image and (d) element distribution of MNO–550. e XRD patterns of MoO3 and Mo2N/MoO2-x samples at different annealing temperatures. f Mo 3p and N 1 s XPS spectra of Mo2N, MoO2-x and Mo2N/MoO2-x samples at different annealing temperatures. The orange, blue, green and magenta peaks are attributed to MoO3, MoO2, Mo 3p and N 1 s of Mo2N components, respectively. g XANES spectra of Mo2N, MoO2-x and MNO–550 at Mo K-edge.
To look into the electronic structures and chemical states of Mo2N/MoO2-x, X-ray photoelectron spectroscopy (XPS) and X-ray absorption near-edge structure spectroscopy (XANES) are employed to examine different Mo-based catalysts27,28. The coordination information for Mo atoms is provided by the Mo K-edge of MNO-550 in the Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra (Supplementary Fig. 1d), revealing the co-existence of Mo-N and Mo-O. XPS characterization shows that the intensity of N 1 s component increases with the annealing temperature (Fig. 1f), consistent with the XRD results. Meanwhile, there is a discernible drop in the intensity of Mo 3p component at higher binding energy (398.7 and 416.2 eV), indicating a gradual reduction of MoO2-x by ammonia when increasing the annealing temperature (Supplementary Table 1). The adsorption edge position of MNO-550 nanosheets is observed to locate between those of reference samples in the XANES spectra (Fig. 1g), suggesting that it possesses composite valence states. As a result, a series of molybdenum-based catalysts (MoO3, MoO2-x, and Mo2N/MoO2-x) with tunable component ratios of nitride and oxygen vacancy have been obtained by the one-step annealing method.
Photothermal CO2 hydrogenation performance of Mo2N/MoO2-x catalyst
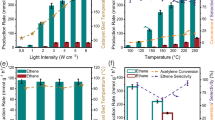
Prior to photothermal performance assessment, we first examine the ability of effectively utilizing light for photothermal catalysis. The UV-vis-NIR absorption spectra (Fig. 2a) show that Mo2N/MoO2-x exhibits an obvious broad-spectrum light adsorption capacity (250–1800 nm) in contrast to its counterparts (i.e., Mo2N and MoO2 components, which are reported as nonmetallic plasmonic materials)24,29. The enhanced LSPR performance of Mo2N/MoO2-x can be attributed to its distinctive nano-architecture. On the one hand, the broad grain size distribution ranging from 15 to 270 nm (Supplementary Fig. 2) can result in the overlapping and widening of plasmonic charaters30,31. On the other hand, a large number of interfaces exist between Mo2N and MoO2-x, and their interfacial interaction will influence the strength of LSPR. Consequently, a larger photocurrent response was observed (Supplementary Fig. 3) in Mo2N/MoO2-x nanosheet owing to the synergistic contribution of plasmonic Mo2N and the oxygen vacancies in MoO2-x c23,24. In addition to the effective promotion of photogenerated charge kinetics, it is noteworthy that the accumulation of hot electrons excited by LSPR effect at the interface can rapidly raise the surface temperature of MNO-550 up to 250 °C without external heat supply (Supplementary Fig. 4)32. The LSPR generated in the catalysts strengthens light utilization efficiency and converts photon energy into thermal energy locally, thereby facilitating catalytic reactions9. Finite-difference time-domain (FDTD) simulations were further conducted to confirm the existence of a pronounced LSPR effect in Mo2N/MoO2-x (Fig. 2b). An increase in the strength of local electric field is observed in Mo2N/MoO2-x, approximately 3 times higher than those of individual component materials33,34.

a UV-vis-NIR absorption spectra and (b) spatial distribution of electric fields enhanced by LSPR effect under the excitation wavelength of 497 nm of MoO2-x, Mo2N and Mo2N/MoO2-x. c The production rate and selectivity of CO evolution in a batch reactor by Mo-based catalysts under 3 W·cm−2 full-spectrum light irradiation. d Long-term stability of MNO-550 in a flow reactor under 3 W·cm−2 full-spectrum light irradiation with the flow rate of 20 SCCM (H2/CO2 1:1). e The CO2 conversion rate and CO selectivity of MNO–550 at various temperatures in thermal catalysis. f The comparison of catalytic performance with state-of-the-art catalysts for photothermal catalytic RWGS reaction, and the specific test parameters, such as light intensity and stability test time, are displayed in Supplementary Table 312,25,27,33,35,36,37,38,39,40,41,42,43,44. Serial numbers 1–16 represent Pt/HxMoO3-y24, CuNi/CeO228, Ni3N44, Au/TiO2 (DP)18, Au/TiO220, Black In2O336, Mo2NHx27, Fe3O437, Ni12P5/SiO238, Pd@Nb2O512, Cu/2D-Si39, Ni@SiO240, CF-Cu2O41, Ru/Mo2TiC242, Ga-Cu/CeO243 and TiN@TiO2@In2O3-x(OH)y33, respectively.
Upon acknowledging the LSPR effect, we proceed to investigate the photothermal catalytic properties of catalysts. The performance of Mo-based catalysts for RWGS reaction is evaluated by feeding mixed gas of CO2 and H2 (1:1) under 3.0 W·cm−2 full-spectrum illumination without external heat supply. The predominant product of these Mo-based catalysts is CO with ~99% selectivity. Notably, the catalytic efficiency of Mo2N/MoO2-x samples is substantially higher than that of MoO3 precursor, commercial MoO2-x, MoN and Mo2N when subjected to identical reaction conditions (Fig. 2c). Among the catalysts, MNO-550 obtains the optimal photothermal RWGS activity in a batch reactor attributing to its optimized synergic component (Supplementary Figs. 5, 6). The CO yield rate of MNO-550 is 389 mmol·gcat−1·h−1 (selectivity > 99%) for 20 min, which is 208 times and 130 times higher than that of MoO2-x and Mo2N, respectively. In contrast, without in-situ constructed interfacial interactions of Mo2N and MoO2-x, the synergistic enhancement of catalytic activity cannot be observed in the mechanically mixed samples (Supplementary Fig. 7).
Impressively, during continuous testing in a flow reactor with the flow rate of 20 SCCM (mL·min−1) (Supplementary Fig. 8), MNO-550 remains highly active and stable for at least one week, achieving an average CO production rate at 355 mmol·gcat−1·h−1 and 13.1% CO2 conversion rate (selectivity > 99.9%) (Fig. 2d and Supplementary Fig. 9). The CO generation rate has merely decreased by 12% after 190 h. In successive light on and off conditions, the catalytic performance of MNO-550 also remains steady (Supplementary Fig. 10). After a long period of catalytic process, the morphology and phase of MNO-550 remain consistent (Supplementary Fig. 11), which proves the stability of this catalyst. As previously mentioned, LSPR-induced photothermal effect elevates surface temperature of MNO-550 up to 250 °C (Supplementary Fig. 4). In contrast, to attain the same CO2 conversion rate in thermal catalysis, it would require a dramatically higher reaction temperature at ~500 °C for MNO-550 (Fig. 2e and Supplementary Fig. 12a) even at lower flow velocity. The reduced temperature requirement for highly efficient RWGS reaction in our system also signifies that the introduction of LSPR effect not only induces a localized photothermal effect, but also promotes the charge dynamics to actuate the reactions. Meanwhile, the surface temperature and CO generation rate of the catalyst are positively correlated with light intensity (Supplementary Fig. 12b), which indicates the excellent photothermal conversion ability of Mo2N/MoO2-x nanosheets7. In addition, the energy conversion efficiency of photothermal catalysis in our system has a significant advantage over thermal catalysis for RWGS reaction (Supplementary Table 2). Specifically, the conversion efficiency of thermal energy to chemical energy by MNO-550 in photothermal catalysis is 4 ~ 5 times higher than that in thermal catalysis, while the required reaction temperature can be reduced by 230 ~ 300 °C in photothermal catalysis compared to thermal catalysis. Consequently, our Mo2N/MoO2-x nanosheets exhibit excellent performance in terms of both stability and activity even under mild conditions, as illustrated in Fig. 2f, in comparison with the state-of-the-art oxide or nitride catalysts for photothermal catalytic RWGS reaction (Supplementary Table 3)12,25,27,33,35,36,37,38,39,40,41,42,43,44.
Spectroscopic characterizations for catalytic mechanism
To elucidate the catalytic mechanism, comprehensive characterizations are performed to gain insight into the origin of remarkably enhanced photothermal driven RWGS activity on Mo2N/MoO2-x. As previously reported, Vo often acts as the active site for the adsorption and activation of CO223,25,36. We thus examine whether the high-temperature ammoniation annealing can introduce oxygen vacancies in our catalysts. The electron paramagnetic resonance (EPR) spectra in Fig. 3a show a response at g = 2.0003 indicating molybdenum-oxygen vacancies embedded in the lattice45. Specifically, the strongest signal is detected from MoO2-x, while MNO-550 exhibits the signal implying its moderate number of oxygen vacancies among the three Mo2N/MoO2-x samples. In addition, XPS can also be applied as an effective technique to investigate Vo25,46. The intensities of peaks at 531.5 eV attributed to Vo exhibit the same variation tendency as EPR spectra (Fig. 3b). It turns out that the concentration of oxygen vacancies is determined by the annealing temperature. As such, we can modulate the proportion of MoO2-x in Mo2N/MoO2-x by adjusting the annealing temperature.

a EPR spectra and (b, c) O 1 s XPS spectra of different samples (b) before and (c) after the reaction. d, e In-situ NAP-XPS spectra of (d) Mo 3p & N 1 s and (e) O 1 s and C 1 s for MNO–550 under full-spectrum light irradiation in 0.3 mbar CO2 + 0.3 mbar H2. f In-situ DRIFTS measurements of MNO–550 under full-spectrum light irradiation in a mixed atmosphere of 50% CO2 and 50% H2. In Fig. 3b, c, e, the orange, purple, blue and brown peaks are attributed to gaseous CO2, adsorbed oxygen species, oxygen vacancy and metal oxygen, respectively. In Fig. 3d, the orange, blue, green and magenta peaks are attributed to MoO3, MoO2, Mo 3p and N 1 s of Mo2N components, respectively.
We also look into the possible changes of catalysts along with photothermal RWGS reactions. After the reactions, a new peak emerges at 533.3 eV in the XPS spectra of MoO2-x, MNO-450 and MNO-550 (Fig. 3c), which can be assigned to adsorbed oxygen species25. Impressively, these three samples still possess adequate oxygen vacancies after the reactions. The C 1 s spectra of Mo-based catalysts before and after the reactions are also compared (Supplementary Fig. 13), suggesting the strong adsorption of carbon oxide species on the oxygen vacancies45,47. Meanwhile, temperature-programmed desorption (TPD) profiles combined with in-situ XPS in CO2 atmosphere indicate that MNO-550 exhibits a moderate adsorption capacity among the three Mo2N/MoO2-x samples (Supplementary Figs. 14 and 15), which is consistent with XPS results. A positive correlation can be observed between the amount of adsorbed CO2 species and the concentration of oxygen vacancies. This correlation suggests that oxygen vacancies primarily function as the active sites for CO2 adsorption and activation on the Mo2N/MoO2-x catalyst surface.
To gain a comprehensive understanding of the photothermal driven CO2 reduction reaction process, in-situ spectroscopic characterizations are employed to investigate the dynamic change of reaction intermediates and the light-induced charge transfer path on the catalyst surface. In-situ EPR spectroscopy results show that the typical signal at g = 2.0003 increases significantly under illumination (Supplementary Fig. 16). This feature proves the presence of photogenerated oxygen vacancies during RWGS reaction, which is beneficial for the catalysis reaction process. In-situ near-ambient-pressure XPS (NAP-XPS) is further performed in the reaction atmosphere (Fig. 3d, e). When the surface of MNO-550 is exposed to 0.3 mbar CO2 and 0.3 mbar H2 without illumination, an identifiable C 1 s peak emerges at 285.6 eV. This new peak is attributed to the C = O configuration, considered as a surface reaction intermediate. Simultaneously, two O 1 s peaks at 533.3 and 537.2 eV, ascribed to the adsorbed C = O species and gaseous CO2, respectively, can also be recognized47. Upon illumination, the N 1 s peak encounters a shift toward lower binding energy by 0.2 eV, while the Mo 3p doublet peaks maintain their original position, indicating the migration of photogenerated electrons onto surface N sites27. At the same time, the intensity of C 1 s peak at 285.6 eV (C = O peak) increases considerably (Fig. 3e), suggesting that the activation of CO2 is promoted by the strengthened local electric field under light illumination47. When light is turned off, the N 1 s peak returns to the pristine position due to the consumption of photogenerated electrons by reactions, and the strength of C = O peak decreases illustrating the decline of reaction rate without illumination.
In-situ diffuse reflection Fourier transform infrared spectroscopy (DRIFTS) is also conducted to detect the evolution of surface species. There are no evident peaks observed within the designated measurement range in a mixed atmosphere of CO2 and H2 during the initial 20 min period without illumination (Fig. 3f). Once exposed to full spectrum illumination, a series of peaks appear promptly and reach a consistent state within 20 minutes. The peaks at 3566, 3550 and 3523 cm−1 are attributed to the vibrations of NHx species attached to Mo atoms, indicating the heterolysis of H2 by Mo-N sites27,48. The evident change of kinetic isotope effects (KIE) value reflects the transformation of hydrogen-involved behavior under different driving conditions for RWGS reaction, which proves the role of photothermal catalysis to promote H2 activation (Supplementary Fig. 17)27. Moreover, the intermediates for CO2 hydrogenation can be identified under illumination, including *COOH (1287 and 1362 cm−1), *CO (2047 and 2017 cm−1), *CO3 (1375 and 1322 cm−1) and *CO2 (1339 cm−1). In addition, the sharply increasing signal of *COOH intermediates on Mo2N/MoO2-x catalyst is also observed through time-of-flight secondary ion mass spectroscopy (TOF-SIMS) following the catalytic reaction (Supplementary Fig. 18). The observed DRIFTS signals of these significant intermediates indicate the pathway of CO2 activation, which exhibits a rise in intensity with prolonged illumination time and aligns well with the findings of NAP-XPS results49,50. Based on the above spectroscopic characterizations, we propose that oxygen vacancies act as the active sites for CO2 activation. In the meantime, the Mo-N sites, which receive photogenerated electrons, have the ability to rapidly dissociate H2 to produce active H atoms for CO2 hydrogeneration due to the LSPR-induced surface electric field.
Theoretical calculation for catalytic mechanism
With the information gleaned from spectroscopic characterizations, we further conduct DFT calculations to study the adsorption and activation process of CO2 by Mo2N/MoO2-x. Mo2N and MoO2-x are selected as contrastive samples to investigate the synergistic effect between two separate active sites. The calculated Gibbs free energy of H2 adsorption over Mo2N/MoO2-x is −2.18 eV (Fig. 4a), suggesting that H2 molecules prefer to be adsorbed on the surface of Mo2N/MoO2-x due to the synergistic effect at the interface51. It should be noted that H2 molecules exhibit considerable activity on the surfaces of both Mo2N/MoO2-x and Mo2N (Supplementary Fig. 19); however, the H-H bond should be dissociated prior to the adsorption of H atoms onto the surface. As a result, the H atoms exhibit enhanced migratory properties over Mo2N/MoO2-x, thereby having a greater propensity to establish chemical interactions with adsorbed CO2 and other intermediates. On the other hand, the free energies of CO2 adsorption over three model samples are −1.71, −1.31 and −2.52 eV, respectively (Fig. 4b). Interestingly, the adsorption strength of CO2 on Mo2N/MoO2-x lies at a moderate level between Mo2N and MoO2-x. This result agrees with the Sabatier’s rule, indicating that Mo2N/MoO2-x can serve as an efficacious catalyst52. The reaction free energy and the corresponding structural configuration of each intermediate are also simulated to illustrate the RWGS reaction mechanism over Mo-based catalysts. Figure 4c shows that the free energies for CO2 protonation to form *COOH on Mo2N/MoO2-x, Mo2N and MoO2-x are −1.80, −1.16 and −2.14 eV, respectively. Based on these simulations, we infer that MoO2-x component is prone to drive catalytic CO2 reduction and promote the formation of *COOH intermediate, which aligns with the findings from NAP-XPS. For the subsequent fundamental step, the free energies of *COOH dehydroxylation to form *CO on Mo2N/MoO2-x, Mo2N and MoO2-x are −4.12, −0.62 and −1.79 eV, respectively (Supplementary Fig. 20 and Table 4). The results demonstrate that *CO prefers to evolve on Mo2N/MoO2-x catalyst, consistent with its impressive catalytic performance for photothermal RWGS reaction. Taken together, the Mo2N component in Mo2N/MoO2-x is active for H2 dissociation, and CO2 reduction is driven by the MoO2-x component. The uniform distribution of Mo2N and MoO2-x and their sufficient combination provide adequate active sites for the synergistic activation of reactant molecules. Overall, the dual active sites of Mo2N/MoO2-x catalyst exhibit a synergistic effect, together with its excellent LSPR effect, hence rendering Mo2N/MoO2-x a promising photothermal catalyst for CO2 hydrogenation.

a Calculated H2 and (b) CO2 adsorption energy, (c) free energy of Mo2N/MoO2-x, MoO2-x and Mo2N. The purple, red, grey, brown and champagne balls represent Mo, O, N, C, and H atoms, respectively.
In summary, we report the successful construction of a nonmetallic plasmonic catalyst (Mo2N/MoO2-x) with dual active sites for actuating the RWGS reaction. The comprehensive characterizations and theoretical calculations demonstrate that H2 and CO2 can be adsorbed and activated on the N sites and oxygen vacancies simultaneously. The synergistic effect of these active sites plays a crucial role in a significant reduction of the reaction energy barrier. Meanwhile, the existence of LSPR effect in the noble-metal-free catalyst efficiently enhances the conversion of photon energy to thermal energy and optimizes the localized energy regulation, further promoting the activation of reactant molecules and facilitating the reaction. These factors result in the exceptional catalytic activity, selectivity and durability as well as energy conversion efficiency of Mo2N/MoO2-x for photothermal catalysis in the RWGS reaction. This work employs a practicable design strategy to establish tunable synergistic sites with LSPR effect, and provides methodological support for understanding the mechanism of photothermal catalytic CO2 hydrogenation. The energy conversion efficiency of photothermal catalysis demonstrated in our system, which has a significant advantage over thermal catalysis, will aid in advancing the development of innovative photothermal catalysts.
Methods
Material synthesis
To obtain MoO3 nanosheets precursor, 576 mg Mo powder was dissolved in 80 mL mixed solution containing 70 mL ethanol and 10 mL H2O2. After magnetic stirring, the transparent solution was transferred into a sealed Teflon-lined autoclave under 160 °C for 24 h. The product was washed with deionized water and ethanol several times, and dried in a vacuum oven for 12 h. The as-prepared MoO3 precursor was annealed in an NH3 atmosphere with a flow rate of 15 SCCM (standard cubic centimeters per minute, mL·min−1) for 6 h. The annealing temperatures were 450, 500, 550, 600, and 650 °C, respectively, to prepare Mo2N/MoO2-x nanosheets with different proportions of Mo2N and MoO2-x. To create oxygen vacancies in MoO2 (MoO2-x), commercial MoO2 was thermally treated at 550 °C for 5 h under the Ar/H2 (5%) mixture gas.
Catalyst characterizations
Powder X-ray diffraction (XRD) patterns were recorded on a Bruker AXS D8 Advance X-ray diffractometer with a Cu Ka radiation target (λ = 0.154178 nm). Scanning electron microscopy (SEM) was conducted on Hitachi S-4800 scanning electron microscope at 5 kV. High-resolution transmission electron microscopy (HRTEM) images, energy-dispersive X-ray spectroscopy (EDS) mapping profiles and selected area electron diffraction results were collected on a JEOL-2100F system. Atomic-level high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and the corresponding STEM-EDS elemental mapping profiles were taken on an FEI Titan Themis Z 3.1 equipped with a SCOR spherical aberration corrector and a monochromator. Measurements of time-of-flight secondary ion mass spectroscopy (TOF-SIMS) were conducted on a TOF-SIMS 5–100 instrument (ION-TOF GmbH). UV-vis-IR diffuse reflectance spectra were measured by a Shimadzu SolidSpec-3700 spectrophotometer in the spectral region of 200–2500 nm. A JEOL JES-FA200 electron spin resonance spectrometer was used to collect the electron paramagnetic resonance (EPR) spectra at room temperature (9.062 GHz)53.
Gas-adsorption analysis
Temperature-programmed desorption (TPD) measurements of reactant gases were collected by a Micromeritics AutoChem II 2920 apparatus. Catalyst powders (70 mg) were pretreated at 200 °C in Ar atmosphere for 1 h and then cooled to room temperature naturally. In order to remove other physisorbed molecules, the sample was heated at 50 °C in H2/Ar (10%) mixture for 1 h and then sluiced with Ar flow. Finally, the H2 desorption was measured in Ar atmosphere (30SCCM) in the temperature range from 50 °C to 600 °C with the heating rate of 10 °C min−1. The CO2 desorption was measured through the same apparatus and the similar procedures54.
In-situ NAP-XPS measurements
In-situ near-ambient-pressure X-ray photoelectron spectroscopy (NAP-XPS) measurements were performed on a SPECS NAP-XPS instrument, where C 1 s, O 1 s, N 1 s and Mo 3p spectra were collected under UHV and 0.6 mbar CO2 + H2 (1:1) conditions. The catalyst powder was pressed onto copper foam, and then the sample was fixed onto the XPS sample holder by tantalum strips55. A 300 W Xenon lamp served as the white light source and was fixed outside the UHV chamber to illuminate the sample via the quartz window.
In-situ DRIFTS measurements
In-situ diffuse reflection Fourier transform infrared spectroscopy (DRIFTS) measurements were performed through a Bruker IFS 66 v Fourier transform spectrometer equipped with a Harrick diffuse reflectance accessory at the Infrared Spectroscopy and Microspectroscopy Endstation (BL01B) of NSRL. Each spectrum was recorded by averaging 256 scans at a resolution of 4 cm−1. The catalysts were placed in an infrared in-situ chamber sealed with ZnSe windows, which was specifically designed to examine highly scattered powder samples in diffuse reflection mode. Then the chamber was purged with Ar flow for 30 min. The spectrum was collected as a background spectrum56. During the in-situ characterization, CO2/H2 mixture gas was continually introduced into the chamber at 250 °C under visible light irradiation conditions.
Performance evaluation of CO2 hydrogenation
5 mg of catalyst was put into a quartz batch tube reactor after being loaded on a glass fiber. The quartz tube was sealed up after fulling of CO2/H2 mixed gas with a volume ratio of 1:1. Then the reactor was illuminated with 300 W Xe lamp (Perfect Light PLS-SEX 300D) at a specific power density (3 W·cm−2) for 20 min. The products were detected by gas chromatography (GC-2014AF, Shimadzu) equipped with a flame ionization detector (FID). The long-term catalytic performance testing was conducted in the flow reactor under the illumination of 3 W·cm−2, and the flow rates of hydrogen and CO2 were both 10 SCCM. A K-type micro thermocouple was used to monitor the reaction temperature in real time (Supplementary Figs. 21 and 22). CO2 hydrogenation reactions in thermal catalysis were tested using a homemade fixed-bed micro-reactor. 20 mg catalysts were loaded in a quartz tube with an inner diameter of 4 mm. The reactant gas consists of 24% CO2 and 24% H2 (volume ratio), balanced with Ar, and the flow rate was 20 SCCM. The effluent gas was online analyzed by Agilent GC6890N equipped with a TDX-1 column and thermal conductivity detector (TCD).
Kinetic isotope effect (KIE) measurements
The KIE tests followed the same procedures as “Performance evaluation of CO2 hydrogenation”, replacing H2 with D2. The KIE value refers to the ratio of the reaction rate constant (k) in the H2 atmosphere to that in the D2 atmosphere under the same reaction conditions (kH/kD). As mentioned above, the KIE value can be directly replaced by the ratio of the CO formation rates (rH/rD) due to the high concentration of CO2 and H2/D2.
Photoelectrochemical measurements
All photoelectrochemical measurements were conducted using an electrochemical workstation (CHI 760E, Shanghai Chenhua, China) in a three-electrode system. A 300 W Xe lamp with a power density of 100 mW·cm−2 served as the light source during the measurements. For the photoelectrode, the prepared samples were drop-coated onto FTO glass. The counter electrode was a Pt foil, while an Ag/AgCl electrode functioned as the reference electrode. These three electrodes were carefully inserted into a quartz cell, which was pre-filled with a 0.5 M Na2SO4 electrolyte solution (pH = 6.6).
Computational details
All the calculations were performed in the framework of the density functional theory with the projector-augmented plane-wave method, as implemented in the Vienna ab initio simulation package57. The generalized gradient approximation proposed by Perdew, Burke, and Ernzerhof was selected for the exchange-correlation potential58. The long-range van der Waals interaction was described by the DFT-D3 approach59. The cut-off energy for the plane wave was set to 400 eV. The energy criterion was set to 10−6 eV in the iterative solution of the Kohn-Sham equation. A vacuum layer of 15 Å was added perpendicular to the sheet to avoid artificial interaction between periodic images60. The k-mesh used in the calculations was chosen according to the size of the structures. 1 × 1 × 1, 3 × 3 × 1 and 2 × 4 × 1 k-meshes were used in the calculations of Mo2N/MoO2-x, Mo2N and MoO2-x, respectively. All the structures were relaxed until the residual forces on the atoms had declined to less than 0.03 eV·Å−1.
The absorption energies of H2/CO2 on different catalysts were evaluated by the following equation:
Where \({E}_{{total}}\), \({E}_{{slab}}\), and \({E}_{{mol}}\) represent to the electron’s energy of the H2/CO2 absorbed on the surface of catalysts, the catalysts, and H2/CO2, respectively. Besides, the stability of all the structures with H2/CO2/intermediates absorbed on the catalysts were evaluated by the frequency calculations.
Data availability
All data that support the findings in this paper are available within the article and its Supplementary Information or are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Pacala, S. & Socolow, R. Stabilization wedges: Solving the climate problem for the next 50 years with current technologies. Science 305, 968–972 (2004).
Bushuyev, O. S. et al. What should we make with CO2 and how can we make It? Joule 2, 825–832 (2018).
Zhu, Z., Tang, R., Li, C., An, X. & He, L. Promises of plasmonic antenna-reactor systems in gas-phase CO2 photocatalysis. Adv. Sci. 10, 2302568 (2023).
Velty, A. & Corma, A. Advanced zeolite and ordered mesoporous silica-based catalysts for the conversion of CO2 to chemicals and fuels. Chem. Soc. Rev. 52, 1773–1946 (2023).
Ghoussoub, M., Xia, M., Duchesne, P. N., Segal, D. & Ozin, G. Principles of photothermal gas-phase heterogeneous CO2 catalysis. Energ. Environ. Sci. 12, 1122–1142 (2019).
Zhou, J., Liu, H. & Wang, H. Photothermal catalysis for CO2 conversion. Chin. Chem. Lett. 34, 107420 (2023).
Shao, T. et al. A stacked plasmonic metamaterial with strong localized electric field enables highly efficient broadband light-driven CO2 hydrogenation. Adv. Mater. 34, 2202367 (2022).
Gao, W., Li, Y., Xiao, D. & Ma, D. Advances in photothermal conversion of carbon dioxide to solar fuels. J. Energy Chem. 83, 62–78 (2023).
Dong, Y., Hu, C., Xiong, H., Long, R. & Xiong, Y. Plasmonic catalysis: New opportunity for selective chemical bond evolution. ACS Catal 13, 6730–6743 (2023).
Lorber, K. & Djinovic, P. Accelerating photo-thermal CO2 reduction to CO, CH4 or methanol over metal/oxide semiconductor catalysts. iScience 25, 104107 (2022).
Song, C., Wang, Z., Yin, Z., Xiao, D. & Ma, D. Principles and applications of photothermal catalysis. Chem Catal 2, 52–83 (2022).
Jia, J. et al. Visible and near-infrared photothermal catalyzed hydrogenation of gaseous CO2 over nanostructured Pd@Nb2O5. Adv. Sci. 3, 2198–3844 (2016).
Ge, H., Kuwahara, Y., Kusu, K., Bian, Z. & Yamashita, H. Ru/HxMoO3-y with plasmonic effect for boosting photothermal catalytic CO2 methanation. Appl. Catal. B 317, 121734 (2022).
Ge, H., Kuwahara, Y., Kusu, K., Kobayashi, H. & Yamashita, H. Enhanced visible-NIR absorption and oxygen vacancy generation of Pt/HxMoWOy by H-spillover to facilitate photothermal catalytic CO2 hydrogenation. J. Mater. Chem. A 10, 10854–10864 (2022).
Li, Y. et al. Pd@HyWO3-x nanowires efficiently catalyze the CO2 heterogeneous reduction reaction with a pronounced light effect. ACS Appl. Mater. Interfaces 11, 5610–5615 (2019).
Li, Y. et al. Predominance of subsurface and bulk oxygen vacancies in reduced manganese oxide. J. Phys. Chem. C 125, 7990–7998 (2021).
Lin, L. et al. Reversing sintering effect of Ni particles on gamma-Mo2N via strong metal-support interaction. Nat. Commun. 12, 6978 (2021).
Upadhye, A. A. et al. Plasmon-enhanced reverse water gas shift reaction over oxide-supported Au catalysts. Catal. Sci. Technol. 5, 2590–2601 (2015).
Wang, K. et al. Operando DRIFTS-MS investigation on plasmon-thermal coupling mechanism of CO2 hydrogenation on Au/TiO2: The enhanced generation of oxygen vacancies. Appl. Catal. B 296, 120341 (2021).
Molina, P. M. et al. Low temperature sunlight-powered reduction of CO2 to CO using a plasmonic Au/TiO2 nanocatalyst. Chem. Cat. Chem. 13, 4507–4513 (2021).
Sayed, M., Yu, J., Liu, G. & Jaroniec, M. Non-noble plasmonic metal-based photocatalysts. Chem. Rev. 122, 10484–10537 (2022).
Lu, C. et al. Full-spectrum nonmetallic plasmonic carriers for efficient isopropanol dehydration. Nat. Commun. 13, 6984 (2022).
Wu, X. et al. Identification of the active sites on metallic MoO2-x nano-sea-urchin for atmospheric CO2 photoreduction under UV, visible, and near-infrared light illumination. Angew. Chem. Int. Ed. 62, e202213124 (2022).
Liu, X. et al. Oxygen vacancy-regulated metallic semiconductor MoO2 nanobelt photoelectron and hot electron self-coupling for photocatalytic CO2 reduction in pure water. Appl. Catal. B 319, 121887 (2022).
Ge, H., Kuwahara, Y., Kusu, K. & Yamashita, H. Plasmon-induced catalytic CO2 hydrogenation by a nano-sheet Pt/HxMoO3-y hybrid with abundant surface oxygen vacancies. J. Mater. Chem. A 9, 13898–13907 (2021).
Ma, J. et al. Rational design of N-doped carbon coated cobalt nanoparticles for highly efficient and durable photothermal CO2 conversion. Adv. Mater. 35, e2302537 (2023).
Sun, M. et al. Photoinduced H2 heterolysis to form Mo2NHx active species for CO2 reduction. ACS Energy Lett. 6, 2024–2029 (2021).
Kim, G. et al. Growth and characterization of chloronitroaniline crystals for optical parametric oscillators I. XPS study of Mo-based compounds. Appl. Surf. Sci. 152, 35–43 (1999).
Song, X. et al. Selective preparation of Mo2N and MoN with high surface area for flexible SERS sensing. Nano Lett. 21, 4410–4414 (2021).
Chang, C. et al. Highly plasmonic titanium nitride by room-temperature sputtering. Sci. Rep. 9, 15287 (2019).
Rej, S. et al. Colloidal titanium nitride nanobars for broadband inexpensive plasmonics and photochemistry from visible to mid-IR wavelengths. Nano Energy 104, 107989 (2022).
Guo, Y. et al. Efficient interfacial electron transfer induced by hollow-structured ZnIn2S4 for extending hot electron lifetimes. Energ. Environ. Sci. 16, 3462–3473 (2023).
Nguyen, N. T. et al. Plasmonic titanium nitride facilitates indium oxide CO2 photocatalysis. Small 16, 2005754 (2020).
Zhang, H. et al. Surface-plasmon-enhanced photodriven CO2 reduction catalyzed by metal-organic-framework-derived iron nanoparticles encapsulated by ultrathin carbon layers. Adv. Mater. 28, 3703–3710 (2016).
Yue, X. et al. Visible light-regulated thermal catalytic selectivity induced by nonthermal effects over CuNi/CeO2. Chem. Eng. J. 458, 141491 (2023).
Wang, L. et al. Black indium oxide a photothermal CO2 hydrogenation catalyst. Nat. Commun. 11, 2432 (2020).
Song, C. et al. Photothermal conversion of CO2 with tunable selectivity using Fe-based catalysts: From oxide to carbide. ACS Catal. 10, 10364–10374 (2020).
Xu, Y. et al. High-performance light-driven heterogeneous CO2 catalysis with near-unity selectivity on metal phosphides. Nat. Commun. 11, 5149 (2020).
Su, Y. et al. High surface area siloxene for photothermal and electrochemical catalysis. Nanoscale 15, 154–161 (2022).
Wang, S. et al. Grave-to-cradle upcycling of Ni from electroplating wastewater to photothermal CO2 catalysis. Nat. Commun. 13, 5305 (2022).
Wan, L. et al. Cu2O nanocubes with mixed oxidation-state facets for (photo)catalytic hydrogenation of carbon dioxide. Nat. Catal. 2, 889–898 (2019).
Wu, Z. et al. Mo2TiC2 MXene-supported Ru clusters for efficient photothermal reverse water-gas shift. ACS Nano. 17, 1550–1559 (2023).
Deng, B., Song, H., Peng, K., Li, Q. & Ye, J. Metal-organic framework-derived Ga-Cu/CeO2 catalyst for highly efficient photothermal catalytic CO2 reduction. Appl. Catal. B 298, 120519 (2021).
Singh, S. et al. Surface plasmon-enhanced photo-driven CO2 hydrogenation by hydroxy-terminated nickel nitride nanosheets. Nat. Commun. 14, 2551 (2023).
He, Y. et al. Operando identification of dynamic photoexcited oxygen vacancies as true catalytic active sites. ACS Catal. 13, 191–203 (2023).
Jin, B., Ye, X., Zhong, H., Jin, F. & Hu, Y. Enhanced photocatalytic CO2 hydrogenation with wide-spectrum utilization over black TiO2 supported catalyst. Chin. Chem. Lett. 33, 812–816 (2022).
Hu, C. et al. Near-infrared-featured broadband CO2 reduction with water to hydrocarbons by surface plasmon. Nat. Commun. 14, 221 (2023).
Zhao, B. et al. Unveiling the activity origin of iron nitride as catalytic material for efficient hydrogenation of CO2 to C2+ hydrocarbons. Angew. Chem. Int. Ed. 60, 4496–4500 (2021).
Qi, Y. et al. Fabrication of black In2O3 with dense oxygen vacancy through dual functional carbon doping for enhancing photothermal CO2 hydrogenation. Adv. Funct. Mater. 31, 2100908 (2021).
Yan, T. et al. Bismuth atom tailoring of indium oxide surface frustrated Lewis pairs boosts heterogeneous CO2 photocatalytic hydrogenation. Nat. Commun. 11, 6095 (2020).
Wang, Y. et al. Exploring the ternary interactions in Cu-ZnO-ZrO2 catalysts for efficient CO2 hydrogenation to methanol. Nat. Commun. 10, 1116 (2019).
Norskov, J. K., Bligaard, T., Rossmeisl, J. & Christensen, C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37–46 (2009).
Ma, J. et al. Cu and Si co-doping on TiO2 nanosheets to modulate reactive oxygen species for efficient photocatalytic methane conversion. Nanoscale Horiz. 8, 63–68 (2022).
Ma, J. et al. Tuning the selectivity of photothermal CO2 hydrogenation through photo-induced interaction between Ni nanoparticles and TiO2. Appl. Catal. B 345, 123600 (2024).
Li, X., Yu, J., Jaroniec, M. & Chen, X. Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem. Rev. 119, 3962–4179 (2019).
Ma, J. et al. Sustainable methane utilization technology via photocatalytic halogenation with alkali halides. Nat. Commun. 14, 1410 (2023).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Xiang, L. et al. Constructing a 1T-MoS2/Ni3S4 heterostructure to balance water dissociation and hydroxyl desorption for efficient hydrogen evolution. Catal. Sci. Technol. 13, 3901–3909 (2023).
Acknowledgements
This work was financially supported in part by the National Key R&D Program of China (2020YFA0406103), NSFC (U23A2091, 22279128, 22122506, 22075267), Jiangsu Funding Program for Excellent Postdoctoral Talent, Youth Foundation of Jiangsu Province (BK20220290), Gusu Innovation and Entrepreneurship Leading Talents Program (ZXL2022386), and Science and Technology Program of Suzhou (SWY2022003). The in-situ DRIFTS measurements were performed at beamline BL01B in the NSRL. The authors thank the support from USTC Center for Micro- and Nanoscale Research and Fabrication. This work was partially carried out at the Instruments Center for Physical Science, University of Science and Technology of China.
Author information
Authors and Affiliations
Contributions
D.L. and Y.X. supervised the project. X.W., Y.L., and Y.C. conceived and designed the experiments. X.W. performed the key experiments and analyzed the results. X.W., Y.L., Y.L., J.M., Y.L., Y.C., R.L., N.L., and K.L. assisted to carry out the in-situ EPR, DRIFTS, NAP-XPS characterization, and DFT simulations. E.Z., Y.G., Y.Z. assisted to perform the chemical synthesis. R.T.L. assisted to test the thermal catalytic performance. J.M., Y.L., D.L., and Y.X. co-wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Vivek Polshettiwar and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wan, X., Li, Y., Chen, Y. et al. A nonmetallic plasmonic catalyst for photothermal CO2 flow conversion with high activity, selectivity and durability. Nat Commun 15, 1273 (2024). https://doi.org/10.1038/s41467-024-45516-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-45516-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
