
Abstract
A general approach to promote IR light-driven CO2 reduction within ultrathin Cu-based hydrotalcite-like hydroxy salts is presented. Associated band structures and optical properties of the Cu-based materials are first predicted by theory. Subsequently, Cu4(SO4)(OH)6 nanosheets were synthesized and are found to undergo cascaded electron transfer processes based on d-d orbital transitions under infrared light irradiation. The obtained samples exhibit excellent activity for IR light-driven CO2 reduction, with a production rate of 21.95 and 4.11 μmol g−1 h−1 for CO and CH4, respectively, surpassing most reported catalysts under the same reaction conditions. X-ray absorption spectroscopy and in situ Fourier-transform infrared spectroscopy are used to track the evolution of the catalytic sites and intermediates to understand the photocatalytic mechanism. Similar ultrathin catalysts are also investigated to explore the generality of the proposed electron transfer approach. Our findings illustrate that abundant transition metal complexes hold great promise for IR light-responsive photocatalysis.
Similar content being viewed by others


Introduction
In the pursuit of global carbon neutrality, artificial photocatalysis offers a powerful solution to simultaneously address the issues of the greenhouse effect and energy shortage. This process involves the direct transformation of carbon dioxide (CO2) and water into useful fuels and oxygen under ambient conditions1. The proper photocatalysts with well-matched band edge positions are the prerequisite requirement to achieve this objective. In principle, the conduction band minimum (CBM) of the catalysts should be more negative than the potential for CO2 reduction, while the valance band maximum (VBM) needs to be more positive than the potential for water oxidation. For this purpose, the band gaps of the photocatalysts should ideally fall within the range of 1.8–2.0 eV2. Recently, the infrared (IR) light-driven redox reactions have gained widespread attention among researchers. Despite the low energy of IR light and its tendency to generate localized heat, its relatively high proportion in the solar spectrum (ca. 50%) has prompted people to explore ways to utilize it. In principle, IR light is theoretically precluded for photocatalytic CO2 reduction owing to its low photonic energy of less than 1.55 eV3. Fortunately, band structure engineering provides a viable strategy to fine-tune the energy band distribution and optimize the transfer behavior of activated electrons4. In this scenario, the step-by-step electron transition mode has proven to be the most attractive strategy to effectively utilize the low-energy photons, which could trigger the concurrent transformation of CO2 and water into hydrocarbons and oxygen under IR light irradiation5,6.
Recently, some ultrathin metallic catalysts (such as CuS and CoS2 nanosheets) have been reported to offer an activity for IR light-driven CO2 photoreduction, in which the intraband and interband electron transitions are coupled to realize the absorption of IR light while enabling the redox potential requirements of electron-hole pairs (Fig. 1a)7,8. Besides, some bismuth-based catalysts, like metallic Bi19Br3S27 nanowires9, has also been reported to exhibit excellent activity for CO2 reduction under near-infrared (NIR) light irradiation. However, metallic conductors usually suffer from severe recombination of the photoexcited charge carriers, resulting in low efficiency for CO2 photoreduction. Meanwhile, the serious electron scattering effect and localized plasmon effect of the metallic conductor can give rise to an energy loss of the active electrons and localized lattice heating, leading to the deactivation of the catalysts10,11. On the other hand, the defect-induced semiconductors with intermediate bands (e.g., WO3 nanosheets with oxygen vacancies) also exhibit certain performance for IR light-driven CO2 reduction, in which the reasonably regulated intermediate band could serve as a step for the cascaded electron transfer processes under IR light excitation (Fig. 1b)3. Nevertheless, the limited materials selection and harsh processes of defect regulation retard their further development. As such, there is an urgent need to investigate photocatalytic systems that follow a universal principle for the transition of charge carriers under IR light. Such systems would expand the range of photocatalysis to the IR spectrum and enable the preparation of full-spectrum-response catalysts by combining infrared photoactive catalysts with conventional catalysts.

a Metallic catalysts (the energy bands below the Fermi level with fully occupied electrons are regarded as CB and the first band above the Fermi level without electron occupation is called to B1). b Semiconductors with the intermediate band; c Hydrotalcite-like hydroxy salts with d-d orbital transition. Green ball: electrons; White ball: holes; Red lightning: IR light irradiation; Red arrows: electron transfer pathway; CB: conduction band; VB: valence band; B1: Lowest empty band; IB Intermediate band; — Fermi level.
Transition metal ion complexes are diverse and can be easily prepared, usually exhibiting a high IR light absorption capacity due to the d-d interband transitions12,13. In general, the transitions of electrons in d orbitals are parity-forbidden because of the same angular momentum quantum number. However, in some transition metal complexes with tetrahedral or octahedral coordination, like hydrotalcite-like hydroxy salts, the strong p-d orbital coupling between the coordinating groups and the metal ion can result in the degeneracy of valence band d orbitals of transition metals14. Therefore, some empty d orbitals appear in the middle of the band gap, which could act as a “cushion step” to make the d-d transition possible and thus induce a strong IR light absorption (Fig. 1c). Nevertheless, like most bulk materials, the excited carriers under IR light irradiation recombine rapidly and cannot be utilized for the catalytic reaction. On account that two-dimension (2D) materials possess ultrathin thickness in one dimension, they could have great potential for electron-hole separation benefiting from the shorter distance for carrier transfer15,16,17. To this end, the hydrotalcite-like hydroxy salts with 2D nanosheet structure could be a good candidate for IR light absorption and charge carrier separation so that the activated electrons participate in the photocatalytic reactions.
In this work, we first predict the existence of splitting d orbitals in 2D hydrotalcite-like hydroxy salts and their potential IR response-ability by the simulated density of states (DOS), band structures, and optical absorption spectra. Then, ultrathin basic copper sulfate (Cu4(SO4)(OH)6) nanosheets (CSON) were prepared as the prototype, and an IR light-driven CO2 reduction reaction was taken as an example to verify the possibility of d-d transition-induced photocatalytic reactions. The basic OH groups on the surface of Cu4(SO4)(OH)6 nanosheets provide abundant sites for initial CO2 adsorption. Meanwhile, the low-coordination tetrahedral Cu in the CSON is confirmed by X-ray absorption near edge structure (XANES) analyses and Auger electron spectroscopy, which could act as the active site for CO2 activation and continuous protonation. Ultraviolet-visible-near-infrared (UV-vis-NIR) spectra and Gibbs free energy diagrams demonstrate that the tetrahedral Cu sites could not only improve the IR light absorption but also reduce the reaction energy barrier for CO2 reduction. Combining Tauc plot, density functional theory (DFT) calculations, and in situ FTIR spectrum, we propose a photocatalytic mechanism of the activated parity-forbidden electron transfer of d-d orbitals under IR light irradiation (Fig. 1c). As a result, the ultrathin CSON exhibit effective production of CO and CH4 with the evolution rate of 21.95 and 4.11 μmol g−1 h−1, respectively, under IR light irradiation, outperforming most reported catalysts under the same reaction conditions (yield < 20 μmol g−1 h−1). We further demonstrate the universality of IR light-driven CO2 reduction based on the d-d electron transfer mechanism by preparing and evaluating various 2D hydrotalcite-like hydroxy salts, like Cu2(NO3)(OH)3 nanosheets (CNON), Cu3(PO4)(OH)3 nanosheets (CPON) and Cu2(CO3)(OH)2 nanosheets (CCON), which provide robust catalyst systems with high performance for photocatalytic CO2 reduction.
Results
Theoretical design and simulations of 2D hydrotalcite-like hydroxy salts
We first construct the model of 2D CSON with the thickness of a single-unit cell for theoretical simulations (Fig. 2a). [100] plane is selected as the exposed surface since it is predicted to have the highest surface energy of 1.03 J m−2 compared with [010] facet (0.78 J m−2) and [001] facet (0.67 J m−2) (Supplementary Fig. 1), suggesting [100] plane has the most excellent activity for catalytic reactions. Moreover, the calculated optical absorption spectra (Fig. 2b) show that there are significant absorption edges in the IR light region (0.6−1.6 eV), which could be caused by the d-d electron transfer. And the intrinsic band gap for photon absorption is around 3.0 eV, indicating that the redox potential of CSON is large enough for simultaneously catalyzing CO2 reduction and water oxidation.

a Theoretical slab model for CSON with the thickness of 1.3 nm along [100] direction. b The simulated optical absorption spectra, (c) DOS and (d) energy band structure of ultrathin 2D CSON slab. Black and red lines in (b) mean the optical absorption along x and y directions of the slab. Black and red lines in (d) represent the spin up and spin down energy band respectively. Source data are provided as a Source Data file.
The DOS and band structure are further calculated to unveil the distribution of orbitals and possible transfer pathways for the excited electrons. As expected, there are apparent splitting d orbitals in the band gap of the spin-down DOS of CSON (Fig. 2c), which is fully consistent with the corresponding band structure shown in Fig. 2d. According to the projected density of state (PDOS) (Fig. 2c), the occurred empty d bands located in the band gap are attributed to the degeneracy of d orbitals in Cu atoms. The hybridization of d orbitals in transition metal atoms and p orbitals in OH ligands leads to the splitting of the metal d electron orbitals, shifting part of the Cu d bands level up to form the empty d bands, which could act as a “cushion step” for d-d electron transition and achieve the cascaded electron transition process. In this respect, valence electrons absorb the IR-light photons and get excited to the middle empty d bands. Subsequently, these activated metastable electrons continue to absorb IR light and transition to the conduction band, enabling them to participate in the CO2 reduction reaction. Meanwhile, the remaining valence band holes can be utilized for the H2O oxidation reaction (Fig. 1c). Therefore, the d-d electron transition mechanism and suitable bandwidth of 2D CSON provide a high potential for IR light absorption and possible activity for IR light-driven CO2 photoreduction. Similarly, theoretical slab models of CNON, CPON and CCON are also analyzed by DFT calculations. According to the obtained DOS and band structures (Supplementary Fig. 2), the splitting empty d orbitals exist in all the Cu-based hydrotalcite-like hydroxy salts (CHHS), indicating the universality of d-d electron transfer mechanism and the huge potential of 2D CHHS for IR light-driven photocatalysis.
Synthesis and characterizations of 2D CSON
Guided by the theoretical simulations, we then synthesized the ultrathin 2D CSON through the solvothermal method (see details in Methods). TEM and HRTEM images in Supplementary Fig. 3a, b show that the obtained CSON have a flake-like morphology with the exposed plane along the [100] direction. The corresponding energy-dispersive spectroscopy (EDS) element mapping images verify the homogeneous distribution of Cu, S and O elements (Supplementary Fig. 3c–f). The thickness of 2D CSON is determined to be ∼1.3 nm by atomic force microscopy (AFM) (Supplementary Fig. 4), which is consistent with the theoretical thickness of its single-unit cell. Compared with its bulk counterpart, CSON exhibit a high ratio of surface Cu sites with tetrahedral coordination in theory (Supplementary Fig. 5).
Previous studies have suggested that the tetrahedral Cu atoms generally exhibit higher extinction coefficients in the IR light region, which are expected to be around 50–100 times larger than that of octahedral Cu13. In this regard, we also prepared the calcined Cu4(SO4)(OH)6 nanosheets (c-CSON) with abundant surface vacancies by calcining the pristine Cu4(SO4)(OH)6 nanosheets (p-CSON) at 300 °C in air (see details in Methods), in which the surface -SO4 vacancies will induce the generation of more tetrahedral Cu atoms. Thermogravimetry-differential scanning calorimetry (TG-DSC) and thermogravimetry-differential thermal gravity (TG-DTG) curves were conducted to explore the structural stability of CSON during the calcination process (Supplementary Fig. 6). The similar TG-DSC and TG-DTG curves of p-CSON and c-CSON confirm that the crystal phase and composition of the two samples are almost identical before and after calcination. Meanwhile, the DSC curves of both p-CSON and c-CSON show that there is no recrystallization or melting process before 300 °C, and only a recrystallization peak occurs at 552–560 °C, followed by a melting peak at 717–718 °C. TEM images in Fig. 3a directly show the clear micro-porous structure on the surface of c-CSON. Compared with the p-CSON, there is no obvious change in the whole 2D morphology, crystallinity, and element distributions, as displayed in Fig. 3b, c. AFM images in Fig. 3d and Supplementary Fig. 7 confirm that the c-CSON maintain the ultrathin but a porous surface with uneven height profiles. Besides, the X-ray diffraction (XRD) pattern, Fourier-transform infrared spectroscopy (FTIR) spectrum (Supplementary Fig. 8) and X-ray photoelectron spectroscopy (XPS) analysis (Supplementary Fig. 9) illustrate that the phase and structure of 2D CSON after calcination are well retained. According to the EDS (Supplementary Fig. 10) and XPS composition analyses (Supplementary Table 1), the c-CSON exhibit reduced S content compared with p-CSON, which further corroborates the existence of surface -SO4 vacancies.

a TEM image. b HRTEM image, in which the exposed facet can be inferred along [100] direction because 0.215 nm and 0.233 nm interplanar distances match well with the d003 and d040 spacings, and the corresponding dihedral angle of 90◦ agrees well with the calculated angle between the (003) and (040) planes. c Annular dark-field TEM images and corresponding elemental mapping images, scale bar: 200 nm; (d) AFM images, in which the obtained nanosheets is confirmed to be ca. 1.31 nm with a hole-rich surface. Insert: the corresponding height profiles of i area (unit: nm). e Cu K-edge XANES spectra, the insert is a zoomed-in view of the part of the green circle. f Cu LMM Auger electron spectroscopy in p-CSON and c-CSON, in which the ratio of Cu (I) component of the calcined samples is remarkably increased after calcination. Source data are provided as a Source Data file.
To further demonstrate the tetrahedral-coordination Cu sites, X-ray absorption near-edge structure (XANES) analyses were performed to determine the valence and coordination environment. As depicted in Fig. 3e and the insert, compared with p-CSON, the Cu K-edge position of the c-CSON shifts to a lower energy and the intensity of the white line at round 8996 eV for c-CSON is much weaker. These verify a lower coordination of Cu atoms in c-CSON18,19. The Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) profiles (Supplementary Fig. 11) show a main peak at about 1.89 Å in both c-CSON and p-CSON, which could be attributed to the Cu-O scattering path, fairly agreeing with the Cu-O bonds (1.91 Å) in theoretical models. The Cu LMM Auger electron spectroscopy was further conducted to reveal the Cu coordination information in p-CSON and c-CSON. As shown in Fig. 3f, the ratio of low-coordination Cu atoms in c-CSON is significantly increased compared to that in the pristine one, indicating the increased tetrahedral Cu sites after the calcination. Moreover, the calculated optical absorption spectra (Supplementary Fig. 12a) confirm that c-CSON also exhibit IR light absorption edges between 0.6 and 1.6 eV. The corresponding DOS and band structure of c-CSON (Supplementary Fig. 12b, c) show that there are some similar splitting d orbitals in the band gap as in p-CSON. Based on the DFT predictions and experimental characterizations presented above, it is evident that both the synthesized p-CSON and c-CSON have the ability to absorb IR light through the d-d electron transition mechanism. This makes it possible to drive CO2 reduction using IR light.
d-d orbital transition processes and IR light-driven CO2 reduction performance
UV-vis-NIR diffuse reflectance spectra were conducted to evaluate the IR light response of p-CSON and c-CSON (Fig. 4a). The strong absorption of IR light by both p-CSON and c-CSON is evident, with the calcined sample exhibiting a higher absorption intensity. This observation is consistent with the color change of 2D CSON from light blue to dark green after calcination (inset of Fig. 4a). The corresponding Tauc plots in Fig. 4b, c illustrate that there are two obvious IR absorption edges around 1.09 and 1.17 eV in p-CSON, and 1.07 and 1.18 eV in c-CSON, fairly agreeing with the theoretical optical absorption spectra. Such IR light absorption is attributed to the electron transfer from the valence band to the splitting empty d band and subsequent continuous transfer to the conduction band, indirectly confirming the d-d transition and cascaded electron transfer processes. To reveal the precise band structure, ultraviolet photoelectron spectroscopy (UPS) was further conducted. As displayed in Fig. 4d–f and Supplementary Figs. 13, 14, for electrode potential (V vs. NHE) at pH = 0, the positions of the CBM for p-CSON (−0.47 V) and c-CSON (−0.31 V) are more negative than the potential of CO2 reduction to CO (−0.11 V) and CH4 (0.16 V), while the VBM for p-CSON (2.70 V) and c-CSON (2.42 V) are more positive than the potential for H2O oxidation to O2 (1.23 V). These results are well-consistent with those obtained from Mott-Schottky plots presented in Supplementary Fig. 15, clearly suggesting the ability for CO2 conversion and oxygen generation over p-CSON and c-CSON.

a UV-vis-NIR diffuse reflectance spectra; Tauc plots for (b) p-CSON and (c) c-CSON. d, e SRPES valence-band and secondary electron cutoff spectra of c-CSON, in which VBM is located at 3.09 eV below the Fermi level determined from Au (called as Eedge) and the energy of secondary electron cutoff is 36.17 eV. f Schematics illustrating the electronic band structures of c-CSON; blue arrows represent the electron transition process. CB, conduction band; VB, valence band. g Yields of photocatalytic CO2 reduction to CO and CH4 over different catalysts and conditions, error bars represent the standard deviation (s. d.) of three independent measurements using 5 mg fresh sample for each measurement. h SVUV-PIMS spectrum of the products after 13CO2 photoreduction for c-CSON at hν = 14.5 eV. i Cycling measurements for CO2 photoreduction to CH4 and CO on c-CSON (using 5 mg sample for this measurement; when a new catalytic cycle begins, the reactor is pumped and refilled with pure CO2). Source data are provided as a Source Data file.
To evaluate the catalytic activity of p-CSON and c-CSON for IR light-driven CO2 photoreduction, photocatalytic experiments were performed under IR light irradiation (simulated by the solar simulator with the AM1.5 and CUT800 filters) without any sacrificial agent at ambient temperature (see more details in the Methods section). The corresponding spectrum of our solar simulator is displayed in Supplementary Fig. 16, in which the IR light component is obtained by using the CUT800 filters. Since the IR light tends to generate heat and raise the temperature of the photocatalyst and reactor20,21, a water-cooled gas-solid reaction system (Supplementary Fig. 17a, b) was designed and applied in this work to avoid the effects of thermocatalysis. The average temperature of CSON-based thin film during IR light irradiation has proved to be almost unchanged by the in situ thermographic photographs (Supplementary Fig. 17c–h). The performance evaluations in Fig. 4g and Supplementary Fig. 18 demonstrate that both p-CSON and c-CSON exhibit excellent activity for CO and CH4 production under IR light irradiation, in which the calcined sample shows better performance with the CO and CH4 evolution rate of 21.95 and 4.11 μmol g−1 h−1 respectively, outperforming most reported catalytic systems (yield < 20 μmol g−1 h−1) under the similar conditions (Supplementary Table 2). The product selectivity of CO and CH4 is calculated to 84.2% and 15.8% for c-CSON, 85.7% and 14.3% for p-CSON; while the electron selectivity is 57.2 % and 42.8% for c-CSON, 59.9 % and 40.1 % for p-CSON. Moreover, the apparent quantum yield of c-CSON for IR light-driven CO2 reduction (Supplementary Fig. 18b) is calculated up to 0.122% under the monochromatic light of 800 nm wavelength, which is quite high compared with the previously reported IR-light catalysts, such as WO3 and CuS3,7. As for the oxidation end, the remaining holes in the valence band are consumed by water to generate O2 (Supplementary Fig. 19). No other liquid or gas products are detected by gas chromatography (GC) (Supplementary Fig. 20) and nuclear magnetic resonance (NMR) spectrum (Supplementary Fig. 21).
In addition, several controlled experiments were conducted to explore the IR light-driven CO2 reduction reaction over the CSON, including experiments conducted in the dark, without CO2, and without catalysts. As shown in Fig. 4g, these experiments confirm that IR light, CO2 reactant, and catalysts are all necessary for IR light-driven CO2 reduction. The isotope-labeled 13CO2 mass spectrometry for c-CSON (Fig. 4h and Supplementary Fig. 22a, b) and p-CSON (Supplementary Fig. 22c) was performed to further unveil the source of CO and CH4 production, in which the photon energy of 14.5 eV is selected for distinguishing the CO and CH4 according to the absolute photoionization cross sections for CO2, CO and CH4 in Supplementary Fig. 23. As a result, only 13CO and 13CH4 species are detected after using the isotope-labeled 13CO2 as reactants, implying that the products indeed originate from IR light-driven CO2 reduction for both of them. It is worth noting that c-CSON do not show any significant performance degradation after 96 h long-term photocatalysis (Fig. 4i). The excellent stability of the catalysts is further confirmed by the TEM, HRTEM, XRD patterns, and FTIR spectra as the morphology and crystal structure for c-CSON remain unchanged before and after 96 h continuous photocatalysis (Supplementary Fig. 24).
Mechanistic insights into the CO2 photoreduction processes
To reveal the electron transition processes by the photoexcitation, the corresponding carrier dynamics characterizations were performed as shown in Supplementary Fig. 25. The transient photocurrent response spectra (Supplementary Fig. 25a) demonstrate that p-CSON and c-CSON exhibit significant photocurrent density under IR light irradiation, indicating that both samples are capable of achieving IR light-driven excitation of electron-hole pairs and charge carrier separation. It is notable that c-CSON show higher photocurrent density and smaller transfer resistance (Supplementary Fig. 25a, b), illustrating the associated higher photoexcited carrier density and rapid electron transition processes. On the other hand, the longer fluorescence lifetime and lower intensity of photoluminescence (PL) spectroscopy indicate that the charge carriers in c-CSON could be easier to separate and participate in the catalytic reactions instead of recombination (Supplementary Fig. 25c, d). The excellent carrier dynamics observed in c-CSON can mainly be attributed to the presence of more surface Cu sites with tetrahedral coordination and the ultrathin 2D porous configuration. These characteristics not only enhance IR light absorption but also reduce the transfer distance, thereby accelerating charge carrier separation. This, in turn, is beneficial for subsequent IR light-driven CO2 reduction.
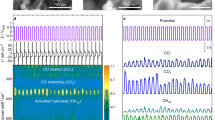
To further understand the reaction mechanism, in situ FTIR spectroscopy was performed to identify the reaction intermediates during IR light-driven CO2 reduction in real-time. As displayed in Fig. 5a for c-CSON and Supplementary Fig. 26a for p-CSON, a series of new infrared peaks at around 1570 cm−1 were detected for both samples, which are attributed to the COOH* group, a crucial intermediate for CO2 reduction to CO or CH422,23. At the same time, the absorption bands near 1116 and 1028 cm−1 belong to the CH3O* group, and the peaks at 1064 cm−1 are assigned to the characteristic bands of CHO*; both the CH3O* and CHO* groups are pivotal intermediates of CO2 photoreduction to CH424,25,26,27. Moreover, the peaks at 2042 and 2175 cm−1 could be attributed to the adsorbed and free CO products respectively28,29,30. It is notable that the intensities of all these peaks for reaction intermediates and products gradually increased with extension of the irradiation time, suggesting the continuous photocatalytic reaction progresses. Besides, the peaks at 1305 and 1208 cm−1 are inferred to the symmetric and asymmetric stretching of HCO3* group, while the peaks at 1411 cm−1 are indexed to the CO3* group22,31. Based on the analysis of in situ FTIR, the most likely reaction pathway for this IR light-driven CO2 reduction could be proposed as follows:
where the asterisks * represent the possible active sites in the catalysts32.

a In situ FTIR spectroscopy characterization for co-adsorption of a mixture of CO2 and H2O vapor under light irradiation over c-CSON. b Gibbs free energy diagrams and (c) intermediate structures of c-CSON during CO2 reduction under IR light irradiation. RDS: rate-determining step. Source data are provided as a Source Data file.
Transition metals are noteworthy for their half-filled d orbitals and high charge density, which allow them to not only provide additional orbitals for bonding with reactants but also participate in catalytic reactions by providing electrons33. In particular, Cu site has been extensively studied as an active center for CO2 reduction reactions34. In comparing p-CSON with defective c-CSON, we observed a significant increase in low-coordinated Cu (tetrahedral Cu) in c-CSON. As a result, the product yield also increased, but the product selectivity remained largely unchanged. This suggests that c-CSON and p-CSON have similar active centers, with differences only in the amount of catalytic sites. Therefore, tetrahedral Cu is regarded as the active site for CO2 reduction. According to the detected intermediates of in situ FTIR, the theoretical models of different reaction intermediates were constructed and the corresponding Gibbs free energy was also calculated to determine the reaction pathway and energy barrier of c-CSON (Fig. 5b, c) and p-CSON (Supplementary Fig. 26b, c) during IR light-driven CO2 reduction. The energy of each elementary reaction is provided in Supplementary Tables 3–5. As the first step, CO2 adsorption plays an important role in promoting the catalytic performance35,36. Given that the basic groups, like -OH and -NH2 ligands, are usually regarded as the efficient active site for CO2 adsorption benefiting from the acid-base neutralization effect37,38, we first calculated the CO2 adsorption energy on OH and Cu sites in CSON. As shown in Supplementary Fig. 27, CO2 molecules prefer to be adsorbed on the surface OH sites than Cu sites due to the lower adsorption energy of the former (−0.02 eV for OH sites and 0.08 eV for Cu sites). More importantly, compared with p-CSON (14.4 m2 g−1), c-CSON with abundant surface vacancies possess about 2-times higher specific surface area (24.2 m2 g−1) according to the Brunauer-Emmet-Teller (BET) plots in Supplementary Fig. 28a, which is beneficial for CO2 adsorption, as further confirmed by the CO2 adsorption isotherms in Supplementary Fig. 28b. Subsequently, the adsorbed CO2* would move to the nearby Cu sites to be activated and protonated for the formation of COOH* intermediates, which is considered as the rate-determining step (RDS) according to the calculated Gibbs free energy in Fig. 5b.
Moreover, the calculated Bader charge (Supplementary Fig. 29) and charge density distribution (Supplementary Fig. 30) demonstrate that the surface Cu sites in c-CSON have higher electron density than that in the pristine sample, which is helpful for CO2 activation and continuous protonation processes. We further calculated the adsorption energy and charge density difference (CDD) of COOH* intermediates on p-CSON and c-CSON. As shown in Supplementary Fig. 31, c-CSON exhibit stronger interaction with the key intermediate of COOH* radicals, in which the adsorption energy on c-CSON is −1.55 eV, much stronger than on p-CSON (−1.16 eV). The corresponding CDD shows that electron transfer between COOH* intermediate and c-CSON is more than that in p-CSON (Supplementary Fig. 32). This means that the surface Cu sites in c-CSON are more favorable for adsorbing COOH* intermediates and thus lower the reaction energy barrier. This result fairly agrees with the calculated Gibbs free energy - the energy barrier of the RDS reduces from 1.925 eV for p-CSON to 1.535 eV for c-CSON. As displayed in Supplementary Fig. 33, different surface Cu sites in c-CSON show similar adsorption energy for COOH* intermediates, which suggests that the surface -SO4 vacancies can significantly reduce the steric hindrance and provide more active Cu sites for CO2 conversion, therefore promoting the performance of IR light-driven CO2 reduction.
The generality of d-d electron transfer approach for IR light-driven CO2 reduction
We demonstrate that the d-d electron transition processes and the underlying reaction mechanisms for IR light-driven CO2 reduction can also be extended to various hydrotalcite-like hydroxy salts. Therein, we prepared CNON, CPON and CCON samples. TEM and HRTEM images confirm their flake-like 2D thin sheet structures (Supplementary Fig. 34a, b, Supplementary Fig. 37a, b, and Supplementary Fig. 40a, b). The corresponding elemental mapping images illustrate the uniform distribution of each element in the counterpart nanosheets (Supplementary Fig. 34c–f, Supplementary Fig. 37c–f, and Supplementary Fig. 40c–f). According to XRD patterns, their phase is indexed to standard PDF card No. 75-1779 for CNON (Supplementary Fig. 35b), No. 72-0258 for CPON (Supplementary Fig. 38b) and No. 72-0075 for CCON (Supplementary Fig. 41b), respectively. Theoretical slab models show that all these nanosheets possess octahedral (in bulk) and tetrahedral (in surface) Cu coordination structures (Supplementary Fig. 35a, Supplementary Fig. 38a, and Supplementary Fig. 41a), indicating their potential IR light absorption abilities. As a result, UV-vis-NIR diffuse reflectance spectra (Supplementary Fig. 35c, Supplementary Fig. 38c, and Supplementary Fig. 41c) clearly demonstrate their similar optical absorption properties with CSON. More impressively, the occurred low-energy absorption edges located around 1.10 eV in the Tauc plots for CNON (Supplementary Fig. 35c), CPON (Supplementary Fig. 38c) and CCON (Supplementary Fig. 41c) are all attributed to the IR light-driven d-d electron transition, while the high-energy absorption edges (higher than 3 eV) represent the intrinsic electron transition from the valence band to the conduction band. To evaluate their catalytic activity for IR light-driven CO2 photoreduction, photocatalysis experiments were performed under the same condition as that for CSON. As displayed in Supplementary Figs. 36, 39, and 42, the obtained CHHS delivered pronounced catalytic activity for IR light-driven CO2 photoreduction, with the CO and CH4 yields of 5.49 and 15.34 μmol g−1 h−1 for CNON, 1.81 and 1.03 μmol g−1 h−1 for CPON, 2.11 and 0.43 μmol g−1 h−1 for CCON, respectively.
Discussion
In summary, we have certified a cascaded d-d electron transition mode over the 2D hydrotalcite-like hydroxy salts, which can be used as the general strategy for IR light-driven CO2 reduction. Various ultrathin Cu-based hydrotalcite-like hydroxy salts are firstly synthesized, for which theoretical and experimental analysis confirm their catalytic performance for IR light-driven CO2 photoreduction based on d-d electron transition mode. In situ characterizations and DFT calculations unveil the evolutions of active sites, charge carriers, and reaction intermediates. As the result, c-CSON exhibit excellent performance for IR light-driven CO2 reduction to CO and CH4 with the rate of 21.95 and 4.11 μmol g−1 h−1 respectively, outperforming most reported IR-responsive catalysts. This work has thus illustrated that the 2D transition metal hydrotalcite-like hydroxy salts have tremendous potential for IR light-driven CO2 reduction, providing an opportunity for the development of IR-light-responsive catalysts and paving the way for the design of full-spectrum catalytic systems.
Methods
Materials
CuSO4 ∙ 5H2O (ACS reagent, ≥ 98%), NaOH (ACS reagent, ≥ 97.0%, pellets), Cu(NO3)2 ∙ 3H2O (puriss. p.a., 99–104% (RT)), Na2HPO4 (ACS reagent, ≥ 99.0%), CuCl2 ∙ 2H2O (ACS reagent, ≥ 99.0%), Na2CO3 (powder, ≥ 99.5%, ACS reagent), Hexadecyltrimethylammonium bromide (CTAB) (≥ 98%), Ethanol (absolute, suitable for HPLC, ≥ 99.8%) and Ammonium hydroxide solution (puriss. p.a., reag. ISO, reag. Ph. Eur., ≥ 25% NH3 basis) are all acquired from Sigma-Aldrich and were used without any further purification. Deionized (DI) water with resistivity of 18.2 MΩ.cm is obtained by the ultra-pure water system from Stakpure GmbH, Germany.
Catalysts synthesis
Synthesis of p-CSON
A 360 mg portion of CuSO4 ∙ 5H2O was added into the mixture of 8 mL absolute ethanol and 10 mL deionized (DI) water. After vigorous stirring for 30 min, 18 mg hexadecyltrimethylammonium bromide (CTAB) was added into the above solution followed by stirring for another 30 min. Subsequently, 18 mL ammonia solution with a 1% volume concentration and 0.4 mL 1.9 M NaOH solution was slowly dropped into the mixture system. Afterwards, the solution was transferred into a 50 mL Tefon-lined autoclave, sealed and heated at 80 °C for 30 min, and allowed to cool to room temperature naturally. The final product was collected by centrifuging the mixture, washed with ethanol and deionized (DI) water for several times until the organic residuals were completely removed, and then dried in vacuum oven at 60 °C overnight. The light blue powder was obtained for further usage.
Synthesis of c-CSON
In a typical procedure, the as-obtained ultrathin 2D p-CSON were calcined at 300 °C with a heating rate of 10 °C min−1 for 1 h under the air atmosphere and then cooled to room temperature. The obtained black green powder was collected for further characterization.
Synthesis of CNON
2 mmol (483.2 mg) Cu(NO3)2 ∙ 3H2O was added into 30 mL DI water. After vigorous stirring for 10 min, 30 mL NaOH solution with a concentration of 0.1 M was dropwise added into the above mixture followed by stirring for 10 min. Afterwards, the mixture solution was further ultrasonicated for 45 min. The final product was collected by centrifuging the mixture, washed with ethanol and deionized (DI) water for several times until the organic residuals were completely removed, and then dried in vacuum oven at 60 °C overnight. The light blue powder was obtained for further usage.
Synthesis of CPON
2 mmol (483.2 mg) Cu(NO3)2 ∙ 3H2O and 190 mg Na2HPO4 was added into 30 mL DI water. After vigorous stirring for 10 min, 15 mL NaOH solution with a concentration of 0.1 M was dropwise added into the above mixture followed by stirring for 1 h. The final product was collected by centrifuging the mixture, washed with ethanol and deionized (DI) water for several times until the organic residuals were completely removed, and then dried in vacuum oven at 60 °C overnight. The light blue powder was obtained for further usage.
Synthesis of CCON
0.9 mmol (153.5 mg) CuCl2 ∙ 2H2O was added into 30 mL DI water (A solution). 0.1 M NaOH and 0.2 M Na2CO3 aqueous solution was mixed and named as B solution. Then B solution was slowly added into A solution until the pH was around 8.2. Afterwards, the solution was transferred into a 50 mL Tefon-lined autoclave, sealed and heated at 80 °C for 12 h, and allowed to cool to room temperature naturally. The final product was collected by centrifuging the mixture, washed with ethanol and deionized (DI) water for several times until the organic residuals were completely removed, and then dried in vacuum oven at 60 °C overnight. The light blue powder was obtained for further usage.
Characterization
TEM and HRTEM images were performed with a JEOL Jem F-200C TEM with an acceleration voltage of 200 kV. XRD patterns were obtained from a Philips X’Pert Pro Super diffractometer with Cu Kα radiation (λ = 1.54178 Å). X-ray photoelectron spectroscopy (XPS) spectra were acquired on an ESCALAB MKII system with Al Kα (hν = 1486.6 eV) as the excitation source. The binding energies obtained in the XPS spectral analysis were corrected for specimen charging by referencing C 1 s to 284.8 eV. UV-vis-NIR diffuse reflectance spectra were measured on a Perkin Elmer Lambda 950 UV-vis-NIR spectrophotometer. In-situ FTIR spectra were obtained by using a Thermo Scientific Nicolet iS50. Synchrotron-radiation photoemission spectroscopy (SRPES) was executed at the National Synchrotron Radiation Laboratory (NSRL) in Hefei, China. AFM measurements was performed using a Veeco DI Nano-scope MultiMode V system. Ultraviolet photoelectron spectroscopy (UPS) was performed at the Catalysis and Surface Science Endstation at the BL11U beamline of the National Synchrotron Radiation Laboratory (NSRL). The workfunction (WF) was determined by the difference between the photon energy and the binding energy of the secondary cutoff edge. To be exact, EB = hν − (EK + 4.3 – 5.0) and WF = hν − (Ecutoff − EF) (EB, binding energy; hν, photon energy; EK, kinetic energy; Ecutoff, secondary cutoff edge; EF, Fermi level; photon energy of 40.0 eV and a sample bias of −5 V applied to observe the secondary electron cutoff). CO2 adsorption isotherms measurements for all the synthetic samples were carried out using an automatic microporous physical and chemical gas adsorption analyser (ASAP 2020 M PLUS). Fluorescence emission decay spectra were recorded with a DeltaFlex-NL (HORIBA Scientific) spectrometer. BET isotherms were conducted by 3 Flex Multiport Surface Characterization Analyzer from Micromeritics.
IR light-driven CO2 reduction tests
Before performing the CO2 photoreduction performance, we fabricated the sample into a thin film: the sample was dispersed in deionized water to gain a concentration of about 1 mg mL−1, and then, through spin-dropping 5 mL of the above dispersion on a quartz glass, followed by heat treatment at 65 °C for 30 min, the catalysts thin film could be achieved. During the CO2 photoreduction process, the catalysts thin film floated on 50 mL of water in a quartz glass vessel with the homothermal condensate water, which could enable the catalysts thin film to retain a constant temperature of 290 ± 0.2 K. A MC-PF-300C Xe lamp with AREF (full spectrum reflectance 200–2500 nm), AM 1.5 G filter and 800 nm cutoff filter was used to simulate infrared light, the corresponding illumination spectrum of which in comparison with sunlight is displayed in Supplementary Fig. 16. Note that the distance from the lamp to the sample was ~10 cm, and the irradiation area of sample is around 9.62 cm2 with an output light density of ~71 mW cm−2.The instrument was initially evacuated for three times, afterwards, pumped by high-purity CO2 to reach an atmospheric pressure. The gas products were quantified by the Agilent GC-8860 gas chromatograph equipped with TDX-01 column, thermal conductivity detector (TCD) and flame ionization detector (FID) while ultrahigh-purity argon was used as a carrier gas (FID detector for carbon-based products and TCD detector for H2). The liquid products were quantified by nuclear magnetic resonance (NMR) (Bruker AVANCE AV III 400) spectroscopy, in which dimethyl sulfoxide (DMSO, Sigma, 99.99%) was used as the internal standard. A recirculating cooling water system was used to keep the photocatalytic system at 290 ± 0.2 K under light irradiation.
The product selectivity for CO2 reduction to CO and CH4 has been calculated using the following equation:
The electron selectivity for CO2 reduction to CO and CH4 (2e− for CO, 8e− for the formation of CH4) has been calculated using the following equation:
where n(CH4) and n(CO) are the amounts of produced CH4 and CO.
Apparent quantum efficiency measurement
The apparent quantum efficiency was defined by the ratio the effective electrons to the total input photon flux of different single wavelength3. The number of effective electrons was determined by CO and CH4 yields of 12 h photoreaction under a monochromatic light wavelength at 800, 850, 940 and 1064 nm. And the total input photon flux of different single wavelength was calculated by the incident light intensity, which was determined using a Silicon-UV enhanced actinometer. The quantum yields for the photocatalytic CO2 reduction reactions in our paper were determined using the following equation:
In situ FTIR spectra experiments
All FTIR spectra were recorded on Thermo Scientific Nicolet iS50. The spectra were displayed in transmission units and acquired with a resolution of 4 cm−1, using 64 scans. The dome of the reaction cell had two KBr windows allowing IR transmission and a third window allowing transmission of irradiation introduced through a liquid light guide that connects to the same IR-light lamp. The catalyst powders were first added to the reaction cell and then trace amounts of water were sprayed on the surface of catalysts. After degassed in N2 atmosphere for 20 min, the gas flow was switched to high-purity CO2 until the adsorption is saturated, then the reaction cell was sealed. Next, the FTIR spectra were recorded as a function of time to investigate the dynamics of the reactant adsorption in the dark and desorption/conversion under irradiation.
Photoelectrochemical measurements
Photocurrent and electrochemical impedance spectroscopy (EIS) were performed using an electrochemical workstation (CHI 660E, CH Instruments, Shanghai, China). Specifically, 10 mg of catalysts were dispersed in a mixture containing 950 µL ethanol and 50 µL Nafion solution, then ultrasonic treated to form homogenous catalyst ink. Then the catalyst ink was dipped on a polished FTO glass and dried in air. The photocurrent measurements were conducted in a three-electrode cell system under irradiation of the same light source with that during IR light-driven CO2 reduction. The FTO glass (2.5 × 1.5 cm2) deposited with materials was used as the photoelectrode, a Pt foil was used as the counter electrode, and Ag/AgCl electrode was used as the reference electrode. The three electrodes were inserted in a quartz cell filled with 0.2 M Na2SO4 electrolyte. The Na2SO4 electrolyte was purged with CO2 for 1 h prior to the measurements. EIS was measured in the frequency of 1 to 1000 kHz.
DFT calculation details
Density functional theory (DFT) calculations were carried out on a Vienna Ab initio Simulation Package (VASP)39. The exchange-correlation potential was described by the generalized gradient approximation (GGA) within the framework of Perdew-Burke-Ernzerhof (PBE) functional40. DFT-D3 method was employed to calculate the van der Waals (vdW) interaction41. The parameters of dipole correction were applied for the calculation of slab models. Electronic energies were computed with the tolerance of 1 × 10−4 eV and total force of 0.01 eV/Å. A Monkhorst−Pack k-mesh of 2 × 4 × 1 k-points was used in the structural relaxation, and a kinetic cutoff energy of 450 eV was adopted. The crystal lattice parameters of Cu4(SO4)(OH)6 bulk are as follows: a = 13.087 Å, b = 9.865 Å, c = 6.015 Å (α = 90◦, β = 103.33◦, γ = 90◦) with the P 1 21/a 1 (14) space group. The Cu4(SO4)(OH)6 slabs were modeled by the corresponding exposed surface along (100) direction with the thickness of single unit cell. The theoretical approach is based on the GGA with on-site Coulomb interaction parameter (GGA + U method), in which an effective U-J parameter of 5.2 eV was applied to improve the description Cu 3d states42,43. A vacuum space of 15 Å was inserted in z direction to avoid interactions between periodic images. All calculations were conducted under spin polarization.
The surface energy Es is the energy required to cleave a surface from the corresponding bulk crystal. It can be given by
where A is the area of the surface on the slab models, Es(unrelax) and Es(unrelax) represent the energy of the unrelaxed and relaxed surface slab models, respectively. N is the number of in the slab and Eb is the energy of each atom in the bulk counterpart.
Adsorption energies Eadsorption are given with reference to the isolated surface Esurface relaxed upon removing the molecule from the unit cell using identical computational parameters and the energy of the molecule Emolecule.
The computational hydrogen electrode (CHE)44 model was used to calculate the Gibbs free energy change (ΔG) of CO2 reduction reaction steps:
where EDFT is the electronic energy calculated for specified geometrical structures, EZPE is the zero-point energy, S is the entropy, h is the Planck constant, ν is the computed vibrational frequencies, Θ is the characteristic temperature of vibration, k is the Boltzmann constant, and R is the molar gas constant. For adsorbates, all 3 N degrees of freedom were treated as frustrated harmonic vibrations with negligible contributions from the catalysts’ surfaces. Thus, free energy changes relative to an initial state can be represented by
where * is the substrate, U is the applied overpotential and e is the elementary charge. In this study, U = 0 V versus reversible hydrogen electrode.
Data availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Code availability
The code that supports the findings of this study is available from the corresponding author upon request.
References
Andrei, V. et al. Floating perovskite-BiVO4 devices for scalable solar fuel production. Nature 608, 518–522 (2022).
Xie, S. et al. Photocatalytic and electrocatalytic transformations of C1 molecules involving C–C coupling. Energy Environ. Sci. 14, 37–89 (2021).
Liang, L. et al. Infrared light-driven CO2 overall splitting at room temperature. Joule 2, 1004–1016 (2018).
Pastor, E. et al. Electronic defects in metal oxide photocatalysts. Nat. Rev. Mater. 7, 503–521 (2022).
Zhu, Q. et al. Recent progress of metal sulfide photocatalysts for solar energy conversion. Adv. Mater. 34, 2202929 (2022).
Shangguan, W. et al. Molecular-level insight into photocatalytic CO2 reduction with H2O over Au nanoparticles by interband transitions. Nat. Commun. 13, 3894 (2022).
Li, X. et al. Ultrathin conductor enabling efficient IR light CO2 reduction. J. Am. Chem. Soc. 141, 423–430 (2019).
Xu, J. et al. Efficient infrared-light-driven CO2 reduction over ultrathin metallic Ni-doped CoS2 nanosheets. Angew. Chem. Int. Ed. 60, 8705–8709 (2021).
Li, J. et al. Interfacial engineering of Bi19Br3S27 nanowires promotes metallic photocatalytic CO2 reduction activity under near-infrared light irradiation. J. Am. Chem. Soc. 143, 6551–6559 (2021).
Hong, J. W. et al. Metal–semiconductor heteronanocrystals with desired configurations for plasmonic photocatalysis. J. Am. Chem. Soc. 138, 15766–15773 (2016).
Dutta, S. K. et al. Metal semiconductor heterostructures for photocatalytic conversion of light energy. J. Phys. Chem. Lett. 6, 936–944 (2015).
Wang, G. et al. On structural features necessary for near-IR-light photocatalysts. Chem. Eur. J. 21, 13583–13587 (2015).
Wang, G. et al. Cu2(OH)PO4, a near-infrared-activated photocatalyst. Angew. Chem. Int. Ed. 52, 4810–4813 (2013).
Lancashire, R. Electronic_spectroscopy: Selection rules for electronic spectra of transition metal complexes. https://chem.libretexts.org/.
Jiao, X. et al. Fundamentals and challenges of ultrathin 2D photocatalysts in boosting CO2 photoreduction. Chem. Soc. Rev. 49, 6592–6604 (2020).
Wang, H. et al. Porous two-dimensional materials for photocatalytic and electrocatalytic applications. Matter 2, 1377–1413 (2020).
Li, X. et al. Opportunities for ultrathin 2D catalysts in promoting CO2 photoreduction. In: Photocatalysis using 2D nanomaterials (eds Zhao, Y., Duan, H.). R. Soc. Chem: pp 65-149 (2022).
Huan, T. N. et al. From molecular copper complexes to composite electrocatalytic materials for selective reduction of CO2 to formic acid. J. Mater. Chem. A 3, 3901–3907 (2015).
Abuín, M. et al. XAS study of Mn, Fe and Cu as indicators of historical glass decay. J. Anal. Spectrom. 28, 1118–1124 (2013).
Liu, X. et al. Photothermal effect of infrared light to enhance solar catalytic hydrogen generation. Catal. Commun. 102, 13–16 (2017).
Wang, G. et al. Near-infrared photothermally enhanced catalysis for benzene hydroxylation. J. Mater. Chem. A 10, 21884–21892 (2022).
Firet, N. J. et al. Probing the reaction mechanism of CO2 electroreduction over Ag films via operando infrared spectroscopy. ACS Catal. 7, 606–612 (2017).
Grabow, L. C. et al. Mechanism of methanol synthesis on Cu through CO2 and CO hydrogenation. ACS Catal. 1, 365–384 (2011).
Wu, J. C. S. et al. In situ DRIFTS study of photocatalytic CO2 reduction under UV irradiation. Front. Chem. Eng. China 4, 120–126 (2010).
Liu, Y. et al. Effcient electrochemical reduction of carbon dioxide to acetate on nitrogen-doped nanodiamond. J. Am. Chem. Soc. 137, 11631–11636 (2015).
Yates, J. T. et al. Search for chemisorbed HCO: The interaction of formaldehyde, glyoxal, and atomic hydrogen + CO with Rh. J. Catal. 74, 97–109 (1982).
Ewing, G. E. et al. Infrared detection of the formyl radical HCO. J. Chem. Phys. 32, 927–932 (1960).
Liu, L. et al. Understanding the reaction mechanism of photocatalytic reduction of CO2 with H2O on TiO2-based photocatalysts: A review. Aerosol Air Qual. Res 14, 453–469 (2014).
Li, L. et al. Copper as a single metal atom based photo-, electro-, and photoelectrochemical catalyst decorated on carbon nitride surface for efficient CO2 reduction: A review. Nano Res. Energy 1, e9120015 (2022).
Liu, D. et al. In situ FT-IR study of CO and H2 adsorption on a Pt/Al2O3 catalyst. Catal. Today 68, 155–160 (2001).
Wang, Y. et al. CO2 photoreduction with H2O vapor on highly dispersed CeO2 /TiO2 catalysts: Surface species and their reactivity. J. Catal. 337, 293–302 (2016).
Li, X. et al. Selective visible-light-driven photocatalytic CO2 reduction to CH4 mediated by atomically thin CuIn5S8 layers. Nat. Energy 4, 690–699 (2019).
Ulmer, U. et al. Fundamentals and applications of photocatalytic CO2 methanation. Nat. Commun. 10, 3169 (2019).
Wu, Y. A. et al. Facet-dependent active sites of a single Cu2O particle photocatalyst for CO2 reduction to methanol. Nat. Energy 4, 957–968 (2019).
Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610–7672 (2019).
Navarro-Jaén, S. et al. Highlights and challenges in the selective reduction of carbon dioxide to methanol. Nat. Rev. Chem. 5, 564–579 (2021).
Cheng, Y. et al. Integrated capture and electroreduction of flue gas CO2 to formate using amine functionalized SnOx nanoparticles. ACS Energy Lett. 6, 3352–3358 (2021).
Zhang, S. et al. Polyethylenimine-enhanced electrocatalytic reduction of CO2 to formate at nitrogen-doped carbon nanomaterials. J. Am. Chem. Soc. 136, 7845–7848 (2014).
Kresse, G. et al. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P. et al. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Scanlon, D. O. et al. Understanding conductivity anomalies in CuI-based delafossite transparent conducting oxides: Theoretical insights. J. Chem. Phys. 132, 024707 (2010).
Huang, G.-Y. et al. Detailed check of the LDA + U and GGA + U corrected method for defect calculations in wurtzite ZnO. Phys. Commun. 183, 1749–1752 (2012).
Peterson, A. A. et al. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010).
Acknowledgements
This work was financially supported by European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No 819698 and GrapheneCore3: 881603), Deutsche Forschungsgemeinschaft (COORNETs, SPP 1928 and CRC 1415: 417590517). The Supercomputing Center of Max Planck Computing & Data Facility (MPCDF) is acknowledged for computational support.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
X.F. and X.D.L. conceived the idea and co-wrote the paper. X.D.L., L.L., G.C., X.H.L., and X.C. carried out the sample synthesis, characterization and CO2 reduction measurement. X.D.L. and L.L. discussed the catalytic process. C.N. helped with the BET characterizations. D.P. and M.L. guided the TEM and SEM characterizations. All the authors contributed to the overall scientific interpretation and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ruixiang Li, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Li, L., Chen, G. et al. Accessing parity-forbidden d-d transitions for photocatalytic CO2 reduction driven by infrared light. Nat Commun 14, 4034 (2023). https://doi.org/10.1038/s41467-023-39666-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-39666-0
This article is cited by
-
Atomic reconstruction for realizing stable solar-driven reversible hydrogen storage of magnesium hydride
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
