
Abstract
Cyclohexanone oxime, an important nylon-6 precursor, is conventionally synthesized through cyclohexanone-hydroxylamine (NH2OH) and cyclohexanone ammoxidation methodologies. These strategies require complicated procedures, high temperatures, noble metal catalysts, and toxic SO2 or H2O2 usage. Here, we report a one-step electrochemical strategy to synthesize cyclohexanone oxime from nitrite (NO2−) and cyclohexanone under ambient conditions using a low-cost Cu-S catalyst, avoiding complex procedures, noble metal catalysts and H2SO4/H2O2 usage. This strategy produces 92% yield and 99% selectivity of cyclohexanone oxime, comparable to the industrial route. The reaction undergoes a NO2− → NH2OH→oxime reaction pathway. This electrocatalytic strategy is suitable for the production of other oximes, highlighting the methodology universality. The amplified electrolysis experiment and techno-economic analysis confirm its practical potential. This study opens a mild, economical, and sustainable way for the alternative production of cyclohexanone oxime.
Similar content being viewed by others
Introduction
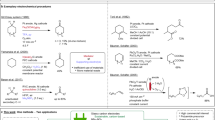
Cyclohexanone oxime is a key precursor for caprolactam production, which is the monomer for the synthesis of nylon-6. Global production of nylon-6 is forecasted to reach 8.9 million tons per year by 2024; thus, the demand for cyclohexanone oxime will increase accordingly1,2. At present, more than 90% of cyclohexanone oxime in the world is produced by the traditional cyclohexanone-hydroxylamine (NH2OH) method (Fig. 1a)3. This strategy includes two steps: (1) NOx is reduced by H2 or SO2 to synthesize NH2OH; (2) NH2OH reacts with cyclohexanone to form cyclohexanone oxime. The former step requires explosive H2 and corrosive SO2 and NOx, causing concerns about safety, cost, and sustainability.

a cyclohexanone-hydroxylamine method, (b) cyclohexanone ammoxidation method, (c) recently reported method and (d) the proposed electrosynthesis of cyclohexanone oxime.
Accordingly, alternative strategies have been developed (Supplementary Fig. 1)4,5. Cyclohexanone ammoxidation is the most promising strategy in industrial production (Fig. 1b). In this method, NH2OH is produced in situ via the oxidation of NH3 by H2O2 and subsequently reacting noncatalytically with cyclohexanone to produce cyclohexanone oxime. Excessive H2O2 is typically required due to its low stability under the associated reaction conditions (e.g., elevated temperatures, high pH), resulting in high cost and easy deactivation problems. Very recently, Lewis et al. successfully used in situ-generated H2O2 from H2 and O2 to replace the performed H2O2 to realize cyclohexanone oxime production (Fig. 1c)6. This approach eliminates the necessity of H2O2 transportation and storage but requires a noble metal catalyst, H2, and an elevated temperature. Therefore, it is highly desirable to develop an alternative strategy to achieve the sustainable, mild, and efficient synthesis of cyclohexanone oxime.
Electrochemistry has emerged as an attractive strategy in synthetic chemistry7,8,9,10,11,12,13,14,15. The electrochemical reduction of nitrogen oxides (NO, NO3−, NO2−, etc.) into the lowest-valence-state ammonia (NH3) has made great advances16,17,18,19,20. In most studies, NH2OH* is revealed as the intermediate during the nitrogen oxide electroreduction process, but NH2OH* is rather unstable and easily reduced to NH3. Thus, the in situ utilization of the generated NH2OH* during nitrogen oxide electroreduction to synthesize organic compounds, especially oxime, is significant but rarely reported21. Inspired by recent advances in nitrogen oxide electroreduction, we speculate that utilizing the NH2OH* formed in situ by electroreduction of nitrogen oxides to react with cyclohexanone to form cyclohexanone oxime can be a promising alternative route for the facile synthesis of cyclohexanone oxime at room temperature. Generally, using gaseous nitrogen oxides as reactants often exhibits a low single-pass conversion rate under ambient conditions; thus, it is desirable to use liquid feedstocks to achieve oxime electrosynthesis with a high conversion rate.
Herein, we report an electrochemical method to synthesize cyclohexanone oxime from nitrite and cyclohexanone over a sulfur-modified Cu (Cu-S) cathode (Fig. 1d). This reaction proceeds in water under ambient conditions and avoids complex procedures and H2O2 usage. A 92% yield and 99% selectivity (in terms of C) of cyclohexanone oxime are achieved at a potential of −0.9 V vs. Ag/AgCl. The high performance can be maintained after 50 cycles of tests, verifying the catalyst’s durability. A series of control experiments, in situ attenuated total reflection Fourier transform infrared (in situ ATR-SEIRAS) spectroscopy, and density functional theory (DFT) calculations reveal a NO2− → NH2OH → oxime pathway. Furthermore, a two-electrode circular flow electrolyzer can deliver 40 mmol of cyclohexanone oxime at a constant current density of 50 mA cm−2 within 6 h, highlighting the application potential.
Results
Cyclohexanone oxime electrosynthesis over a Cu-S cathode
Cu materials have been reported to be highly active in the electrochemical reduction of NO3−/NO2− with H2O to ammonia (NH4+)22. To make the electrochemical reduction reaction stay at the NH2OH step for synthesizing cyclohexanone oxime, the reduction ability of the Cu catalyst should be weakened. Recently, we reported that the introduction of S into Cu (Cu-S) weakens the electroreduction ability of Cu and inhibits the overhydrogenation of alkenes23. Thus, we propose that Cu-S is an electrocatalyst candidate for using NO2- electroreduction to produce NH2OH in situ for cyclohexanone oxime.
The Cu-S electrocatalyst was synthesized and characterized by scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS) (Supplementary Figs. 2, 3 and Supplementary Note 1). The catalytic performance was tested in an H-type cell using 20 mL of 0.5 M phosphate buffer solution (PBS) containing 0.2 mmol cyclohexanone and 2 mmol NaNO2 (Supplementary Fig. 4). The linear sweep voltammetry (LSV) curve shows an enhanced current density after the addition of cyclohexanone and NaNO2 (Supplementary Fig. 5). After 4000 s of electrolysis at different potentials, the products were analysed and quantified by 1H nuclear magnetic resonance (1H NMR), gas chromatography‒mass spectrometry (GC‒MS), and gas chromatography (GC). Interestingly, cyclohexanone oxime was identified as the only organic product. All the peaks at 2.4, 2.1, 1.6, and 1.5 ppm in the 1H NMR spectrum (Fig. 2a, Supplementary Fig. 6 and Supplementary Note 2) and 160.7, 32.1, 26.8, 25.7, 25.5, and 24.4 ppm in the 13C NMR spectrum (Fig. 2b, Supplementary Fig. 7 and Supplementary Note 3) match well with the cyclohexanone oxime standard sample. Additionally, the molecular weight of 113.1 given by GC‒MS further confirms the successful synthesis of cyclohexanone oxime (Fig. 2c and Supplementary Fig. 8). A 92% yield, 99% selectivity (in terms of C), 26% FE, and 0.165 mmol h−1 cm−2 formation rate of cyclohexanone oxime are obtained at the optimum potential of −0.9 V vs. Ag/AgCl, and NH4+ is the major byproduct (Fig. 2d, e, Supplementary Figs. 9–12 and Supplementary Note 4). The performance is better than that of the pure Cu catalyst (Supplementary Figs. 13 and 14 and Supplementary Note 5). Then, the reaction process is monitored (Fig. 2f). Cyclohexanone is consumed completely within 4000 s, and the yield of cyclohexanone oxime shows an opposite tendency compared to cyclohexanone. Then, a preliminary techno-economic analysis (TEA) on plant-gate levelized cost per tonne of cyclohexanone oxime was performed (Supplementary Note 6). The electrosynthesis strategy is much more profitable at the optimum condition of −0.9 V vs. Ag/AgCl at a given electricity price of 10 cents kWh−1 (the price of electricity generated from renewable sources is 3 cents kWh−1) (Fig. 2g)24, showing the industrial application potential of this electrosynthesis strategy. Notably, 10-fold molar equivalents of NaNO2 were used to realize the rapid synthesis of cyclohexanone oxime, and the cost should be further decreased by improving the utilization of NaNO2 (Supplementary Fig. 15). Subsequently, the durability of the catalyst was assessed, and the performance and catalyst structure were well maintained during 50 cyclic tests, showing the good stability of the Cu-S catalyst (Fig. 2h) (Supplementary Figs. 16, 17).

a 1H NMR, (b) 13C NMR, and (c) GC‒MS detection of cyclohexanone oxime product. d Potential-dependent cyclohexanone oxime yield and selectivity. e Potential-dependent cyclohexanone oxime yield rates. f Time-dependent cyclohexanone conversion and cyclohexanone oxime yield. g Plant-gate levelized cost per tonne of cyclohexanone oxime from TEA at −0.9 V vs. Ag/AgCl. h Durability test at −0.9 V vs. Ag/AgCl.
Mechanistic studies of cyclohexanone oxime electrosynthesis
The reaction mechanism was investigated. To confirm the origin of cyclohexanone oxime production, D2O and Na15NO2 were used as the H and N sources to replace H2O and NaNO2, respectively (Entries 1, 2 in Table 1). 1H NMR, 15N NMR, and GC‒MS (Fig. 3a–d) demonstrate the acquisition of deuterated and 15N-labeled cyclohexanone oxime. Meanwhile, no cyclohexanone oxime is detected when removing electricity, cyclohexanone, and NaNO2. These results demonstrate that cyclohexanone oxime production is an electrically driven process with NaNO2 and cyclohexanone as the N and C sources, respectively (Entries 3–5 in Table 1).

a 1H NMR spectra of cyclohexanone oxime (red) and deuterated cyclohexanone oxime (blue). b GC‒MS spectrum of deuterated cyclohexanone oxime. c 15N NMR spectrum of 15N-cyclohexanone oxime. d GC‒MS detection of the 15N-cyclohexanone oxime product. Time-dependent in situ ATR-SEIRAS using (e) 14NO2− as the N-source and H2O as the H source, (f) 14NO2− as the N-source and D2O as the D source, (g) 15NO2− as the N-source and H2O as the H source and (h) 15NO2− as the N-source and D2O as the D source. i Schematic illustration of the cyclohexanone oxime generation pathway. j Yield and selectivity of different substrates. k Photograph of the circular flow electrolyzer. l Time-dependent cyclohexanone oxime yield at a current density of −50 mA cm−2 (the inset is a photograph of the produced cyclohexanone oxime).
The reaction pathway was elucidated by control experiments and in situ ATR-SEIRAS. At a reaction potential of −0.9 V vs. Ag/AgCl, NH2* (1260 cm−1) and NH2OH* (1199 cm−1) were detected by in situ ATR-SEIRAS using cyclohexanone and NaNO2 as the raw materials (Fig. 3e)25,26. Because the wide H2O peak at approximately 1650 cm−1 overlaps with the peaks of C = N and NO, we conducted the test in D2O. The vibration bands at 1690 cm−1, 1573 cm−1, and 1481 cm−1, assigned to the stretching vibrations of C = N, NO, and O-H in oxime, appear (Fig. 3f). Furthermore, isotope-labeling in situ ATR-SEIRAS experiments using Na15NO2 as the N source were conducted to verify the above analysis. The vibrations of 15NO* (1558 cm−1), 15NH2* (1232 cm−1), 15NH2OH* (1168 cm−1), and C = 15N (1654 cm−1) shift to lower wavenumbers by 20–40 cm−1, while the vibration of O-H remains unchanged (Fig. 3g, h). These blueshifts are attributed to the isotope effect27. These results confirm the successful synthesis of cyclohexanone oxime and the formation of NO*, NH2*, and NH2OH* during the electroreduction process, which may serve as the active intermediate for oxime formation.
To verify the N-containing active species for cyclohexanone oxime formation, control experiments using cyclohexanone as the C source and NO, NH2OH, and NH4+ as the N sources were carried out under standard conditions. No cyclohexanone oxime was detected when using NH4+ as the N source, excluding the involvement of NH3 in cyclohexanone oxime formation (Entry 6 in Table 1). However, when using NO or NH2OH as the N source, both can produce cyclohexanone oxime products (Entries 7 and 8 in Table 1). Considering that NH2OH is the more reduced intermediate than NO in NO2− electroreduction, it is reasonable to regard NH2OH as the active N-containing species for cyclohexanone oxime formation. Control experiments reveal that when NH2OH and cyclohexanone are mixed at room temperature, cyclohexanone oxime is immediately generated even without electricity, indicating that the condensation of NH2OH and cyclohexanone is a spontaneous process (Entry 9 in Table 1). This inspired us to explore whether cyclohexanone oxime can be formed by adding cyclohexanone at the end of NO2− electroreduction. As a result, no cyclohexanone oxime was detected. We speculate that the adsorbed hydroxylamine is difficult to desorb from the catalyst surface into the electrolyte solution to react with cyclohexanone. To prove this hypothesis, we performed an electrolysis experiment of NO2− electroreduction without the addition of cyclohexanone. The concentration of NH2OH was determined by ion chromatography. As a result, no NH2OH was detected during and after the reaction. Theoretical calculations were further conducted to explain this phenomenon. As shown in Supplementary Figs. 18 and 19 and Supplementary Note 7, NH2OH* desorption is an endothermic process, while its further reduction is an exothermic process. This indicates that the formed NH2OH* is easier to further reduce to NH3 rather than desorbing from the catalyst surface. This result explains why NH2OH was not detected in the electrolyte. Therefore, cyclohexanone oxime is formed by condensation of NH2OH* and cyclohexanone on the catalyst surface rather than in the electrolyte solution.
Thus, the reaction pathway is proposed based on the above discussion (Fig. 3i). The electroreduction of NO2− first proceeds on the Cu-S surface solely in the following reaction pathway: NO2− → NO* → NHO* → NH2O* → NH2OH*(Ref. 28). Then, the adsorbed cyclohexanone is rapidly attacked by nucleophilic NH2OH* to yield cyclohexanone oxime (Path I). Meanwhile, the further electroreduction of NH2OH* to NH4+ also proceeds as a competing reaction. Delightedly, NH4+ can be converted to NO2− by the electrooxidation cycle29 (Supplementary Fig. 20 and Supplementary Note 8) or collected as ammonium phosphate (a valuable fertilizer) (Path II).
Universality and application evaluation of the electrosynthesis strategy
The universality of our method was evaluated. Other substrates of ketones and aldehydes, such as furfural, cyclopentanone, and cycloheptanone, are transformed to the corresponding oximes with high yields (72%–97%) and selectivity (>99%) (Fig. 3j, Supplementary Figs. 21–33 and Supplementary Notes 9–21). To evaluate its application potential, a circular flow electrolyzer was adopted to perform two-electrode tests using the Cu-S catalyst as the cathode and titanium mesh as the anode (Fig. 3k, Supplementary Fig. 34 and Supplementary Note 22). Constant current tests were performed at current values of 30, 40, 50, and 60 mA cm−2 for 6 h of electrolysis (Fig. 3l and Supplementary Fig. 35). The optimal productivity of 40 mmol (4.52 g) cyclohexanone oxime with a production rate of 6.6 mmol h−1 is obtained at a current value of −50 mA cm−2. These results suggest promising applications of this electrocatalytic strategy in the production of various oximes with good substrate tolerance.
Discussion
In conclusion, we report an electrochemical strategy to synthesize cyclohexanone oxime by utilizing NH2OH* generated in situ by NO2− electroreduction. This strategy avoids the use of H2O2, H2, SO2, high temperature, and noble metal catalysts that are required for the conventional approach. Up to 92% yield and 99% selectivity (in terms of C) of cyclohexanone oxime are obtained over a Cu-S cathode. The catalytic performance can be maintained well during 50 cycles of tests. The combined results of in situ ATR-SEIRAS, control experiments, and DFT calculations reveal that the reaction undergoes the processes of NO2− RR to NH2OH* and the condensation of NH2OH* with cyclohexanone to cyclohexanone oxime. In addition, this method is suitable for synthesizing other oximes, highlighting the universality. Furthermore, the application potential of this strategy is elucidated by an amplified electrolysis experiment and TEA. This work opens a door for the mild, economical, and sustainable production of cyclohexanone oxime, which may be an alternative/complementary to the current industrial production of cyclohexanone oxime. Furthermore, our method may have wider applications in other industrial processes that require the use of NH2OH.
Methods
Synthesis of self-supported Cu(OH)2 nanowire arrays (NAs)
Cu(OH)2 NAs were synthesized with slight modification according to a previous report30. Commercial copper foam (CF) was cut into a rectangular shape with a size of 1.0 × 3.0 cm2. Then, the small pieces of CF were carefully washed with 3.0 M acid, acetone, and deionized water, respectively. Cu(OH)2 NAs supported on CF were self-grown by simple oxidization of the Cu substrate in an alkaline environment. NaOH (3.0 g) and 0.68 g (NH4)2S2O8 were dissolved in 30 mL deionized (DI) water under vigorous stirring, and the precleaned Cu substrate was immersed in it. After 20 minutes, a blue hydroxide layer was observed on the surface of Cu. The Cu substrate covered with nanowires was removed from the solution, rinsed repeatedly with DI water, and then dried at room temperature.
Synthesis of self-supported CuS nanowire arrays (CuS NAs)
CuS NAs were prepared by the reported hydrothermal sulfidation method30. Thiourea (0.22 g) was dissolved in 30 mL ethylene glycol under magnetic stirring at room temperature. Then, the solution was loaded into a 50 ml Teflon-lined autoclave, and a piece of freshly treated CF was also added into the autoclave, sealed, heated to 80 °C and kept at this temperature for 60 min. After the reaction cooled naturally, the CF with products on it was removed, washed with water, and dried naturally.
Synthesis of Cu-S nanowire sponges via in situ electroreduction of CuS NAs
Cu-S was synthesized with slight modification according to our previous report23. The electroreduction of CuS was conducted on an Ivium-n-Stat electrochemical workstation (Ivium Technologies B.V.) in a typical three-electrode system in 1.0 M KOH. A Hg/HgO with 1.0 M KOH as the inner reference electrolyte was used as the reference electrode. A carbon rod was used as the counter electrode. CuS NAs/CF was sealed in advance with epoxy to ensure an exposed area of 1.0 cm2 and then used as the working electrode. Linear sweep voltammetry (LSV) was recorded in the voltage range −0.7 ~ −1.5 V vs. Hg/HgO at a scan rate of 5 mV s−1 until the reductive peaks disappeared.
Synthesis of Cu nanowire arrays (Cu NAs) via in situ electroreduction of Cu(OH)2 NAs
Cu(OH)2-derived Cu NAs were synthesized by a similar in situ electroreduction strategy to those of Cu-S NSs, where CuS NAs were replaced by Cu(OH)2 NAs.
Characterization
The in situ ATR-FTIR was performed on a Nicolet 6700 FTIR spectrometer with silicon as the prismatic window. 1H and 13C NMR were recorded on a Bruker AVANCE III 400 M NMR instrument. GC‒MS was measured on an Agilent 5977B mass spectrometer with an Agilent Technologies 8860 GC system.
Electrochemical measurements
The electrochemical measurement was carried out on an Ivium-n-Stat electrochemical workstation (Ivium Technologies B.V.) with an H-type cell (Supplementary Fig. 2). Meanwhile, the Cu or Cu-S, carbon rod, and Ag/AgCl electrode were adopted as the working electrode (the working area is 1.0 cm2), the counter electrode, and the reference electrode, respectively. A Nafion 117 proton exchange membrane was applied to separate the anode and cathode compartments of the H-type cell. 20 mL of 0.5 M pH 5.8 PBS with 0.2 mmol cyclohexanone and 2 mmol NaNO2 was used as the catholyte, while 20 mL of 0.5 M pH 5.8 PBS was used as the anolyte. For the linear sweep voltammetry (LSV) test, the constant potential was set as −0.3 V to −1.0 V (vs. Ag/AgCl) with a scan rate of 10 mV s−1. For the constant potential electrolysis test, the potential range was set as −0.6 to −1.0 V (vs. Ag/AgCl), and the reaction time was 4000 s. The products were analysed and quantified by NMR spectroscopy, GC‒MS and GC.
Amplified electrolysis experiments
The amplified electrolysis experiments were conducted on a flow electrolytic cell (Fig. 3k, Supplementary Fig. 27). The electrolyzer is a two-electrode divided cell, and the anode and cathode are separated by a Nafion membrane. Cu-S catalyst and stainless-steel mesh were used as the cathode and anode, respectively. Before the reaction for cyclohexanone oxime electrosynthesis, 800 mL PBS (pH=5.8) was added to the anode and cathode reservoirs, and the cathodic CuS catalyst was electroreduced at a constant current of −30 mA cm−2 for 20 min to obtain a Cu-S cathode. After that, 15 g cyclohexanone was first dissolved in 20 mL methanol and subsequently added to the cathode reservoir. The flow rate of the electrolyte was 100 L/h, and the current densities were 30, 40, 50, and 60 mA cm−2. The products were analysed and quantified by GC‒MS and GC.
Product identification and quantification
The products in the electrolyte were identified by NMR spectroscopy and GC‒MS. Cyclohexanone oxime production was quantified by GC with cetane as the internal standard, and NH4+ was quantified by 1H NMR with maleic acid as the internal standard. The amount of the analyte was calculated based on the area ratio of the analyte peak to that of the internal standard. For the identification and quantification of organic products, after electrolysis, after the reactions finished, the products were extracted by dichloromethane (DCM) and analysed by NMR spectroscopy and GC‒MS. For the identification and quantification of NH4+ from NO2− electroreduction, an extra 20 μL of 4.0 M H2SO4 was added to the as-prepared NMR sample to reach a pH value of ~3, and the concentration was calculated based on the area ratio of the NH4+ peak (NH4+, ~ 6.90 ppm, double peak) to that of maleic acid using calibration curves.
The yield was calculated by below equation:
The yield rate was calculated by below equation:
where t is the reaction time and s is the geometric area of the electrode.
The conversion was calculated by below equation,
The Faradaic efficiency (FE) is the ratio of the number of electrons transferred for the formation of each product to the total amount of electricity passing through the circuit. The FE for the products was calculated using below equation:
where F is the Faraday constant, Q is the electric quantity, n is the mole of generated products, and b is the electron transfer number.
In this paper, error bars correspond to the standard deviation of three independent measurements.
Electrochemical in situ ATR-FTIR spectra measurements
The in situ ATR-FTIR was performed on a Nicolet 6700 FTIR spectrometer equipped with an MCTA detector with silicon as the prismatic window. First, CuS ink (pure ethanol as a dispersant) was carefully dropped on the surface of the gold film, which was chemically deposited on the surface of the silicon prismatic before each experiment. Then, the deposited silicon prismatic served as the working electrode. The Pt foil and Ag/AgCl electrode containing saturated KCl solution were used as the counter and reference electrodes, respectively. The 0.5 M PBS H2O/D2O solution (pH = 5.8) with cyclohexanone and NaNO2/Na15NO2 was employed as the electrolyte. Spectra were recorded at −0.9 V vs. Ag/AgCl. The background spectrum of the catalyst electrode was acquired at an open-circuit voltage before each systemic measurement.
Data availability
The data that support the plots within this paper are available from the corresponding author upon reasonable request. The source data underlying Figs. 2 and 3 are provided as a Source Data file. Source data are provided with this paper.
References
HDIN Research, Global nylon 6 production capacity to reach 8.86 million tons in 2024, 2019. www.hdinresearch.com/news/56.
Thomas, J. M. & Raja, R. Design of a “Green” One-step catalytic production of ε-caprolactam (precursor of nylon-6). Proc. Natl. Acad. Sci. USA 102, 13732–13736 (2005).
Mokaya, R. & Poliakoff, M. A cleaner way to nylon? Nature 437, 1243–1244 (2005).
Roffia, P. et al. Gervasutti, cyclohexanone ammoximation: A break through in the 6-caprolactam production process. Stud. Surf. Sci. Catal. 55, 43–52 (1990).
Sivasanker, A. S. & Ratnasamy, P. Catalytic properties of crystalline titanium silicalites III. Ammoximation of cyclohexanone. J. Catal. 131, 394–400 (1991).
Lewis, R. J. K. et al. Highly efficient catalytic production of oximes from ketones using in situ-generated H2O2. Science 376, 615–620 (2022).
Tang, C., Zheng, Y., Jaroniec, M. & Qiao, S. Z. Electrocatalytic refinery for sustainable production of fuels and chemicals. Angew. Chem. Int. Ed. 60, 19572–19590 (2021).
Li, L. Q. et al. Efficient nitrogen fixation to ammonia through integration of plasma oxidation with electrocatalytic reduction. Angew. Chem. Int. Ed. 60, 14131–14137 (2021).
Jouny, M. et al. Formation of carbon-nitrogen bonds in carbon monoxide electrolysis. Nat. Chem. 11, 846–851 (2019).
Ko, B. H., Hasa, B., Shin, H., Zhao, Y. R. & Jiao, F. Electrochemical reduction of gaseous nitrogen oxides on transition metals at ambient conditions. J. Am. Chem. Soc. 144, 1258–1266 (2022).
Li, J., Zhang, Y., Kuruvinashetti, K. & Kornienko, N. Construction of C−N bonds from small-molecule precursors through heterogeneous electrocatalysis. Nat. Rev. Chem. 6, 303–319 (2022).
Li, J. & Kornienko, N. Electrochemically driven C−N bond formation from CO2 and ammonia at the triple-phase boundary. Chem. Sci. 13, 3957–3964 (2022).
Wu, Y., Jiang, Z., Lin, Z., Liang, Y. & Wang, H. Direct electrosynthesis of methylamine from carbon dioxide and nitrate. Nat. Sustain. 4, 725–730 (2021).
Tao, Z., Rooney, C. L., Liang, Y. & Wang, H. Accessing organonitrogen compounds via C–N coupling in electrocatalytic CO2 reduction. J. Am. Chem. Soc. 143, 19630–19642 (2021).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Wang, Y., Yu, Y., Jia, R., Zhang, C. & Zhang, B. Electrochemical synthesis of nitric acid from air and ammonia through waste utilization. Natl Sci. Rev. 6, 730–738 (2019).
Rooney, C. L., Wu, Y., Tao, Z. & Wang, H. Electrochemical reductive N‑methylation with CO2 enabled by a molecular catalyst. J. Am. Chem. Soc. 143, 19983–19991 (2021).
Shi, J. et al. Promoting nitric oxide electroreduction to ammonia over electron-rich Cu modulated by Ru doping. Sci. China Chem. 64, 1493–1497 (2021).
Han, S. et al. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism Nat. Catal. https://doi.org/10.1038/s41929-023-00951-2 (2023).
Wang, Y. et al. Structurally disordered RuO2 nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia. Angew. Chem., Int. Ed. 61, e202202604 (2022).
Zhang, X. et al. Direct electro-synthesis of valuable C=N compound from NO. Chem. Catal. 2, 1807–1818 (2022).
Wang, Y., Zhou, W., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem., Int. Ed. 59, 5350–5354 (2020).
Wu, Y. et al. Converting copper sulfide to copper with surface sulfur for electrocatalytic alkyne semi-hydrogenation with water. Nat. Commun. 12, 3881 (2021).
Shin, H., Hansen, K. U. & Jiao, F. Techno-economic assessment of low-temperature carbon dioxide electrolysis. Nat. Sustain. 4, 911–919 (2021).
Yao, Y., Zhu, S., Wang, H., Li, H. & Shao, M. A spectroscopic study on the nitrogen electrochemical reduction reaction on gold and platinum surfaces. J. Am. Chem. Soc. 140, 1496–1501 (2018).
Pérez-Gallent, E., Figueiredo, M. C., Katsounaros, I. & Koper, M. T. M. Electrocatalytic reduction of nitrate on copper single crystals in acidic and alkaline solutions. Electrochim. Acta 227, 77–84 (2017). 26.
Zhu, S., Jiang, B., Cai, W. B. & Shao, M. Direct observation on reaction intermediates and the role of bicarbonate anions in CO2 electrochemical reduction reaction on Cu surfaces. J. Am. Chem. Soc. 139, 15664–15667 (2017).
Rosca, V., Duca, M., Groot, M. T. M. & Koper, M. T. M. Nitrogen cycle electrocatalysis. Chem. Rev. 109, 2209–2244 (2019).
Liu, H. Y. et al. Electrocatalytic, Homogeneous ammonia oxidation in water to nitrate and nitrite with a copper complex. J. Am. Chem. Soc. 19, 8449–8453 (2022).
Liu, X. L. & Zhu, Y. J. CuS nanotubes prepared using Cu(OH)2 nanowires as self-sacrificial template. Mater. Lett. 65, 1089–1091 (2011).
Acknowledgements
We acknowledge the National Natural Science Foundation of China (grant no. 22271213 to B.Z.) and the National Postdoctoral Science Foundation of China (grant no. 2022M722357 to Y.W.) for financial support. We also appreciate the kind help from Ms. Yang Liu for ATR-FTIR measurements.
Author information
Authors and Affiliations
Contributions
B.Z. and Y.W. conceived the idea and designed the research. J.Z. and Y.W. conducted experiments and data analysis. Z.S. assisted in some experiments. C.W. conducted the calculations. T.L. and Y.W. performed the in situ ATR-FTIR measurements. B.H.Z. performed the techno-economic analysis. C.L. contributed to the discussion. Y.W. wrote the manuscript. B.Z. revised the paper with comments from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Richard Lewis and Shizhang Qiao for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, Y., Zhao, J., Wang, C. et al. Electrosynthesis of a nylon-6 precursor from cyclohexanone and nitrite under ambient conditions. Nat Commun 14, 3057 (2023). https://doi.org/10.1038/s41467-023-38888-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-38888-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
