
Abstract
Renewable energy-based electrocatalytic oxidation of organic nucleophiles (e.g.methanol, urea, and amine) are more thermodynamically favourable and, economically attractive to replace conventional pure water electrooxidation in electrolyser to produce hydrogen. However, it is challenging due to the competitive oxygen evolution reaction under a high current density (e.g., >300 mA cm−2), which reduces the anode electrocatalyst’s activity and stability. Herein, taking lower energy cost urea electrooxidation reaction as the model reaction, we developed oxyanion-engineered Nickel catalysts to inhibit competing oxygen evolution reaction during urea oxidation reaction, achieving an ultrahigh 323.4 mA cm−2 current density at 1.65 V with 99.3 ± 0.4% selectivity of N-products. In situ spectra studies reveal that such in situ generated oxyanions not only inhibit OH− adsorption and guarantee high coverage of urea reactant on active sites to avoid oxygen evolution reaction, but also accelerate urea’s C − N bond cleavage to form CNO − intermediates for facilitating urea oxidation reaction. Accordingly, a comprehensive mechanism for competitive adsorption behaviour between OH− and urea to boost urea electrooxidation and dynamic change of Ni active sites during urea oxidation reaction was proposed. This work presents a feasible route for high-efficiency urea electrooxidation reaction and even various electrooxidation reactions in practical applications.
Similar content being viewed by others

Introduction
Abundant urea in wastewater and human urine is a practically promising alternative energy carrier and (indirect) hydrogen (H2) storage chemical1,2,3,4. The six-electron transferred electrocatalytic urea oxidation reaction (UOR) is an important anode half-reaction in energy-related applications including, direct urea /urine fuel cells (DUFC)5,6,7 and urea-assistant water electrolysers8,9,10. The thermodynamic potential for UOR coupled with cathode hydrogen evolution reaction is theoretically 0.37 V (vs. reversible hydrogen electrode, RHE), a value significantly less than that of 1.23 V for water electrolysis8,10. Therefore, UOR has a significant potential to reduce voltage input for H2 production by replacing traditional oxygen evolution reaction (OER) in the anode of a water electrolyser. Recently, nickel (Ni)-based electrocatalysts have been reported for UOR11,12,13,14,15. In most cases, these catalysts undergo surface reconstruction into nickel oxyhydroxide (NiOOH) species during UOR because of the anodic potential and electrolyte’s hydroxyl anions, which serve as the active species for UOR11,14,16. However, the derived NiOOH are also highly active species for OER, an undesired side reaction toward UOR17,18. More importantly, OER will over-oxidize NiOOH to high-valent nickel19,20, thereby degrading the electrode stability and reducing further UOR efficiency. This competition between UOR and OER is more significant in practical UOR-related applications, especially with high operating potential and large current density devices3,14,21.
The competition between UOR and OER originates from the adsorption of OH−. UOR in an alkaline environment needs OH– to proceed through a series of proton-coupled electron transfers and to cleavage the C–N bonds in urea molecule21,22, however, excessive OH– adsorption on the active sites will block adsorption of urea reactant and accelerate competing OER. Although this kind of competition has been mentioned in some anodic reactions including, methanol electrooxidation and 5-hydroxymethylfurfural electrooxidation23,24, it has not been explored in UOR. Importantly, there is no efficient strategy for catalysts to overcome this competition. Practically, given UOR and OER is six-electron and four-electron transferred reaction, respectively3,16,25, competitive OER will, theoretically, reduce anode current density by >30%. Therefore, the design for catalysts to obviate OER in the UOR at a high potential to achieve a large apparent current density and excellent electrode stability is essential for the development of the next generation of urea-assisted industrial water electrolysers.
Here, we realized highly selective and active UOR on oxyanions (sulfur, phosphorus, and selenium) coordinated nickel catalyst. Electrochemical kinetics measurements, combined with in situ spectroscopy characterizations and potential dependent density function theory (DFT) calculations indicate that the critical roles of such in situ generated oxyanions: (1) it protects Ni active sites with high coverage of urea reactant via inhibiting adsorption of OH− anion to obviate OER; (2) it promotes the C − N bond cleavage of urea molecules to generate more CNO − intermediates for boosting UOR. As a result, the optimized sulfur oxyanion nickel (Ni-SOX) sample exhibits an ultrahigh current density of 323.4 mA cm–2 and a 99.3 ± 0.4% UOR selectivity at 1.65 V (vs. RHE). This versatile and feasible oxyanion-engineered strategy solves competitive adsorption of organic reactant and hydroxyl anion on the active sites and opens a fresh avenue for the design of high-performance electrocatalysts under large current density operations, and therefore, is expected to be extended to other organic electrooxidation reactions proceeding in aqueous electrolyte to obviate competing OER.
Results
Catalyst preparation and characterization
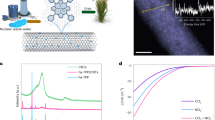
Pre-catalyst NiS2 was prepared via a sulfuration of Ni(OH)2 precursors (Supplementary Fig. 1, and Methods section). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and X-ray diffraction (XRD) confirmed a pure phase of NiS2 (PDF: 03-065-3325) with nanoparticles morphology. Energy-dispersive X-ray (EDX) mapping images evidenced that Ni and S atoms are uniformly distributed (Fig. 1a and Supplementary Fig. 2). After an electrochemical activation at a potential of 1.45 V for 600 s in urea-containing alkaline solution (1 M KOH + 0.33 M urea, Supplementary Fig. 3), derived electrocatalyst was obtained, which exhibited a core-shell structure with a crystalline NiS2 core and a 2–5 nm thick amorphous shell (Fig. 1b). EDX mapping images and line analyses showed that the Ni and O elements present mainly in the surface layer with small amount of S, evidencing that the sulfur species exist with nickel (oxy)hydroxide in the shell (Fig. 1c and Supplementary Fig. 4)26. The derived electrocatalyst was denoted therefore Ni-SOX. Compared with fresh NiS2, Ni-SOX exhibited weaker peaks for Ni-S binding and a stronger S-O peak in the X-ray photoelectron spectroscopy (XPS) patterns (Fig. 1d), demonstrating that partial S in the matrix was oxidized to sulfate interacting with nickel (oxy)hydroxide in the shell of Ni-SOX26,27. This finding is also confirmed by the Near-edge X-ray absorption fine structure spectroscopy (NEXAFS) for S K-edge, in which Ni-SOX exhibited a weaker peak for S22− and a stronger peak for SO42− compared with NiS2 (Supplementary Fig. 5)28,29. For comparison, NiOX was also synthesized via electrooxidation of pristine Ni(OH)2 catalysts without sulfuration, which presented typical nickel (oxy)hydroxide on the surface shell (Supplementary Figs. 6 and 7).

HRTEM image of a NiS2 and b Ni-SOX catalyst. c EDX elemental mapping (upper) and linear scan (lower) for Ni-SOX. d S 2p XPS curves for NiS2 and Ni-SOX catalyst.
Electrocatalytic UOR performance
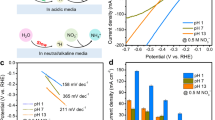
UOR performance for Ni-SOX and NiOX powder electrodes were tested in 1 M KOH with 0.33 M urea solution via a typical three-electrode system. Both the Ag/AgCl (saturated KCl) with a salt bridge and Hg/HgO were used as the reference electrodes. As is shown in the electrochemically active surface area (ECSA) normalized linear sweep voltammetry (LSV) curve (Fig. 2a and Supplementary Figs. 8, 9), both Ni-SOX and NiOX exhibit higher anodic current toward UOR (solid line) than that for OER (dash line). Overall OER performance for Ni-SOX and NiOX is similar, however UOR performance for the former is significantly better than that for the latter (confirmed by a smaller Tafel curves in Supplementary Fig. 10). At 1.65 V, UOR current density on Ni-SOX is as high as 323.4 mA cm−2, which is ca. 3 times greater than that on NiOX. Significantly, this finding is amongst ‘best’ reported performance for powder Ni-based UOR electrocatalysts (Fig. 2b and Supplementary Table 1). To exclude the effect of different nanostructures on the final UOR performance, we also synthesized Ni(OH)2* nanoparticles precursors with similar morphology to NiS2 nanoparticles precursors (Supplementary Fig. 11). After the electrochemical activation, the ECSA-normalized UOR performance of Ni-SOX remained superior to NiOX* (Supplementary Figs. 12, 13). This finding confirms that the significant UOR current density for Ni-SOX is because of the critical roles of sulfur oxyanion dopant and, importantly, not the nanostructure of the catalyst itself.

a ECSA-normalized LSV curves in 1 M KOH solution with (solid line), or without (dash line) 0.33 M urea. b Comparison of current density on Ni-SOX with reported powder catalysts under a 1.50 − 1.65 V potential zone for UOR. c In situ evaluation of O2 on Ni-SOX and NiOX using RRDE in 1 M KOH with 0.33 M urea. d Multi-step chronopotentiometric test at different potentials from 1.30 to 1.70 V. e Long-term chronopotentiometric stability determined at current density 50 mA cm −2. f Comparison of Ni3+ reduction peaks for Ni-SOX and NiOX in 1 M KOH with (solid line) or without (dash line) 0.33 M urea.
It is noteworthy that the UOR current density for NiOX exhibits current passivation at a high potential (>1.50 V, yellow-colour zone) (Fig. 2a), while that for Ni-SOX exhibits a potential-current linear relationship. To exclude the effect of diffusion limit, we performed the polarization tests at different rotating speeds of 400, 800, and 1600 rpm in 1 M KOH with 0.33 M urea electrolyte (Supplementary Fig. 14). The LSV curves for Ni-SOX and NiOX exhibited almost constant current densities with an increasing rotating speed. This finding evidences that UOR behaviors for Ni-SOX and NiOX are diffusion-independent. Because this current passivation in NiOX is exhibited near the OER zone, we hypothesized that it is current consumption by the 4e − transferred OER rather than the 6e − transferred UOR. This was confirmed via a rotating ring-disk electrode (RRDE) where UOR /OER occurred on the disk and generated O2 (if any) was reduced on the Pt ring electrode. As is shown in Fig. 2c, compared with the significant reduction current for NiOX under potential >1.50 V, no ring current is exhibited with Ni-SOX in the test range. This difference demonstrates that UOR selectivity on Ni-SOX was ca. 100%, whereas a certain amount of OER exhibits on NiOX under a high potential.
As expected, the partial OER influences the stability of the whole electrocatalyst. As is shown in Fig. 2d, the current density for NiOX decays significantly from 1.50 V, and the decay rate exceeds 20% at 1.70 V, in contrast to Ni-SOX which remains stable at all potentials. This difference is evidenced also in the long-term stability of Ni-SOX and NiOX via chronopotentiometry at 50 mA cm −2 (Fig. 2e), in which NiOX remained stable for only 3 h, significantly less than the 80 h for Ni-SOX. The performance degradation for NiOX is likely because of the generation of high valance Ni species (> +3) associated with OER (Fig. 2f). Specifically, in 1 M KOH medium, Ni-SOX and NiOX have similar high state Ni3+ reduction peaks because of continuous oxidation potentials and OER. However, in 1 M KOH with 0.33 M urea, Ni-SOX exhibited a much smaller Ni3+ reduction peak than NiOX, evidencing that the Ni species maintain a relatively low valance state on Ni-SOX because of UOR (we will discuss the reason in later sections), which leads to it exhibiting greater stability than NiOX.
UOR/OER selectivity quantification
In situ differential electrochemical mass spectrometry (DEMS) was used to determine the potential dependent UOR/OER selectivity on the electrocatalysts (Fig. 3a, b). For Ni-SOX, from 1.31 to 1.65 V, the N2 signal was detected without attenuation, with O2 negligible. In comparison, the N2 signal weakened and O2 signal gradually increased for NiOX from 1.50 V. This finding is in good agreement with experimental results of RRDE. The potential dependent Faradic efficiency (FEs) for each ion and gaseous product from UOR and OER (if any) were quantified via ion chromatography (IC) and gas chromatography (GC) (Fig. 3c, d and Supplementary Fig. 15). Error bars indicate the standard deviation based on three independent measurements. The N-containing products from UOR are nitrite (NO2−), N2, nitrate (NO3−), and cyanate (CNO − ), of which NO2− is main. The generated N2 may follow the equation: CO(NH2)2 + OH− → CO2 + N2 + H2O + 6e−, and the typical reaction pathway: urea adsorption; dehydrogenation of N − H; C − N bond breakage; N − N coupling; N2 and CO2 desorption10,12,16,21. The C-containing products from UOR are CNO − and carbonate (CO32 − ). The FEs for UOR products are computed based on N-containing products (N2, NO2−, and NO3−). For Ni-SOX, the FEs for N-containing products (FEN-products) from UOR remained above 95 ± 4% at all potentials with few OER products (O2). In contrast, for NiOX, the FEN-products monotonically decreased, and \({{{\rm{FE}}}}_{{{{\rm{O}}}}_{2}}\) increased with increasing applied potential. Specifically, UOR selectivity was reduced to 82.6 ± 0.7% at 1.65 V for NiOX, whilst that for Ni-SOX remained up to 99.3 ± 0.4%. This difference in OER /UOR selectivity for NiOX and Ni-SOX leads to the disparity in the apparent current (Fig. 2a). Significantly, this UOR selectivity on Ni-SOX is one of the highest reported Ni-based electrocatalysts under large current density operation conditions (Supplementary Table 2).

In situ detection of the representative gaseous products of UOR (N2) and OER (O2) via DEMS (upper) and the corresponding current acquired in 1 M KOH with 0.33 M urea (lower, without iR correction) on a Ni-SOX and b NiOX. FEs for different UOR /OER products under different potentials on c Ni-SOX and d NiOX. Error bars correspond to the standard deviation of three independent measurements.
Dynamic state change of Ni during UOR
To determine the origin for the high UOR activity, selectivity, and stability on Ni-SOX, we performed in situ X-ray absorption near-edge structure (XANES) and in situ Raman spectra to gain some insights into the influence of oxyanion incorporation on active Ni sites during UOR. With applied potential increasing, the adsorption edge of Ni-SOX is positively shifted towards higher energy than that of Ref NiO, but still lower than that of Ref LaNiO3, which is evidence that the state of Ni in Ni-SOX increased from +2 and remained below +3 during UOR up to 1.60 V (Fig. 4a). And a linear relationship between the Ni K-edge absorption edge and the Ni oxidation states in Ni-SOX@1.4 V, Ni-SOX@1.5 V, Ni-SOX@1.6 V, Ref NiO, and Ref LaNiO3 is plotted (Supplementary Fig. 16)30,31. The in situ Raman spectra for Ni-SOX show that, with 0.33 M urea in the electrolyte, there is not a high state of NiOOH accumulation (note that the peak at 475 cm − 1 is attributed to Ag mode of NiS2 in the core of Ni-SOX32,33 and remained almost unchanged at different voltage) (Fig. 4b). NiOOH species were detected in pure 1 M KOH electrolyte in which OER is dominated (Fig. 4c), which is in good agreement with the results from the CV curves (Fig. 2f). This can be explained that when there is urea in the electrolyte, the potential induced NiOOH rapidly catalyzes the nucleophile urea molecule to oxidation products and spontaneously reduces to Ni2+ 21,22,34. Conversely, accumulation of abundant NiOOH is found on NiOX in 1 M KOH with 0.33 M urea electrolyte (Fig. 4d). This evidences that NiOX has ‘slow’ UOR kinetics, causing accumulation of NiOOH, which then acts as active species for OER, leading to O2 evolution.

a In situ Ni K-edge XANES spectra for Ni-SOX under varying potential during UOR. Inset, magnified absorption edge region. In situ Raman spectra for Ni-SOX electrode in 1 M KOH b with, and c without, 0.33 M urea at applied potentials. d In situ Raman spectra for NiOX electrode in 1 M KOH with 0.33 M urea.
UOR mechanism analyses
In situ ATR-IR spectroscopy was used to explore how sulfur oxyanion dopant promotes UOR kinetics and inhibits OER on Ni-SOX. As is shown in Fig. 5a, b, a urea’s C − N stretching vibration peak at ∼1504 cm−1 was found in both Ni-SOX and NiOX in the initial stage35,36.

a–d In situ ATR-IR spectra in potential window 1.20 to 1.60 V for Ni-SOX and for NiOX in different ranges. e, f Schematic for competing adsorption of urea and hydroxyl on Ni-SOX and NiOX surfaces.
With the increase in potentials, this band on Ni-SOX decreased more than that on NiOX, evidencing that the C − N bond can be more readily cleaved on Ni-SOX. This is evidenced also by a stronger CNO − intermediate vibration peak at ∼ 2168 cm−1 on Ni-SOX37,38. Then, we constructed relatively matched NiOOH-SO4 and NiOOH models to simulate active sites of Ni-SOX and NiOX during UOR and speculated possible pathways of urea oxidation to main product NO2− based on the observed intermediate (Supplementary Fig. 17). The computational results show that C − N bond cleavage is promoted on NiOOH-SO4 compared with NiOOH, which is consistent with in situ ATR-IR data (Supplementary Fig. 18). To further explore the origin of this C-N bond cleavage promotion on Ni-SOX, the surface adsorption behaviour of the two catalysts was investigated. In the initial stage, an identical stretching mode for the O–H group in H2O molecule was found for both Ni-SOX and NiOX (Fig. 5c, d)39,40. As the potential increased to 1.50 V, these peaks exhibited a red-shift from 3606 to 3563 cm − 1 on NiOX, whilst there was no change for Ni-SOX. This red-shift, together with potentials confirms generation of OH* as a surface-adsorbed species, according to the Stark tuning phenomenon40,41. Importantly, given that the adsorption of OH* on the active sites of catalysts is the first step in OER, this atomic level observation explains the selectivity of OER on NiOX at a high potential. However, the surface of Ni-SOX is covered by urea molecules which then readily are attacked by OH − species from the alkaline electrolyte, thereby promoting the C-N cleavage of urea for boosted UOR and obviating adsorption of OH* for suppressed OER (Fig. 5e, f). An experimental kinetics study exhibits that the reaction order of UOR for NiOOH-SO4 with respect to OH − concentration is 0.65, a value significantly less than that for NiOOH of 1.87, further confirming that UOR for NiOOH-SO4 exhibits a weaker dependence on OH − concentration (Supplementary Fig. 19)8,42.
To validate the inhibiting role of oxyanion toward OH − adsorption on more Ni-based electrocatalysts, we further tested the UOR selectivity of other Ni compounds including NiSe2 and Ni5P4 (Supplementary Fig. 20), which was derived to Ni-SeOx and Ni-POx during UOR. HAADF-STEM images and XPS spectra evidence that selenium and phosphorus oxyanion doped amorphous nickel (oxy)hydroxides are also formed on the surface of Ni-SeO4 and Ni-PO4 (Supplementary Figs. 21–23). As expected, selenium and phosphorus oxyanion doping boosts UOR activity and inhibits OER as sulfur (Supplementary Fig. 24).
UOR /OER Competition Mechanism
To manipulate the competing adsorption of urea and OH − , electrochemical tests were performed in electrolytes containing different concentrations of reactants. As is shown in Fig. 6a, a greater OH − concentration in electrolyte leads to more significant OER competition for NiOx, whilst having no apparent effect for Ni-SOX (Supplementary Fig. 25). The increase in urea concentration slightly attenuates but does not eliminate competition between UOR and OER on NiOX (Fig. 6b), evidencing that the surface of NiOX is mainly covered by OH − species but not urea reactant, leading to dominant OER selectivity.

a RRDE curves for NiOX obtained in different concentration of KOH with 0.33 M urea. b RRDE curves for NiOX obtained in 1 M KOH with different concentrations of urea. c Representation for UOR mechanism on Ni-SOX accompanying adsorption of reactants and dynamic evolution of active sites under high potential (>1.50 V). d Representation of UOR and OER mechanism on NiOX accompanying adsorption of reactants and dynamic evolution of active sites under high potential (>1.50 V).
Based on above electrochemical tests and in situ spectroscopic studies, a possible UOR mechanism on two kinds of electrocatalysts including adsorption of reactants and dynamic evolution of active sites is proposed. When the applied potential is low (<1.50 V), Ni-SOX and NiOX are derived to, respectively, NiOOH-SO4 and NiOOH on the surface through electrochemical oxidation (Supplementary Fig. 26). The generated NiOOH-SO4 and NiOOH catalyze the nucleophile urea molecules into N products and spontaneously reduced to low-valent Ni-SOX and NiOX, maintaining a good balance of derivatization and reduction. Compared with NiOOH, NiOOH-SO4 makes the C − N of urea more readily cleaved, resulting in boosted UOR. Once the potential is applied over 1.50 V (Fig. 6c, d), derived NiOOH is attacked by OH − to generate high valance Ni species (≥ 3). These over-oxidized Ni species significantly participate in competing OER, which makes less NiOOH oxidizing urea, and spontaneous reduction to NiOX is slowed, leading to UOR-OER competition. However, derived NiOOH-SO4 can inhibit the adsorption of OH − , making it covered by sufficient nucleophile urea molecules, thereby obviating the generation of higher-valent OER-active species. Simultaneously, the repulsion of OH − on NiOOH-SO4 promotes cleavage of C − N of urea molecules and generates more CNO − intermediates, resulting in faster UOR kinetics and more facile spontaneous reduction into Ni-SOX as a clean active site for the next derivatization and reduction cycle.
Discussion
In summary, we achieved ultrahigh activity, selectivity, and stability of UOR via constructing oxyanion-engineered nickel catalysts (Ni-SOX, Ni-POX, and Ni-SeOX). Notably, the optimal Ni-SOX exhibited ultrahigh 323.4 mA cm − 2 UOR current density at 1.65 V with nearly 100% selectivity of N-products. The combination of diverse in situ spectroscopic measurements and DFT calculations evidenced the essential roles of oxyanion for UOR. On one hand, it inhibits the competitive adsorption of OH − on the Ni active sites to avoid OER. On the other hand, it accelerates urea’s C − N bond cleavage to form CNO − intermediates for boosting UOR. Correspondingly, we proposed a comprehensive mechanism for competitive adsorption behaviour between OH − and urea to boost UOR and dynamic change of Ni active sites. We expect that this strategy will aid future research in practical urea electrolysis, and other multi-electron organic molecule oxidation coupled with cathodic hydrogen evolution for overall atomic economy and additional green energy production.
Methods
Synthesis of Ni(OH)2, NiS2, Ni5P4, and NiSe2
In typical synthesis for Ni(OH)2 catalyst, 5 mmol nickel (II) nitrate hexahydrate (Ni(NO3)2·6H2O) and 10 mmol hexamethylenetetramine (HMT) were dissolved in 35 mL of deionized (DI) water under vigorous stirring for 30 min to form a transparent solution. The mixture was transferred to a 50 mL Teflon-lined autoclave, sealed, and heated at 120 °C for 12 h. The resulting powder was washed with ethanol /water and dried overnight to obtain Ni(OH)2 powder. NiS2 catalyst was obtained from Ni(OH)2 via a simple calcination. In detail, 20 mg of Ni(OH)2 and 0.8 g sulfur (S) powder were placed in two separate porcelain boats and put into a tube-furnace, with S powder on the upstream. After flushing with Ar for ~30 min, the sample was heated to 350 °C at a rate 2 °C min−1 in Ar for 2 h and cooled naturally to room temperature (RT). The Ni5P4 catalyst was obtained via replacing 0.8 g S with 0.8 g hypophosphite monohydrate (NaH2PO2•H2O). The NiSe2 catalyst was obtained via replacing 0.8 g S with 0.4 g Selenium (Se) powder heated to 450 °C at a rate 20 °C min−1 in Ar for 1 h.
Synthesis of NiOX, Ni-SOX, Ni-POX and Ni-SeOx
NiOX, Ni-SOX, Ni-POX and Ni-SeO4 catalysts were prepared via electrochemical activation of Ni(OH)2, NiS2, Ni5P4 and NiSe2 at a potential of 1.45 V for 600 s in 1 M KOH with 0.33 M urea aqueous solution.
Characterizations
X-Ray Powder Diffraction (XRD) data were collected on a Rigaku MiniFlex 600 X-Ray Diffractometer. HAADF-STEM images were obtained using an FEI Titan G2 80–300 microscope at 300 kV equipped with a probe corrector. EDX imaging was carried out with an FEI Titan Themis 80–200 microscope equipped with a SuperX detector. X-ray photoelectron spectroscopy (XPS) analyses were conducted under ultra-high vacuum on a Kratos Axis Ultra with a Delay Line Detector photoelectron spectrometer using an aluminum monochromatic X-ray source. XPS data were corrected using the C1s line at 284.8 eV. In situ XAS data were collected on XAS beamline of Australian Synchrotron (ANSTO, Melbourne) using a home-made cell and processed via the Athena program. In situ Raman spectroscopy data were obtained using a Via-Reflex spectrometer (Renishaw) with a laser excitation wavelength of 532 nm. The measured potential was in the range of 1.20 to 1.60 V vs. RHE controlled by a CHI 760E electrochemical workstation.
In situ DEMS measurement
A 60 nm gold-sputtered PTFE membrane was used as working electrode substrate. Electrocatalyst ink was added onto the gold-sputtered membrane and dried at RT to a uniform layer. Real-time generated gaseous products (N2 and O2) during the Cyclic voltammetry (CV) test were pumped to in situ DEMS system (HPR-40, Hiden Analytical Ltd.), and the signal with mass-to-charge ratio of 28, 32, was obtained. CV test was performed at a rate 10 mV s −1 under 1 M KOH with 0.33 M urea.
In situ Attenuated Total Reflectance Infrared (ATR-IR) spectroscopy measurement
In situ ATR-IR measurements were performed on a Thermo-Fisher Nicolet iS20 equipped with a liquid nitrogen-cooled HgCdTe (MCT) detector and a VeeMax III ATR accessory (Pike Technologies). A silicon prism coated with Au film (60°, PIKE Technologies) was mounted in a PIKE electrochemical three-electrode cell with an Ag/AgCl reference electrode (Pine Research) and a Pt counter electrode. Catalysts were sprayed onto the Au film as a working electrode. During testing, the electrolyte (1 M KOH with 0.33 M urea solution) was purged with Ar continuously. A CHI 760E electrochemical workstation was used to perform chronoamperometric tests from 1.20 to 1.60 V vs. RHE. ATR-IR curves were concurrently collected with 64 scans and a spectral resolution of 4 cm−1.
Electrochemical measurement
Electrochemical data were recorded via a CHI 760E electrochemical workstation. Tests were conducted in a gas-tight three-electrode H-cell at 25 °C (with Ar purge). Anode and cathode compartments were separated by a proton exchange membrane (Nafion 117). For the working electrode, 3.5 mg catalyst, 1 mg carbon black, and 10 μL 5 wt% Nafion solution were dispersed in 400 μL ethanol. Catalyst dispersion was dropped on a carbon-fibre paper or a rotating disk electrode as working electrode (1 × 0.5 cm −2, loading mass: ~0.5 mg cm−2). The Ag/AgCl (saturated KCl) with a salt bridge and Hg/HgO were used as the reference electrodes, while a graphite rod was used as the counter electrode. The electrolyte was 1 M KOH (or 2 M KOH, 4 M KOH) with and without 0.33 M (or 1.32 M) urea. The linear sweep voltammetry (LSV) and cyclic voltammetry (CV) tests were collected at a scan rate of 5 mV s−1 with Ar purging. The estimated electrochemical double layer capacitances (Cdl) were obtained via CV testing in a non-Faradaic potential region, 1.09 ~ 1.17 V vs. RHE at a scan rate of 20, 40, 60, 80, and 100 mV s−1. Potentials were converted to reversible hydrogen electrode (RHE) and corrected with 90% iR-compensation, except however for stability experiments.
Product analyses
The gas products (N2, O2) formed during urea oxidation were analyzed every 15 minutes via on-line gas chromatograph (GC, 8890, Agilent). The GC was fitted with HP-PLOT Q and CP-Molsieve 5 Å PT column (both Agilent), together with thermal conductivity (TCD) and flame ionization (FID) detectors. Liquid nitrate and nitrite (NO3− and NO2− ) were determined via ion chromatography (IC, Thermo Scientific Dionex Integrion RFIC) equipped with a Dionex IonPac AS19-4 µm 2 × 250 mm column. Calibration-curve standards, containing 5–200 ppm NO3−/NO2 − were prepared from commercially available solution of the anions (1000 ppm, Sigma-Aldrich). FE for the formation of the product was computed from:
in which e is the number of transferred electrons for each product, F Faraday constant, Q charge, I applied current, t reaction time, and n, total product (mole).
Measurements for all products were repeated at least 3 times during each applied potential to exclude any possible errors.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data that support findings from this study are available from the corresponding author upon reasonable request. The source data underlying Figs. 1–6 are provided as a Source Data file. Source data are provided in this paper.
References
Yao, S. J., Wolfson, S. K., Ahn, B. K. & Liu, C. C. Anodic oxidation of urea and an electrochemical approach to de-ureation. Nature 241, 471–472 (1973).
Chen, S., Duan, J., Vasileff, A. & Qiao, S.-Z. Size fractionation of two-dimensional sub-nanometer thin manganese dioxide crystals towards superior urea electrocatalytic conversion. Angew. Chem. Int. Ed. 55, 3804–3808 (2016).
Han, W.-K. et al. Activating lattice oxygen in layered lithium oxides through cation vacancies for enhanced urea electrolysis. Angew. Chem. Int. Ed. 61, e202206050 (2022).
Wang, G. et al. Solar driven hydrogen releasing from urea and human urine. Energy Environ. Sci. 5, 8215–8219 (2012).
Senthilkumar, N., Gnana, kumarG. & Manthiram, A. 3D hierarchical core–shell nanostructured arrays on carbon fibers as catalysts for direct urea fuel cells. Adv. Energy Mater. 8, 1702207 (2018).
Lan, R., Tao, S. & Irvine, J. T. S. A direct urea fuel cell-power from fertiliser and waste. Energy Environ. Sci. 3, 438–441 (2010).
Zhu, X. et al. Metallic nickel hydroxide nanosheets give superior electrocatalytic oxidation of urea for fuel cells. Angew. Chem. Int. Ed. 55, 12465–12469 (2016).
Geng, S.-K. et al. Nickel ferrocyanide as a high-performance urea oxidation electrocatalyst. Nat. Energy 6, 904–912 (2021).
Kumar, A. et al. Discovering ultrahigh loading of single-metal-atoms via surface tensile-strain for unprecedented urea electrolysis. Energy Environ. Sci. 14, 6494–6505 (2021).
Tatarchuk, S. W., Medvedev, J. J., Li, F., Tobolovskaya, Y. & Klinkova, A. Nickel-catalyzed urea electrolysis: from nitrite and cyanate as major products to nitrogen evolution. Angew. Chem. Int. Ed. 61, e202209839 (2022).
Wang, L. et al. Regulating the local charge distribution of Ni active sites for the urea oxidation reaction. Angew. Chem. Int. Ed. 60, 10577–10582 (2021).
Ji, Z. et al. Pathway manipulation via Ni, Co, and V ternary synergism to realize high efficiency for urea electrocatalytic oxidation. ACS Catal. 12, 569–579 (2022).
Pei, C. et al. Nanostructured metallic glass in a highly upgraded energy state contributing to efficient catalytic performance. Adv. Mater. 34, 2200850 (2022).
Forslund, R. P. et al. Nanostructured LaNiO3 perovskite electrocatalyst for enhanced urea oxidation. ACS Catal. 6, 5044–5051 (2016).
Boggs, B. K., King, R. L. & Botte, G. G. Urea electrolysis: direct hydrogen production from urine. Chem. Commun. 4859–4861 (2009).
Daramola, D. A., Singh, D. & Botte, G. G. Dissociation rates of urea in the presence of NiOOH catalyst: a DFT analysis. J. Phys. Chem. A 114, 11513–11521 (2010).
Song, J. et al. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 49, 2196–2214 (2020).
Menezes, P. W. et al. Facile access to an active γ-NiOOH electrocatalyst for durable water oxidation derived from an intermetallic nickel germanide precursor. Angew. Chem. Int. Ed. 60, 4640–4647 (2021).
Xu, K. et al. Metallic nickel nitride nanosheets realizing enhanced electrochemical water oxidation. J. Am. Chem. Soc. 137, 4119–4125 (2015).
Rao, R. R. et al. Spectroelectrochemical analysis of the water oxidation mechanism on doped nickel oxides. J. Am. Chem. Soc. 144, 7622–7633 (2022).
Chen, W. et al. Unveiling the electrooxidation of urea: intramolecular coupling of the N−N bond. Angew. Chem. Int. Ed. 60, 7297–7307 (2021).
Li, J. et al. Deciphering and suppressing over-oxidized nitrogen in nickel-catalyzed urea electrolysis. Angew. Chem. Int. Ed. 60, 26656–26662 (2021).
Qi, Y. et al. Insights into the activity of nickel boride/nickel heterostructures for efficient methanol electrooxidation. Nat. Commun. 13, 4602 (2022).
Zhou, P. et al. Heterogeneous-interface-enhanced adsorption of organic and hydroxyl for biomass electrooxidation. Adv. Mater. 34, 2204089 (2022).
Patil, S. J. et al. Fluorine engineered self-supported ultrathin 2D nickel hydroxide nanosheets as highly robust and stable bifunctional electrocatalysts for oxygen evolution and urea oxidation reactions. Small 18, 2103326 (2022).
Li, S. et al. Coordination environment tuning of nickel sites by oxyanions to optimize methanol electro-oxidation activity. Nat. Commun. 13, 2916 (2022).
Zhao, C.-X. et al. An anionic regulation mechanism for the structural reconstruction of sulfide electrocatalysts under oxygen evolution conditions. Energy Environ. Sci. 15, 3257–3264 (2022).
Jalilehvand, F. Sulfur: not a “silent” element any more. Chem. Soc. Rev. 35, 1256–1268 (2006).
Yang, J. et al. Highly efficient oxygen evolution from CoS2/CNT nanocomposites via a one-step electrochemical deposition and dissolution method. Nanoscale 9, 6886–6894 (2017).
Cheng, W., Xi, S., Wu, Z.-P., Luan, D. & Lou, X. W. In situ activation of Br-confined Ni-based metal-organic framework hollow prisms toward efficient electrochemical oxygen evolution. Sci. Adv. 7, eabk0919 (2021).
Bai, L., Hsu, C.-S., Alexander, D. T. L., Chen, H. M. & Hu, X. Double-atom catalysts as a molecular platform for heterogeneous oxygen evolution electrocatalysis. Nat. Energy 6, 1054–1066 (2021).
Shi, Y. et al. Unveiling the Promotion of surface-adsorbed chalcogenate on the electrocatalytic oxygen evolution reaction. Angew. Chem. Int. Ed. 59, 22470–22474 (2020).
Marini, C. et al. Combined Raman and infrared investigation of the insulator-to-metal transition in NiS2−xSex Compounds. Phys. Rev. B 84, 235134 (2011).
Zhu, B., Liang, Z. & Zou, R. Designing advanced catalysts for energy conversion based on urea oxidation reaction. Small 16, 1906133 (2020).
Climent, V., Rodes, A., Orts, J. M., Aldaz, A. & Feliu, J. M. Urea adsorption on Pt(111) electrodes. J. Electroanal. Chem. 461, 65–75 (1999).
Wang, L. et al. Insight into the synergistic effect between nickel and tungsten carbide for catalyzing urea electrooxidation in alkaline electrolyte. Appl. Catal. B 232, 365–370 (2018).
Williams, D. The infrared spectrum of potassium cyanate solutions. J. Am. Chem. Soc. 62, 2442–2444 (1940).
Zhang, L. et al. A lattice‐oxygen‐involved reaction pathway to boost urea oxidation. Angew. Chem. Int. Ed. 131, 16976–16981 (2019).
Ye, N. et al. Understanding the high performance of PdSn–TaN(tantalum nitride)/C electrocatalysts for the methanol oxidation reaction: coupling nitrides and oxophilic elements. J. Mater. Chem. A 10, 266–287 (2022).
Zhu, S., Qin, X., Yao, Y. & Shao, M. pH-dependent hydrogen and water binding energies on platinum surafaces as directly probed through surface-enhanced infrared absorption spectroscopy. J. Am. Chem. Soc. 142, 8748–8754 (2020).
Yang, X., Nash, J., Oliveira, N., Yan, Y. & Xu, B. Understanding the pH dependence of underpotential deposited hydrogen on platinum. Angew. Chem. Int. Ed. 58, 17718–17723 (2019).
Vedharathinam, V. & Botte, G. G. Understanding the electro-catalytic oxidation mechanism of urea on nickel electrodes in alkaline medium. Electrochim. Acta 81, 292–300 (2012).
Acknowledgements
We gratefully acknowledge the financial support provided by the Australian Research Council through the Discovery Project and Linkage Project Programs [FL170100154, FT200100062, DP220102596, DP190103472, and LP210301397 (S.-Z.Q.)]. In situ XAS was undertaken on the X-ray Absorption Spectroscopy beamlines at the Australian Synchrotron, ANSTO, Melbourne. X.G. was supported by the Chinese CSC Scholarship Program. DFT computations were undertaken with the support of supercomputing resources provided by the Phoenix HPC service at The University of Adelaide.
Author information
Authors and Affiliations
Contributions
Y.Z. and S.-Z.Q. conceived and supervised the research. X.G. and P.W. designed and conducted the experiments. X.G., Y.Z., and S.-Z.Q. performed data analyses. X.B. and Y.J. performed the DFT computations. X.G., Y.Z., K.D., and S.-Z.Q. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, X., Bai, X., Wang, P. et al. Boosting urea electrooxidation on oxyanion-engineered nickel sites via inhibited water oxidation. Nat Commun 14, 5842 (2023). https://doi.org/10.1038/s41467-023-41588-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-41588-w
This article is cited by
-
Boosting alkaline urea oxidation with a nickel sulfide/cobalt oxide heterojunction catalyst via interface engineering
Advanced Composites and Hybrid Materials (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
