
Abstract
Oxidation of renewable polyol/sugar into formic acid using molecular O2 over heterogeneous catalysts is still challenging due to the insufficient activation of both O2 and organic substrates on coordination-saturated metal oxides. In this study, we develop a defective MnO2 catalyst through a coordination number reduction strategy to enhance the aerobic oxidation of various polyols/sugars to formic acid. Compared to common MnO2, the tri-coordinated Mn in the defective MnO2 catalyst displays the electronic reconstruction of surface oxygen charge state and rich surface oxygen vacancies. These oxygen vacancies create more Mnδ+ Lewis acid site together with nearby oxygen as Lewis base sites. This combined structure behaves much like Frustrated Lewis pairs, serving to facilitate the activation of O2, as well as C–C and C–H bonds. As a result, the defective MnO2 catalyst shows high catalytic activity (turnover frequency: 113.5 h−1) and formic acid yield (>80%) comparable to noble metal catalysts for glycerol oxidation. The catalytic system is further extended to the oxidation of other polyols/sugars to formic acid with excellent catalytic performance.
Similar content being viewed by others


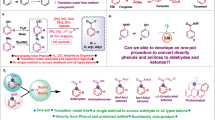
Introduction
Formic acid (FA), as the simplest carboxylic acid, is a valuable chemical and a potential energy carrier1,2. Preferably, FA is obtained from renewable resources such as biomass or CO2 rather than the current route based on fossil derived feedstock (e.g., methanol and CO)3,4,5. In this context, catalytic oxidation of polyol/sugar emerges as a promising alternative for FA production6,7,8,9,10,11,12,13,14,15,16. However, the mainstream homogeneous catalysts including heteropoly acids, NaVO3, VOSO4, and others suffer from poor catalyst recycling, metal contamination, and/or the employment of non-benign oxidants8,17,18,19,20,21,22,23. Recently, several heterogeneous catalysts based on CuOx or VOx have been excavated using O2 as oxidant20,24, but the attainment of a high FA yield with sufficient reaction rate using non-precious metal oxide catalyst remains a challenge1. One limitation is the sluggish O2 activation and subsequent bond cleavage over the saturated metal oxide surface with few accessible sites4,19. Developing defective metal oxide catalysts with enhanced ability for bond activation is desirable in the cascade oxidation of polyol/sugar to FA using molecular oxygen.
Manganese oxides exhibit a wide range of applications in oxidation reactions due to the superior redox property and structural flexibility25,26. Mn-based oxides, mainly MnO2, are amphoteric, containing Mn-induced acidity and nearby O-induced basicity. These co-existing acidic and basic sites in metal oxides could be regarded as frustrated Lewis pairs (FLPs), which promote reactions on the catalyst surfaces that require both acid and base functions, as previously demonstrated on (hydroxylate-)Al2O3, InOx, CeOx, MoOx and CoOx oxides27,28,29,30,31,32,33,34,35. We notice that the cascade oxidation of alcohol using O2 may be regarded an acid-base synergistic reaction: the activation of O2 into reactive oxygen species is accelerated by acid sites36,37, while C–H bond activation and C–C bond cleavage are known to be promoted by basic sites38,39. The characteristics of FLPs match the active site requirements for polyol/sugar oxidation40,41,42,43,44, but the saturated coordination structure in traditional MnO2 catalyst shows limited surface oxygen vacancies and low surface electron density. Due to the tendency of the rigid lattice of traditional metal oxides to form saturated coordination M-O structures, it is still challenging to construct defective metal oxides with FLPs based on existing limited and cumbersome preparation strategies45,46,47. Inspired by the variable coordination structure of transition metals48, rational reduction of the coordination number of the central metal provides a feasible strategy to obtain defective MnO2 with enhanced FLPs for polyol/sugar cascade oxidation to formic acid.
Herein, we present a strategy to construct low-coordinated, defective MnO2 catalyst (MnO2-D) for polyol/sugar oxidation. Specifically, the defective Mnδ+–OV structure with unsaturated tri-coordinated Mn forms FLPs to allow spatially adjacent acid and basic sites to work cooperatively. Mnδ+ species associated with oxygen vacancy serve as Lewis acid to promote the activation of O2 under the assistance of adjacent basic sites, whereas electron reconstructed O nearby Mnδ+ serves as Lewis base to facilitate the bond activation of organic substrate, in synergy with the Lewis acid site. In this manner, MnO2-D with intensified FLPs exhibits superior catalytic activity in converting various substrates into formic acid using O2, surpassing previous reports using heterogeneous catalysts.
Results
Synthesis and characterization of defective MnO2-D with low Mn-O coordination
Two α-MnO2 catalysts (MnO2-P and MnO2-D) were synthesized by hydrothermal methods, in which P and D refer to perfect and defective structures, respectively49,50. Significant differences in composition and crystal structure were observed on Rietveld refinement of powder X-ray diffraction (PXRD) and high-resolution transmission electron microscopy (HRTEM) images. Although only the Bragg peaks corresponding to the α-phase MnO2 (I4/m space group) are present for both samples (Fig. 1a)51,52,53,54, the PXRD pattern of MnO2-D displays a broader diffraction peak than that of MnO2-P, indicating its smaller crystallite size [8.2(1) nm] compared with MnO2-P [36.5(1) nm]. The site occupancies of O and Mn were obtained by refining the PXRD patterns (Supplementary Tables 1 and 2), which were then taken as the starting model for Density functional theory (DFT) calculation described in later sections. The hydrothermal synthesis process was significantly affected by the nature of the reductant and temperature, resulting in a sharp difference in the amount of O and Mn ion defects in the two catalysts. In MnO2-P, Mn is distributed at the octahedral 8 h site with a 99(1)% occupancy, along with the existence of oxygen vacancy [5(2)%] at O1 (8 h) site. The valence state sum of Mn cation was calculated as 3.93+ based on the charging balance. In contrast, in MnO2-D the Mn occupancy of Mn at the octahedral 8 h site is 99(2)% with 13(1)% oxygen vacancy at O1 (8 h) site and 7(2)% oxygen vacancy at O2 (8 h) site, with the valence state of Mn determined at a lower value of 3.64+. The formation of more defect structures in MnO2-D also resulted in the change of Mn–O chemical bond length (Supplementary Fig. 1), which manifests the Jahn–Teller (JT) effect55,56,57. The Mn–O bond lengths in MnO2-P are Mn–O1: 1.64(3) Å and Mn–O2: 1.89(3) Å, respectively, while the ones in MnO2-D are Mn–O1: 1.69(4) Å and Mn-O2: 1.92(4) Å51,58,59. Scanning electron microscopy (SEM) image in Fig. 1b shows that MnO2-P consists of short and thick nanowires with length of ~500 nm and width of ~30 nm, while MnO2-D exhibits longer and thinner nanowire morphology (width: ~10 nm), which are consistent with the crystallinity results of PXRD, and the notion that MnO2-D exposes more defective sites on surface. The HRTEM images and selected area electron diffraction (SAED) pattern indicate that MnO2-P mainly exposes (211) and (200) planes, while MnO2-D mainly exposes (310) and (301) planes (Fig. 1b). The crystallinity results of PXRD also confirms that the ratio of (310) crystal plane to (211) crystal plane on MnO2-D [I(310)/I(211) = 0.78] is significantly higher than that on MnO2-P [I(310)/I(211) = 0.65].

a Rietveld refinement and visualization of the associated crystal structure for a1 MnO2-P and a2 MnO2-D. Unit cell of samples MnO2-P (I4/m (84), a = b = 9.804(1) Å, c = 2.855(1) Å) and MnO2-D (I4/m (84), a = b = 9.818(1) Å, c = 2.847(1) Å) obtained from powder diffraction data based on the Rietveld profile refinement and Stephens peak shape function using the Topas software (Supplementary Tables 1 and 2) with the Rwp are 11.62% and 9.82%, respectively. b SEM/HRTEM images of b1 MnO2-P and b2 MnO2-D. c Fourier transform of Mn K-edge extended EXAFS oscillations. d Schematic diagram of coordination structure of Mnδ+ on MnO2-P and MnO2-D.
To confirm the coordination structure and valence states of Mn, X-ray absorption fine structure (XAFS) were performed. The k3-weighted Fourier-transform Mn K-edge extended XAFS spectra (EXAFS) in Fig. 1c show that both MnO2-P and MnO2-D exhibit typical spectral features of α-MnO2 phase with the intense FT peaks at approximately 1.52 Å, 2.85 Å and 3.40 Å, corresponding to Mn–O, edge-shared Mn–(O)–Mn, and corner-shared Mn–Mn shells, respectively55,56,60,61. Compared with MnO2-P, MnO2-D exhibits lower intensity of Mn-O shell and stronger intensity of corner-shared Mn-Mn shell, manifesting the increase of defective structure49,50,62. Wavelet transform (WT) analysis in Supplementary Fig. 2 shows that the peak with a maximum intensity of approximately 6 Å is observed for bulk α-MnO2, MnO2-P and MnO2-D in the k-space, ascribed to the Mn–O bond. The WT spectrum of MnO2-D also shows a maximum intensity of approximately 12 Å in the k-space and 3.5 Å in the R-space, corresponding to the Mn-Mn shell. EXAFS data fitting suggests that the coordination number of O for bulk α-MnO2 and MnO2-P is 6.0 and 5.2, respectively, while that for MnO2-D is only 3.0, confirming the significant decrease of Mn-O coordination (Supplementary Table 3). Figure 1d provides a cartoon representation of the 5-coordination pentahedron geometry of Mn in MnO2-P, and the 3-coordination plane/tetrahedron structure of Mn in MnO2-D. The latter is an equivalent model consisting of saturated 6-coordination Mn4+ and other defect coordination Mnδ+. Obviously, compared with penta-coordinated Mn in common MnO2-D, the tri-coordinated Mn is successfully constructed in the defective MnO2-D.
Following exhaustive characterizations of MnO2-D and MnO2-P, we proceeded to synthesize tetra-coordinated α-MnO2 (MnO2-T) with an intermediate coordination number of 4.2 (Supplementary Fig. 2 and Supplementary Table 3). In addition, we synthesized and characterized two sets of distinct phases, β-MnO2 and γ-MnO2. Each set comprises a high coordination and a low coordination sample, respectively (Supplementary Table 3 and Supplementary Fig. 2). The intention behind these preparations was to establish a comprehensive comparison on glycerol oxidation efficiency across various MnO2 samples bearing different coordination environment.
Probing the structure of the Frustrated Lewis Pairs in defective MnO2-D
The Mn K-edge X-ray absorption near edge structure (XANES) shows that the absorption edges of MnO2-P and MnO2-D are similar to standard MnO2 (Fig. 2a). Through the first-derivative of absorption edge in normalized XANES spectra, the Mn valence state is quantified as 3.89 and 3.56 for MnO2-P and MnO2-D respectively (Supplementary Fig. 3), consistent with PXRD refinement results. The Mn 2p X-ray Photoelectron Spectroscopy (XPS) spectra (Fig. 2b) clearly demonstrates that the formation of Mnδ+–OV defect structure leads to a significant decrease of the binding energy, confirming that the surface of MnO2-D possesses more electrons63. The XPS peak deconvolution analysis in Supplementary Table 4 shows a higher content of Mn2+ and Mn3+ is observed in MnO2-D. This electronic reconstruction caused by the reduction of coordination number induces Mn to be more prone to get electrons, while O more prone to lose electrons—an essential prerequisite for the formation of FLPs.

a The first-order derivatives of Mn K-edge XANES. b Mn 2p XPS spectra of MnO2-P and MnO2-D. c O2-TPD and d XPS spectra of O 1 s of MnO2-P and MnO2-D. e Aberration-corrected HAADF-STEM image of e1 MnO2-P and e2 MnO2-D. f Structure diagram and Mulliken charge distribution of Frustrated Lewis Pair in MnO2-P and MnO2-D. g NH3-TPD and CO2-TPD of g1 MnO2-P and g2 MnO2-D.
Notably, the generation of oxygen vacancy in the low-coordination Mnδ+-OV structure is crucial for FLPs formation: the reduced oxygen coordination leads to the exposure of the metal atom as Lewis acid sites, while the oxygen vacancies also induce stronger electronegativity of the residual oxygen, thus enhancing the Lewis basicity. As shown in Fig. 2c, the temperature-programmed desorption (TPD) of O2 of the two catalysts reveals the presence of the surface oxygen species (~200 °C, Osur), the active oxygen species near the surface (~300 °C, Osur-sub) and the lattice oxygen species (~600 °C, Olat). MnO2-D exhibits a high Osur-sub content (77.6%) and a minimum Olat content (9.5%), indicating that the defective structure induces more active oxygen species (Supplementary Table 5). Low-temperature electron paramagnetic resonance (EPR) also shows that MnO2-D exhibits a stronger oxygen vacancy feature at a g-factor of 2.00364,65 than MnO2-P (Supplementary Fig. 4). Consistent with O2-TPD and EPR analysis, the XPS spectra of O 1 s region in Fig. 2d and Supplementary Table 4 demonstrates that the MnO2-D catalyst has the highest oxygen vacancy content (27.6% OII) with lower binding energy. Additionally, the existence of oxygen vacancy on the surface of MnO2-D is observed via aberration-corrected high-angle annular dark-field scanning TEM (Fig. 2e).
On these foundations, we analyzed the structure and Mulliken charge of penta-coordinated MnO2-P and tri-coordinated MnO2-D to provide direct evidence for the formation of FLPs. Figure 2f shows that the near saturated coordination O in MnO2-P creates a steric hindrance effect that obstructs the access of partially negatively charged reactant molecules to the Mn Lewis acid site. In sharp contrast, Mn in the low-coordinated MnO2-D being highly unsaturated requires more molecules to coordinate (Lewis acid property). In parallel, the three O atoms in MnO2-D process more electrons due to the electron transfer from Mn to O, which could further transfer electrons to reactant molecule (Lewis base property). Hence, the behavior of defective Mnδ+–OV structure in MnO2-D aligns well with the concept of FLPs30,66,67,68,69,70. To probe the FLPs on MnO2-D, NH3-TPD, and CO2-TPD were further performed. Figure 2g and Supplementary Table 5 show that the MnO2-D exhibits sharply different acid-base properties from MnO2-P. For NH3-TPD, MnO2-D displays more medium strong and strong acid sites, while MnO2-P mainly contains weak acid sites with a small amount of strong acid sites. This indicates that the decrease of coordination number not only increases the amount of Lewis acid sites (exposure of more Mn), but also increases the acid strength. In addition, pyridine infrared results demonstrate that the Lewis acid sites on MnO2-P and MnO2-D are water tolerant (Supplementary Fig. 5). In CO2-TPD, MnO2-D exhibits increased amount of strong base sites compared with MnO2-P, attributable to the stronger adsorption of CO2 on electron-enriched O in FLPs.
Enhanced cascade oxidation of polyol/sugar to formic acid over MnO2-D
Next, catalytic oxidation of glycerol was tested using the prepared Mn catalysts under 1 MPa O2 at 120 °C. Figure 3a and Supplementary Table 6 show that at low conversion (<20%), the initial reaction rate reached 31.3 mmol/h/gcat, almost 20 times higher than that of MnO2-P. Moreover, at a similar glycerol conversion level (~64%), MnO2-D exhibits a high FA selectivity of 83.3%, while MnO2-P displays a total selectivity of 66.9% for various carboxylic acids, including glyceric acid (GLYA), oxalic acid (OA), glycolic acid (GLYOA) and FA. The tetra-coordinated MnO2-T, characterized by its intermediate coordination number, exhibited moderate activity that fell between the performances of MnO2-P and MnO2-D catalysts (20.7 mmol/h/gcat). Consistently, low-coordination number β-MnO2 and γ-MnO2 samples displayed superior catalytic performance in oxidizing glycerol to FA compared to their high coordination number counterparts (Supplementary Table 6). The above results indicates that the saturated coordination structure of MnO2 does not have sufficient activity in C–H and C–C bond activation, implying that an unsaturated structure is key for high activity. Of note, the difference of specific surface area is not the main factor affecting the activity and FA selectivity, as excluded by control experiments (Supplementary Fig. 6), while little activity was observed in the absence of a Mn-based catalyst (Supplementary Table 6). Through further optimization of reaction time, MnO2-D delivered an optimal 99.2% conversion and 83.2% FA selectivity in 6 h (Supplementary Fig. 7).

a Cascade oxidation of glycerol to formic acid over the MnO2-P and MnO2-D [25 mL aqueous phase solution (0.1 M), 0.5 g NaOH, 0.1 g catalyst, 1 MPa O2, MnO2-P for 10 h, MnO2-D first for 2 h, MnO2-D second for 6 h,120oC]. b Oxidation of glycerol to formic acid over MnO2-D and other reported catalysts. c Catalytic stability of the MnO2-P and MnO2-D under multiple cycle test conditions. d Catalytic performance of the MnO2-D for catalytic oxidation of different substrates (polyols/sugars) to formic acid [Formic acid (FA); Glycolic acid (GLYOA); Oxalic acid (OA); Glycerol (GLYA); Tartronic acid (TA); Ethylene glycol (EG); 1,2-propanediol (PG-2); 1,3-propanediol (PG-3); Erythritol (ET); Xylitol (XT); Sorbitol (ST)].
Ethanol pulse adsorption was further conducted to determine the exposed active sites (Supplementary Fig. 8), based on which the TOF of MnO2-D catalyst was determined as 113.5 h−1. This value matches or outperforms the catalytic activities of noble metal catalyst operated under similar conditions (Fig. 3b and Supplementary Table 7). Moreover, MnO2-D exhibits excellent stability for glycerol oxidation, with almost no change in FA selectivity (>80%) and a slight decrease in glycerol conversion (<20%) after 6 catalytic cycles due to the coverage of some surface active sites by carboxylic acid products (Fig. 3c and Supplementary Fig. 5). Characterizations of the spent catalyst using XRD, UV-Vis, XPS, NH3-TPD and CO2-TPD techniques prove that the defective Mnδ+–OV structure and acid-base properties in MnO2-D were well-preserved during the reaction (Supplementary Fig. 5). After calcining the spent catalyst, the catalytic activity of MnO2-D was fully restored. MnO2-D is further extended to the selective oxidation of intermediate substrates derived from glycerol and other polyols/sugars (Fig. 3d). In all cases, high conversion (>75%) and FA selectivity (>60%) were obtained. The excellent catalytic performance encourages us to further explore the structure-activity relationship on the defective Mnδ+–OV structure with strength-intensified FLPs in the polyol/sugar oxidation to FA.
Activation of O2 into hydroxyl radical on Lewis acid site in MnO2-D
In situ EPR spectra were collected to probe the evolution of oxygen vacancy of MnO2-D during catalytic oxidation of glycerol (Fig. 4a). The signal of oxygen vacancy could be regarded as the function strength of FLPs in the Mnδ+-OV structure since the strength-intensified FLPs are originated from the rich oxygen vacancies induced by the low-coordination structure. We observe a symmetrical EPR peak at g = 2.003 in MnO2-D, attributable to unpaired electrons associated with oxygen vacancies of metal oxides. Interestingly, the intensity of the peak shows a progressive increase with rising reaction temperature. Moreover, the peak intensity demonstrates a strong linear correlation with initial catalytic activity (Supplementary Fig. 9), indicating that an increase in the oxygen vacancy signal corresponds to a proportional increase in catalytic activity. The sluggish O2 activation is one of the important reasons that restrict the oxidation activity. Inferentially, the formation of strength-intensified FLPs promotes the activation of O2, and then linearly increases the oxidation activity. To confirm this point, free radical trapping agent was added during glycerol oxidation by MnO2-P and MnO2-D catalyst to provide insights on O2 activation (Fig. 4b). Blank test confirmed that there is no signal of radical in the absence of catalyst. In sharp contrast, both catalysts (especially MnO2-D) display obvious hydroxyl radical signals. The hydroxyl radical quenching experiment, employing organic and inorganic radical scavengers, further established that the produced hydroxyl radical significantly impacts the oxidation reaction (Supplementary Table 6). It is plausible that O2 mainly interacts with H2O to generate hydroxyl radicals that participate in the subsequent glycerol activation, aligning with previous studies71,72,73.

a In situ EPR spectra for the detection of the evolution of oxygen vacancies on MnO2-D. b In situ EPR spectra with free radical trapping agent (DMPO) for the oxidation of glycerol on MnO2-P and MnO2-D (for the capitation of hydroxyl radical). c Electron transition in orbit during O2 adsorption over Mnδ+–OV structure.
Due to the difference in electronegativity between Mnδ+ and OV, the d-π feedback of the anti-bonding orbital weakens the O = O bond, allowing O2 to dissociate into OH* in the presence of H2O (Fig. 4c). The activation of O2 is mainly promoted by Mnδ+ acid site with the assistant of OV basic sites since one O atom is directly anchored by the Mn center, while another O atom is activated by H dissociated from H2O. For H2O activation, H2O is dissociated into OH adsorbed on the Mn center and H adsorbed on adjacent OMn or OV. The latter process well resembles the traditional definition of Lewis acid-base, where one accepts OH and the other accepts H. Finally, the Mnδ+ Lewis acid could easily accept the as-formed OH to formation Mnδ+–OH. Isotope experiments were further conducted to investigate whether lattice oxygen in MnO2-D participates in the oxidation reaction (Mars van Krevelen (MvK) mechanism). Supplementary Fig. 9 shows that labeled 18O in MnO2-D is not observed in the oxidation products of glycerol. This confirms that the liquid-phase oxidation reaction on MnO2-D does not occur via the MvK mechanism. Rather, O2 and H2O dissociate into hydroxyl radical on the defective Mnδ+–OV pairs, before participating in the oxidation reaction.
Enhanced C–C and C–H bond activation over Lewis base sites in MnO2-D
The reaction mechanism of polyol oxidation to formic acid over the Mnδ+–OV pair was further investigated by in situ Fourier transform infrared reflection (in situ FTIR), DFT calculation and reaction kinetic studies. Figure 5a shows that the α interaction (1125–1075 cm−1) and γ interaction (1075–1000 cm−1), attributable to alkoxy bond between primary hydroxyl and metal oxides, gradually increase. ρ(OH) at 1370 cm−1 and δ(OH) at 1440 cm−1 belonged to glyceraldehyde gradually increase, suggesting the activation of primary hydroxyl group in glycerol71,74. Meanwhile, the β interaction at 1200–1125 cm−1, and ω(CH2) at 1250 cm−1 and τ(CH2) at 1300 cm−1 belonging to dihydroxyacetone are also enhanced. These suggest MnO2-P activates primary and secondary hydroxyl groups indiscriminately. Compared with MnO2-P, τ(CH2), ω(CH2) and the β interaction corresponding to the activation of secondary hydroxyl groups are significantly lower on MnO2-D, thus the preferential activation of primary hydroxyl group and C–C bond cleavage promote the generation of formic acid.

a In situ FT-IR spectra of glycerol oxidation to formic acid over a1 MnO2-P and a2 MnO2-D. b Schematic diagram of reaction mechanism for the oxidation of glycerol to formic acid (the gray box represents O-H bond activation, the blue box represents C-H bond activation, and the orange box represents hydroxyl radical reaction and C-C bond cleavage).
It is found that MnO2-D exhibits low activation energies for continuous C–C bond cleavage (Supplementary Fig. 10), but high activation energies for deep oxidation reactions (such as the dihydroxy oxidation and decarboxylation to CO2). In contrast, the activation energies of C–H and C–C bonds activation on MnO2-P is significantly higher than that on MnO2-D, and furthermore, the activation energies of multiple C–C bond cleavage are close to that of decarboxylation to CO2 on MnO2-P. These corroborate in situ FTIR findings that MnO2-P without Mnδ+–OV pairs does not activate glycerol effectively and that the specific selectivity towards formic acid is low.
Figure 5b presents a schematic of the oxidation mechanism of glycerol to FA via MnO2-D. Initially, the Mnδ+–OV structure utilizes Mnδ+ to adsorb oxygen from the O–H bond, while the OV attracts hydrogen from the O–H bond, thereby achieving the dehydrogenation of the O–H bond. In the subsequent step, the synergistic adsorption of oxygen-containing intermediates at the Mnδ+ metal site assists OV in attracting hydrogen from the C–H bond, leading to the formation of glyceraldehyde. Under the attack of hydroxyl radical, the as-formed glyceraldehyde is rapidly transformed into CH2OHCHOHCHOOH, which undergoes C–C bond cleavage over Mnδ+–OV structure. After a series of processes compromising multiple O–H bond dehydrogenations, C–H bond dehydrogenation, free radical attack, and C–C bond cleavage steps mentioned above, the original glycerol is converted into three molecules of FA product. Notably, detailed DFT calculation (Supplementary Figs. 11 and 12) shows that the C–H bond activation in the C1H3O2* (HCHOOH*) intermediate with the highest free energy is the rate-determining step (RDS), and the activation energies of two C–C bond cleavages on MnO2-D (Tri) is less than 1 eV. As compared to MnO2-P (Penta) [εd = 2.63 eV], the d-band center of MnO2-D (Tri) [εd = 2.91 eV] moves away from Fermi energy EF, suggesting the electrons in the Mn 3d orbital could effectively overflow into nearby O (Supplementary Fig. 11c)75,76. Benefiting from the enhancement of O electronegativity (Lewis base) in the Mnδ+–OV pair, the binding ability of C and H is abnormally promoted. This provides a superior stable base for C–C bond cleavage and C–H bond activation, resulting in the low activation energies of C–C cleavage and C–H bond activation on MnO2-D (Tri). Combined with micro-kinetic analysis (Supplementary Fig. 11d), the MnO2-D (Tri) is located in the red region (i.e., high TOF) with ~8.0 eV of EC and ~6.0 eV of EO, close to several noble metal catalysts77.
Discussion
In summary, we developed a defective α-MnO2 catalyst enriched with Frustrated Lewis Pairs (FLPs) to promote the cascade oxidation of various polyols and sugars to formic acid. The reduction of oxygen coordination generates abundant exposed Mn species as Lewis acid sites, and strengthens the electron donating properties of adjacent oxygen to serve as Lewis base sites. During catalytic tests, a positive correlation between the abundance of strength-intensified FLPs in the defective Mnδ+–OV structure and catalytic activity was identified. Further characterization by various techniques provide evidence that the FLPs on defective MnO2-D catalyst promote the O2 as well as C–H and C–C bond activation synergistically on adjacent Lewis acid and base sites. As a result, the low-coordination MnO2-D exhibits superior catalytic activity (TOF: 113.5 h−1) and formic acid yield (formic acid yield >80%) for glycerol oxidation, which are comparable to the performance of previously reported noble metal catalysts. The catalyst is also effective in converting ethylene glycol, 1,2-propanediol, 1,3-propanediol, erythritol, xylitol and sorbitol into formic acid in yields ranging from 51.5 to 94.8%. This work demonstrates that metal oxide catalysts with water tolerant FLPs are promising for the oxidative polyol/sugar transformation.
Methods
Catalyst preparation
The MnO2-P catalyst was prepared by a hydrothermal method50. In a typical process, 0.1 mol HCl and 0.25 mol KMnO4 were added into 90 mL deionized water. Then, the mixed solution was transferred into a Teflon-lined stainless autoclave, which was hydrothermally treated at 140 °C (12 h). The as-formed precipitate was centrifugated and washed to remove excess ions. After drying in air overnight (100 °C), the sample was calcined to obtain the MnO2-P catalyst in muffle furnace at 400 °C for 1 h.
The MnO2-D catalyst was prepared by another hydrothermal method50. In a typical process, 1.58 g KMnO4 was added in 30 mL deionized water, and 20 mL (NH4)2C2O4·H2O was further added in the above solution drop by drop. After 1 h of stirring, the mixture was transferred to a Teflon-lined stainless autoclave for 24 h at 180 °C. The as-formed powder was washed and filtered repeatedly. After drying in air overnight (100 °C), it was calcined to obtain the MnO2-D catalyst in muffle furnace at 400 °C for 1 h.
Catalytic test
Catalytic oxidation of polyol/sugar was carried out in the 50 mL autoclave. In a typical process, 25 mL deionized, 0.5 g substrate and 0.1 g catalyst were added in the autoclave. Through several times of O2 washing, the final pressure was maintained at 1.0 MPa. After this oxidation reaction, the liquid product was analysed by the high performance liquid chromatography (HPLC) equipped with refractive index (RID-10A) and UV detectors (Shimadzu LC-20AT). The Rezex ROA-Organic Acid H+ (8%) was used as the chromatographic column in the mobile phase (0.005 M H2SO4). The gas product was detected by the Chromatography equipped with a FID and TCD detector (Scion 456-GC). The definitions of conversion (X), product selectivity (S), turnover frequency (TOF) and carbon mass balance (CMB) were calculated by the following formula:
Theory calculations
The Dmol3 in the Material Studio 8.0 was employed to perform the DFT calculation. The Perdew–Burke–Ernzerh of (PBE) functional with the generalized gradient approximation (GGA) was selected to determine the correlation energies. Sampling brillouin zone (3 × 3 × 1) via Monkhorst–Pack method and the double numerical plus polarization (DNP) together with effective core potentials were used. The TS parameters for van der Waals dispersion correction was considered, and all the energies were corrected by zero point energy (ZPE). The completed LST/QST method was used to search the transition state. Allowable deviations for displacement, gradient and total energy are 0.005 Ǻ, 0.002 Ha/Ǻ and 1.0 × 10−5 Ha, respectively. The cif document of the MnO2-P and MnO2-D catalysts were obtained from the refined XRD, and detailed information was provided in the supporting data. For the two models, the 211 and 310 surface was cut from the MnO2 bulk crystal. The 3-layer p(2 × 2) MnO2-P and 3-layer p(2 × 2) MnO2-D were constructed to represent the MnO2-P and MnO2-D (T) catalysts respectively. During the geometry optimization, the bottom layer atoms of the two models were fixed and the other atoms were relaxed. The adsorption energy is defined as Eads = EMnO2+substrate-EMnO2-Esubstrate, where EMnO2+substrate, EMnO2 and Esubstrate are the total energy of the calculation system, the isolated energy of calculation model and substrate, respectively.
All energy correction terms are extracted from the normal mode analyses of transition state the optimized reactant at various temperatures. Gibbs free energies correction has contained the ZPE correction. Outputs from Dmol3 calculations include corrections to consider bare Gibbs free energies and electronic energies. The activation barrier (ΔE), the activation barrier with zero-point vibrational energy correction (ΔEZ) and free energy presented (ΔG) are obtained using the following formula:
Where ER and ETS are the electronic ground state energies of the reactant and transition state, respectively. The ZPETS and ZPER are the respective zero point vibrational energy (ZPE) corrections. GR and GTS are the free energy corrections of the reactant and the transition state, respectively.
Characterizations
Rietveld refinements were carried out using TOPAS Academic51,53,59. Firstly, Pawley fitting was performed to refine the lattice parameters. The background was modeled using the Chebyshev function with 12 parameters. The peak shape was modeled via the Stephens peak shape function (an approach to spherical harmonics for hkl dependent peak shapes) considering the strain anisotropy broadening3. The initial atomic coordinates for the Manganese oxides with space group I4/m (87) were generated from the crystal structure in the Pearson’s Crystal Data (#1102438)4. The refined parameters such as lattice parameters, background and sample specimen displacement from the Pawley fitting were kept the same for the Rietveld refinements. The z atomic coordinates of the Oxygen and Manganese ions were set to 0. Then, the following parameters were refined in sequence: (1) scale factor, (2) O, Mn atomic coordinates (x and y), (3) atomic (Oxygen and Manganese ions) occupancies and (4) the overall atomic displacement parameters. All the Rietveld refinements gave satisfactory agreement factors.
Scanning electron microscopy (SEM) was used to obtain the morphology of the MnO2-P and MnO2-D catalysts on a Hitachi S-4800. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was carried out on the Titan 80–300 scanning/transmission electron microscope. X-ray photoelectron spectroscopy (XPS) was measured on the Thermo ESCALAB 250Xi with the correction of C 1 s binding energy of 284.8 eV, and other testing and analysis details were provided in Supporting Information. N2 physisorption was measured on Micromeritics ASAP 2020. X-ray absorption spectroscopy (XAS) were measured on the Advanced Photon Source at Argonne National Laboratory (fluorescence mode on beamline 12-BM). The ATHENA module was employed to deal the data of X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure spectroscopy (EXAFS). CO2 temperature programmed desorption (CO2-TPD), H2-temperature programmed reduction (H2-TPR), NH3-TPD and O2-TPD were measured on the Micromeritics AutoChem II 2920. For the CO2-TPD, O2-TPD and NH3-TPD, 0.1 g sample was treated in the N2 gas at 300 °C for 1 h (60 mL/min). After the cool down of the sample (50 °C), 5 vol% O2 (NH3 or CO2) gas in 95 vol% N2 was introduced for 1 h (30 mL/min). Then, the quartz tube was heated to 1000 °C at a rate of 20 °C/min. For H2-TPR, the sample was reduced in 10 vol% H2 in Ar (60 mL/min) from 50 °C to 1000 °C with a heating rate of 15 °C/min. All the signals were collected by TCD detector. UV–vis absorption spectras were measured on a UV–vis–NIR Cary 5 (Varian) spectrophotometer. NMR was conducted on a Bruker Ascend 400 MHz NMR spectrometer. Mass spectrometric analysis was conducted on a QMS 200 (Balzers) quadrupole mass spectrometer.
Electron paramagnetic resonance (EPR) spectra were measured to obtain oxygen vacancy on a Bruker EMX-6/1spectrometer at 298 K. For the in situ EPR experimental (detection of oxygen vacancy), an in situ cell was loaded by 10 mL mixed solution of glycerol and sodium hydroxide (0.05 mol/L). The system pressure drop was monitored in real time. The sample was heated using a Bruker EMX plus continuous flow temperature control system. During this process, pure O2 (100 mL/min) was purged into the cell. The EPR spectra were collected between 2400 and 3600 G in 83 ms. The microwave frequency was 9.3 GHz with a power of 0.2 mW, and the field was modulated at 100 kHz and with an amplitude of 5 G. For the Operando EPR experimental (detection of hydroxyl radical), three kinds of contrast experiments were designed. Fully put MnO2-P and MnO2-D catalysts into the evaluation conditions for reaction (120 °C for 4 h), and immediately put the samples after the reaction with DMPO into EPR for testing. The other group is a parallel blank experiment. Except that no catalyst is added, the other processes are completely consistent.
In situ, Fourier transform infrared reflection (in situ FTIR) was measured on the Thermo Scientific Nicolet iS50 FT-IR. For the in situ CO and CO2, the samples were pretreated by 50 mL/min of N2 at 300 °C for 1 h. After the sample cools to 80 °C, 5 vol% CO or CO2 in N2 (40 mL/min) was purged for 20 min to realize the adsorption saturation. Then, 40 mL/min N2 was performed to remove the gas phase CO or CO2. The spectra were collected using 128 scans in a resolution of 4 cm−1. Detailed testing processes of the in situ FTIR of glycerol oxidation were provided in Supporting Information.
Data availability
All relevant data that support the findings of this study are presented in the manuscript and supplementary information file. Source data are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Shen, F. et al. Critical assessment of reaction pathways for conversion of agricultural waste biomass into formic acid. Green. Chem. 23, 1536–1561 (2021).
Han, X. et al. Electrocatalytic oxidation of glycerol to formic acid by CuCo2O4 spinel oxide nanostructure catalysts. ACS Catal. 10, 6741–6752 (2020).
Farnetti, E. & Crotti, C. Selective oxidation of glycerol to formic acid catalyzed by iron salts. Catal. Commun. 84, 1–4 (2016).
Xu, J. et al. Selective oxidation of glycerol to formic acid catalyzed by Ru(OH)4/r-GO in the presence of FeCl3. Appl. Catal. B. 154-155, 267–273 (2014).
Wu, L. et al. Highly efficient conversion of carbohydrates into formic acid with a heterogeneous MgO catalyst at near-ambient temperatures. ACS Sustain. Chem. Eng. 10, 15423–15436 (2022).
Oh, L. S. et al. How to change the reaction chemistry on nonprecious metal oxide nanostructure materials for electrocatalytic oxidation of biomass-derived glycerol to renewable chemicals. Adv. Mater. 35, 2203285 (2022).
Li, Y., Wei, X., Han, S., Chen, L. & Shi, J. MnO2 electrocatalysts coordinating alcohol oxidation for ultra-durable hydrogen and chemical productions in acidic solutions. Angew. Chem. Int. Ed. 60, 21464–21472 (2021).
Chen, X., Liu, Y. & Wu, J. Sustainable production of formic acid from biomass and carbon dioxide. Mol. Catal. 483, 110716 (2020).
Maerten, S. et al. Glucose oxidation to formic acid and methyl formate in perfect selectivity. Green. Chem. 22, 4311–4432 (2020).
Albert, J. et al. Formic acid-based Fischer-Tropsch synthesis for green fuel production from wet waste biomass and renewable excess energy. ACS Sustain. Chem. Eng. 4, 5078–5086 (2016).
Lu, T., Hou, Y., Wu, W., Niu, M. & Wang, Y. Formic acid and acetic acid production from corn cob by catalytic oxidation using O2. Fuel Process. Technol. 171, 133–139 (2018).
Lu, T. et al. Catalytic oxidation of cellulose to formic acid in H5PV2Mo10O40 + H2SO4 aqueous solution with molecular oxygen. Green. Chem. 18, 4725–4732 (2016).
Niu, M., Hou, Y., Ren, S., Wu, W. & Marsh, K. N. Conversion of wheat straw into formic acid in NaVO3-H2SO4 aqueous solution with molecular oxygen. Green. Chem. 17, 453–459 (2015).
Wang, C. et al. Room temperature, near-quantitative conversion of glucose into formic acid. Green. Chem. 21, 6089–6096 (2019).
Wang, T. et al. A nanoporous nickel catalyst for selective hydrogenation of carbonates into formic acid in water. Green. Chem. 19, 716–721 (2017).
Jin, F. et al. Hydrothermal conversion of carbohydrate biomass into formic acid at mild temperatures. Green. Chem. 10, 612 (2008).
Chagas, P. et al. Metal-free bifunctional silica for conversion of waste glycerol from biodiesel: sustainable production of formic acid. Chem. Eng. J. 369, 1102–1108 (2019).
Park, J. et al. Sustainable low-temperature hydrogen production from lignocellulosic biomass passing through formic acid: combination of biomass hydrolysis/oxidation and formic acid dehydrogenation. Environ. Sci. Technol. 53, 14041–14053 (2019).
Liu, M. et al. One-pot controlled synthesis of AuPd@Pd core-shell nanocrystals with enhanced electrocatalytic performances for formic acid oxidation and glycerol oxidation. J. Colloid Interface Sci. 508, 551–558 (2017).
Schünemann, S., Schüth, F. & Tüysüz, H. Selective glycerol oxidation over ordered mesoporous copper aluminum oxide catalysts. Catal. Sci. Technol. 7, 5614–5624 (2017).
Lin, Y., Yang, J. & Mou, C. Highly selective conversion of glycerol to formic acid over a synergistic Au/Phosphotungstic acid catalyst under nanoconfinement. ACS Sustain. Chem. Eng. 9, 3571–3579 (2021).
Zhu, J. et al. Facile preparation of nitrogen-doped carbon spheres with wrinkled cage-supported single-atom copper catalysts for selective oxidation of glycerol to formic acid. ACS Sustain. Chem. Eng. 10, 17177–17186 (2022).
Zhong, J., Pérez-Ramírez, J. & Yan, N. Biomass valorisation over polyoxometalate-based catalysts. Green. Chem. 23, 18–36 (2021).
Li, D. et al. Selective aerobic oxidation of glycerol over zirconium phosphate-supported vanadium catalyst. Mol. Catal. 474, 110404 (2019).
Wang, F. et al. Unraveling the effects of the coordination number of Mn over α-MnO2 catalysts for toluene oxidation. Chem. Eng. J. 396, 125192 (2020).
Wang, J. et al. The effect of manganese vacancy in birnessite-type MnO2 on room-temperature oxidation of formaldehyde in air. Appl. Catal. B. 204, 147–155 (2017).
Jing, Y. & Wang, Y. Heterolytic dissociation of H2 and bond activation: spotting new opportunities from a unified view. Chem. Catalysis 3, 100515 (2023).
Chen, H. et al. Defect-regulated Frustrated-Lewis-Pair behavior of boron nitride in ambient pressure hydrogen activation. J. Am. Chem. Soc. 144, 10688–10693 (2022).
Lin, W. et al. Creating frustrated Lewis pairs in defective boron carbon nitride for electrocatalytic nitrogen reduction to ammonia. Angew. Chem. Int. Ed. 134, e202207807 (2022).
Chen, W., Han, J., Wei, Y. & Zheng, A. Frustrated Lewis pair in zeolite cages for alkane activations. Angew. Chem. Int. Ed. 134, e202116269 (2022).
Wischert, R., Laurent, P., Copéret, C., Delbecq, F. & Sautet, P. γ-Alumina: the essential and unexpected role of water for the structure, stability, and reactivity of “Defect” sites. J. Am. Chem. Soc. 134, 14430–14449 (2012).
Wischert, R., Copéret, C., Delbecq, F. & Sautet, P. Optimal water coverage on alumina: a key to generate Lewis Acid-Base Pairs that are reactive towards the C-H bond activation of methane. Angew. Chem. Int. Ed. 50, 3202–3205 (2011).
Ghuman, K. K. et al. Illuminating CO2 reduction on frustrated Lewis pair surfaces: investigating the role of surface hydroxides and oxygen vacancies on nanocrystalline In2O3−x(OH)y. Phys. Chem. Chem. Phys. 17, 14623–14635 (2015).
Stephan, D. W. Frustrated Lewis pairs. J. Am. Chem. Soc. 137, 10018–10032 (2015).
Comas-Vives, A., Valla, M., Copéret, C. & Sautet, P. Cooperativity between Al sites promotes hydrogen transfer and carbon-carbon bond formation upon dimethyl ether activation on alumina. ACS Cent. Sci. 1, 313–319 (2015).
Wang, Q., Zhang, M., Chen, C., Ma, W. & Zhao, J. Photocatalytic aerobic oxidation of alcohols on TiO2: the acceleration effect of a Brønsted acid. Angew. Chem. Int. Ed. 122, 8148–8151 (2010).
Makwana, V. The role of lattice oxygen in selective benzyl alcohol oxidation using OMS-2 catalyst: a kinetic and isotope-labeling study. J. Catal. 210, 46–52 (2002).
Yan, H. et al. Insight into the basic strength-dependent catalytic performance in aqueous phase oxidation of glycerol to glyceric acid. Chem. Eng. Sci. 230, 116191 (2021).
Yan, H. et al. Synergistic Pt/MgO/SBA-15 nanocatalysts for glycerol oxidation in base-free medium: catalyst design and mechanistic study. J. Catal. 370, 434–446 (2019).
Gastelu, G. et al. Autocatalytic O-formylation of alcohols using CO2. ACS Catal. 13, 2403–2409 (2023).
Das, S., Turnell Ritson, R. C., Dyson, P. J. & Corminboeuf, C. Design of frustrated Lewis pair catalysts for direct hydrogenation of CO2. Angew. Chem. Int. Ed. 134, e202208987 (2022).
Wang, T. et al. Frustrated Lewis pair catalyzed hydrodehalogenation of benzyl-halides. Chem. Commun. 58, 1175–1178 (2022).
Holtrop, F. et al. Single‐electron transfer in frustrated Lewis pair chemistry. Angew. Chem. Int. Ed. 132, 22394–22400 (2020).
Jupp, A. R. & Stephan, D. W. New directions for frustrated Lewis pair chemistry. Trends Chem. 1, 35–48 (2019).
Zou, Y. et al. Highly selective transfer hydrogenation of furfural into furfuryl alcohol by interfacial frustrated Lewis pairs on CeO2. J. Catal. 410, 54–62 (2022).
Zhang, S. et al. Hydrogen activation enabled by the interfacial frustrated Lewis pairs on cobalt borate nanosheets. J. Catal. 372, 142–150 (2019).
Zhang, S. et al. Interfacial frustrated Lewis pairs of CeO2 activate CO2 for selective tandem transformation of olefins and CO2 into cyclic carbonates. J. Am. Chem. Soc. 141, 11353–11357 (2019).
Xie, C. et al. Defect chemistry in heterogeneous catalysis: recognition, understanding, and utilization. ACS Catal. 10, 11082–11098 (2020).
Gu, H. et al. Adjacent single-atom irons boosting molecular oxygen activation on MnO2. Nat. Commun. 12, 5422 (2021).
Luo, J. et al. Synthesis of single-crystal tetragonal α-MnO2 nanotubes. J. Phys. Chem. C. 112, 12594–12598 (2008).
Stephens, P. W. Phenomenological model of anisotropic peak broadening in powder diffraction. J. Appl. Crystallogr. 32, 281–289 (1999).
Muraoka, Y. et al. Preparation of α-MnO2 with an open tunnel. J. Solid State Chem. 144, 136–142 (1999).
Coelho, A. A. Whole-profile structure solution from powder diffraction data using simulated annealing. J. Appl. Crystallogr. 33, 899–908 (2000).
John, R. E., Chandran, A., Thomas, M., Jose, J. & George, K. C. Surface-defect induced modifications in the optical properties of α-MnO2 nanorods. Appl. Surf. Sci. 367, 43–51 (2016).
O. Ou, et al. Carbon nitride photocatalysts with integrated oxidation and reduction atomic active centers for Improved CO2 conversion. Angew. Chem. Int. Ed. 134, e202206579 (2022).
Gu, T. H. et al. MnO2 nanowire-anchored highly oxidized cluster as a catalyst for Li-O2 batteries: superior electrocatalytic activity and high functionality. Angew. Chem. Int. Ed. 130, 16216–16221 (2018).
Jiang, Y. et al. Jahn-Teller disproportionation induced exfoliation of unit-cell scale ϵ‐MnO2. Angew. Chem. Int. Ed. 132, 22848–22855 (2020).
Zeng, J. et al. Insight into the effects of oxygen vacancy on the toluene oxidation over α-MnO2 catalyst. Chemosphere 291, 132890 (2022).
Yang, W. et al. Insights into the surface-defect dependence of molecular oxygen activation over birnessite-type MnO2. Appl. Catal. B. 233, 184–193 (2018).
Lin, C. et al. In-situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012–1023 (2021).
Zhang, H., Sui, S., Zheng, X., Cao, R. & Zhang, P. One-pot synthesis of atomically dispersed Pt on MnO2 for efficient catalytic decomposition of toluene at low temperatures. Appl. Catal. B 257, 117878 (2019).
Rong, S., Zhang, P., Liu, F. & Yang, Y. Engineering crystal facet of α-MnO2 nanowire for highly efficient catalytic oxidation of carcinogenic airborne formaldehyde. ACS Catal. 8, 3435–3446 (2018).
Biesinger, M. C. et al. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co, and Ni. Appl. Surf. Sci. 257, 2717–2730 (2011).
Li, S. et al. Rational screening of transition metal single-atom-doped ZSM-5 zeolite catalyst for naphtha cracking from microkinetic analysis. Chem. Eng. J. 445, 136670 (2022).
Fagan, W. P., Villamena, F. A., Zweier, J. L. & Weavers, L. K. In Situ EPR spin trapping and competition kinetics demonstrate temperature-dependent mechanisms of synergistic radical production by ultrasonically activated persulfate. Environ. Sci. Technol. 56, 3729–3738 (2022).
Wang, L. et al. Room-temperature activation of H2 by a surface frustrated Lewis pair. Angew. Chem. Int. Ed. 131, 9601–9605 (2019).
Huang, Z., Zhang, T., Chang, C. & Li, J. Dynamic frustrated Lewis pairs on ceria for direct nonoxidative coupling of methane. ACS Catal. 9, 5523–5536 (2019).
Zhang, S. et al. Solid frustrated-Lewis-pair catalysts constructed by regulations on surface defects of porous nanorods of CeO2. Nat. Commun. 8, 15266 (2017).
Maier, A. F. G. et al. Dehydrierende Oxidation von Indolinen und anderen Heterocyclen durch frustrierte Lewis-Paare. Angew. Chem. Int. Ed. 128, 12407–12411 (2016).
Wischert, R., Copéret, C., Delbecq, F. & Sautet, P. Optimal water coverage on alumina: a key to generate Lewis acid-base pairs that are reactive towards the C-H bond activation of methane. Angew. Chem. Int. Ed. 123, 3260–3263 (2011).
An, Z. et al. Pt1 enhanced C-H activation synergistic with Ptn catalysis for glycerol cascade oxidation to glyceric acid. Nat. Commun. 13, 5467 (2022).
Duan, Z. & Wang, G. A first principles study of oxygen reduction reaction on a Pt(111) surface modified by a subsurface transition metal M (M = Ni, Co, or Fe). Phys. Chem. Chem. Phys. 13, 20178 (2011).
Zope, B. N., Hibbitts, D. D., Neurock, M. & Davis, R. J. Reactivity of the gold/water interface during selective oxidation catalysis. Science 330, 74–78 (2010).
Yan, H. et al. PO43-Coordinated robust single-atom platinum catalyst for selective polyol oxidation. Angew. Chem. Int. Ed. 134, e202116059 (2022).
Li, L. et al. Atom-economical synthesis of dimethyl carbonate from CO2: engineering reactive frustrated Lewis pairs on ceria with vacancy clusters. Angew. Chem. Int. Ed. 61, e202214490 (2022).
Xiang, S. et al. A unique Co@CoO catalyst for hydrogenolysis of biomass-derived 5-hydroxymethylfurfural to 2,5-dimethylfuran. Nat. Commun. 13, 3657 (2022).
Yan, H. et al. Rational screening of metal catalysts for selective oxidation of glycerol to glyceric acid from microkinetic analysis. AIChE J. 69, e17868 (2022).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22108305, 22272199, and 21978325), the Natural Science Foundation of Shandong Province (ZR2021QB076, ZR2020YQ17, ZR2020KB006, ZR2023YQ009, and ZR2022MB015), Fundamental Research Funds for the Central Universities (20CX06073A) and Postdoctoral Research Foundation of China: China Postdoctoral international exchange program (2020072). N.Y. thanks the the National Research Foundation Singapore NRF Investigatorship (NRF-NRFI07–2021–0006) for the financial support.
Author information
Authors and Affiliations
Contributions
H.Y. and N.Y. conceived this work. H.Y. performed the experiments, collected the data, and wrote the paper. B.L. performed the XRD. X.Z., F.M., M.Z., Y.P., J.L., Y.W., and H.Z. conducted the evaluation experiment. Y.L. and X.F. helped with the characterization analysis. X.C., D.C., L.L., and H.S. assisted the XAS characterization and analysis. C.Y. and N.Y. helped with data analyses and discussions. N.Y. revised the paper. All authors contributed to editing the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mark Douthwaite, Yongquan Qu and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, H., Liu, B., Zhou, X. et al. Enhancing polyol/sugar cascade oxidation to formic acid with defect rich MnO2 catalysts. Nat Commun 14, 4509 (2023). https://doi.org/10.1038/s41467-023-40306-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40306-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
