
Abstract
Oxidative carbonylation of methane is an appealing approach to the synthesis of acetic acid but is limited by the demand for additional reagents. Here, we report a direct synthesis of CH3COOH solely from CH4 via photochemical conversion without additional reagents. This is made possible through the construction of the PdO/Pd–WO3 heterointerface nanocomposite containing active sites for CH4 activation and C–C coupling. In situ characterizations reveal that CH4 is dissociated into methyl groups on Pd sites while oxygen from PdO is the responsible for carbonyl formation. The cascade reaction between the methyl and carbonyl groups generates an acetyl precursor which is subsequently converted to CH3COOH. Remarkably, a production rate of 1.5 mmol gPd–1 h–1 and selectivity of 91.6% toward CH3COOH is achieved in a photochemical flow reactor. This work provides insights into intermediate control via material design, and opens an avenue to conversion of CH4 to oxygenates.
Similar content being viewed by others


Introduction
Methane conversion into value-added chemicals under mild condition is a promising strategy for maximizing CH4 utilization and mitigating the greenhouse effect1,2,3,4,5. In particular, the partial oxidation of CH4 at low temperature (<200 °C) is an attractive approach to generate valuable oxygenates (e.g., CH3OH, HCHO, HCOOH and CH3COOH) while reducing energy input and carbon emission in traditional gas-phase CH4 conversion6,7,8,9. Among the products, acetic acid (CH3COOH) is an important feedstock of chemical industries. Generally, the synthesis of CH3COOH from CH4 requires a three-step process involving the production of syngas and methanol, which suffers from extra resource consumption and safety issues10. It is thus imperative to develop a green synthetic approach that can directly convert CH4 to CH3COOH. Although recent reports have demonstrated the oxidative carbonylation of CH4 to CH3COOH in thermocatalytic processes, the requirement for additional oxidants (e.g., O2 and H2SO4) and/or CO limits their further applications11,12. Moreover, multiple side reactions take place in various reactants to generate undesired products such as HCOOH and CO2, and further limit the selectivity to CH3COOH13.
Intuitively, photocatalysis should be a potential approach to the green transformation of CH4, in which the ·OH radical derived from water oxidation is the ideal substitute for additional oxidant14,15. In fact, the metal-decorated semiconductor photocatalysts, which offer synergistic effects between metal and semiconductor on electronic structure, charge separation and intermediate adsorption, have been demonstrated to be effective for CH4 activation16,17,18. For instance, Pd-based photocatalysts have been reported for the conversion of CH4 into C1 oxygenate products (CH3OH, CH3OOH, HCHO, etc.)19. The ·OH radical produced from photocatalytic water oxidation enables the activation of CH4 to generate methyl intermediates (*CH3), which can be stabilized on Pd sites for further reactions20,21. However, it still remains a grand challenge to achieve the photocatalytic production of CH3COOH, mainly due to the difficulties of forming carbonyl intermediates and controlling methyl−carbonyl coupling in photocatalysis. The formation of carbonyl intermediates is the key to the production of CH3COOH using CH4 as the sole carbon source, which raises very high requirements for rational construction of catalytically active sites on photocatalysts. Once carbonyl intermediates can be formed from CH4, the carbonylation of CH4 to CH3COOH would no longer need the addition of CO reagent. As demonstrated in thermocatalysis22, the carbonyl group generated in situ from CH4 oxidation can be coupled with the adsorbed *CH3, leading to CH3COOH formation.
Here, we report that CH4 as the sole carbon source can be directly converted to CH3COOH without additional reagents, by rationally integrating the catalytically active sites for CH4 activation and C–C coupling on material surface. The key is the construction of Pd/PdO heterostructure on WO3 support. The photogenerated holes in WO3 enable oxidation of H2O to ·OH radicals for CH4 activation while Pd sites stabilize *CH3 for further conversion. More importantly, PdO—the active species for CH4 oxidation through the Mar−van Krevelen mechanism23—is regarded as the key component for the transformation of CH4 to carbonyl intermediate (*CO) under light irradiation. As such, the carbonylation of CH4 can be achieved through the coupling of methyl and carbonyl intermediates, forming acetyl (CH3CO*) precursor toward the final product of CH3COOH. To facilitate the continuous reaction between methyl and carbonyl intermediates, we design a photochemical flow reaction device with arc-shaped flow channel, in which the flux of *CH3 can react with *CO intermediate continually by fully utilizing PdO and *CH3, to perform the cascade reaction. As a result, the PdO/Pd–WO3 heterointerface nanocomposite with optimal PdO content enables the remarkable selectivity of 91.6% and production rate of 1.5 mmol gPd–1 h–1 toward CH3COOH, providing a feasible strategy for scale-up CH4 conversion.
Results
Structural characterization of nanocomposites
In the preparation of nanocomposites, Pd nanoparticles (NPs) are loaded on WO3 nanosheets (Pd/WO3), followed by the further thermal annealing process to decorate PdO species on Pd NPs. The obtained samples are denoted as PdO/Pd–WO3-x where x = 1−5 by increasing the annealing temperature (refer to Methods). The Pd contents are kept constant in these samples, which are confirmed by inductively coupled plasma optical emission spectrometry (ICP-OES) (Supplementary Table 1), to exclude the effect of Pd content on CH4 conversion performance. Transmission electron microscopy (TEM) images reveal that the prepared WO3 nanosheets have the edge lengths of ~170 nm (Supplementary Fig. 1), and the nanoparticles in all samples are highly dispersed on WO3 substrate (Supplementary Fig. 2). The sizes of Pd NPs increase from Pd/WO3 to PdO/Pd–WO3-5 with the annealing temperature raised (Supplementary Fig. 3), implying the incorporation of oxygen atoms into the nanoparticles along with their lattice expansion. The samples are further characterized by X-ray diffraction (XRD) as shown in Supplementary Fig. 4. The diffraction peaks of Pd and PdO are absent in the XRD patterns, indicating that the nanoparticles are highly dispersed at a low loading amount.
To look into the detailed structures, the nanoparticles on WO3 supports are examined by high-resolution TEM (HRTEM). The Pd NPs are decorated with PdO with different oxidation degree by controlling the annealing temperature. As shown in Fig. 1a, the pristine Pd nanoparticle only displays the interplanar distance of 2.2 Å, corresponding to the spacing of Pd (111) planes24,25,26. After the annealing process, the new lattice fringes with a spacing of 2.65 Å appear in the nanoparticles (Fig. 1b and Supplementary Fig. 5), which can be assigned to the (101) planes of PdO27,28. Meanwhile, the Pd (111) planes are still observed in the nanoparticles, indicating the existence of Pd/PdO heterostructure in PdO/Pd–WO3-1 to PdO/Pd–WO3-4. As the annealing temperature reaches 450 °C, the Pd NPs are completely transformed to PdO NPs (Fig. 1c). Moreover, the compositions of Pd and PdO species are investigated by X-ray photoelectron spectroscopy (XPS). As shown in Supplementary Fig. 6, the content of Pd2+ increases by elevating the annealing temperature, in agreement with the findings from HRTEM images.

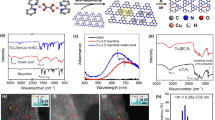
a–c HRTEM images of Pd/WO3 (a), PdO/Pd–WO3-2 (b) and PdO/Pd–WO3-5 (c). d Typical HRTEM image of PdO/Pd–WO3-2 sample showing Pd/PdO heterostructure. e Corresponding FFT patterns of α and β regions in d. f HAADF-STEM image of PdO/Pd–WO3-2 sample. g EELS spectra collected in the regions 1 and 2 marked in f. h Structural illustration of PdO–Pd–WO3 heterointerface.
The Pd–PdO interface is further resolved meticulously to illustrate the active structure for CH4 conversion. Taking PdO/Pd–WO3-2 as an example, abundant Pd/PdO grain boundaries are observed by the distinguishable lattice parameters of Pd and PdO (Fig. 1d and Supplementary Fig. 7). Figure 1e shows the fast Fourier transform (FFT) diffraction patterns obtained from the α and β regions in Fig. 1d. The FFT pattern with the labels of (110), (101) and (1\(\bar{1}\)2) in α region fits the tetragonal structure of PdO along the zone axis of [\(\bar{1}\)11]29. Meanwhile, in β region, we can also obtain the FFT pattern of Pd along the zone axis of [011] direction belonging to the face-centered cubic (fcc) structure with (200), (1\(\bar{1}\)1) and (0\(\bar{2}\)2)30. Furthermore, the Pd/PdO grain boundary is examined by atomic-resolution high-angle annular dark-field scanning TEM (HAADF-STEM), with their compositions further analyzed via electron energy-loss spectroscopy (EELS). As shown in Fig. 1f and g, in addition to the detected signals of Pd–M4,5 edges at both sites 1 and 2, the peak of O–K edge is also recognized at site 1, corresponding to the composition of PdO31,32. Taken together, the aforementioned results demonstrate the existence of Pd/PdO heterostructure on WO3 support. During the annealing process, the PdO component appears on Pd NPs under the cooperation of oxygen and support, as nano-islands rather than core-shell structure33, forming the PdO–Pd–WO3 triple interface (Fig. 1h).
Performance of light-driven CH4 conversion
Upon acquiring the fine structures, we are now in a position to investigate the efficacy of the PdO/Pd–WO3 nanocomposites in light-driven CH4 conversion. The photochemical measurements are conducted in a quartz reactor under xenon arc lamp irradiation. Pure WO3 nanosheets show sluggish properties for CH4 conversion (Supplementary Fig. 8). After Pd NPs deposition, the CH4 conversion activity over Pd/WO3 is enhanced with CH3OH as the primary product (Fig. 2a). Interestingly, the photochemical performance is significantly altered after the incorporation of PdO species into the Pd/WO3 structure, which exhibits a volcano-like relationship with the amount of PdO. Specifically, the addition of PdO to the samples dramatically boosts the production of CH3COOH. Among the samples, the PdO/Pd–WO3-2 achieves the highest production rate and selectivity toward CH3COOH at 62.5 μmol g–1 h–1 and 60.2%, respectively. The outstanding performance of converting CH4 to CH3COOH indicates that the Pd/PdO heterostructure enables an efficient C–C coupling process. However, the excessive PdO in nanocomposites hinders the Schottky contact between Pd and WO3, which further reduces photo-induced charge separation efficiency and substantially suppresses photochemical performance (Supplementary Figs. 9 and 10).

a Production rates for light-driven CH4 conversion over Pd/WO3 and PdO/Pd–WO3-1 to PdO/Pd–WO3-5 samples. b Time-dependent rates of CH3COOH and CH3OH production as well as CH4 conversion over PdO/Pd–WO3-2 nanocomposite. c Reaction-regeneration cycles in CH4 conversion. The error bars represent the standard deviation of the experiments.
To confirm the carbon source of the liquid products, the origin of CH3OH and CH3COOH, as the main products, are traced with 13C nuclear magnetic resonance (13C NMR) spectroscopy by using 13CH4 as the reactant. As shown in Supplementary Fig. 11, the peaks at 20.5 and 176.7 ppm are attributed to 13CH313COOH while the peak at 48.9 ppm is assigned to 13CH3OH. In addition, the control experiments indicate that no product can be detected in the absence of nanocomposite, light irradiation or CH4 reactant (Supplementary Fig. 12). These results provide the evidence that the primary products indeed originate from light-driven CH4 conversion.
It is worth pointing out that differing from the conventional CH4 photooxidation requiring extra oxidant addition (e.g., O2), our reaction system utilizes the reactants of CH4 and H2O, in which the ·OH radical produced from water oxidation is the ideal oxidizer for CH4 activation (Supplementary Figs. 13 and 14)34. As displayed in Supplementary Fig. 15, increasing the concentration of O2 will not promote the generation of liquid products but lead to CO2 production during the CH4 photooxidation over PdO/Pd–WO3-2. The activation of CH4 by ·OH radicals can produce ·CH3 as detected by in situ electron paramagnetic resonance (EPR) measurement (Supplementary Fig. 16).
To better evaluate the efficiency, we conduct time-dependent measurement over PdO/Pd–WO3-2. As shown in Fig. 2b, the production of CH3COOH and CH3OH gradually increases in the first 3 h, achieving an impressive CH4 conversion rate of 181.5 μmol g–1 h–1. However, the performance shows distinct decay when the reaction time exceeds 3 h, which should be ascribed to the consumption of PdO species after the photochemical process (Supplementary Figs. 17 and 18). The constructed Pd/PdO heterostructure is gradually destroyed along with the reaction, which further reduces the efficiency of C–C coupling toward CH3COOH production. In the meantime, negligible H2 detection during the reaction suggests that the lattice oxygen of WO3 is consumed for H2O production, which also leads to performance decay (Supplementary Figs. 19 and 20). To overcome this limitation, we carry out the regeneration process by heating the nanocomposites in air, in which the consumed oxygen (i.e., PdO on Pd and lattice oxygen in WO3) can be replenished to recover activity (Fig. 2c and Supplementary Fig. 21). As such, a durable photochemical CH4 conversion process can be established by recycling the photochemical CH4 conversion and air recovery. The durability measurement indicates that the performance of PdO/Pd–WO3-2 is well maintained for five cyclic tests with each cycle lasting 5 h in such a recycling system (Supplementary Fig. 22). Moreover, the leaching out of Pd during the cyclic tests is also negligible according to the results of mass spectrometry (Supplementary Fig. 23).
Reaction intermediates detection
The information gleaned above has recognized the promising performance for the conversion of CH4 to CH3COOH by modulating Pd/PdO heterostructure. Naturally, a question arises how CH4 evolves into CH3COOH over PdO/Pd–WO3 nanocomposites without additional carbon sources. To this end, we investigate the reaction intermediates over the nanocomposites during the photochemical CH4 conversion process. Figure 3a shows the in situ diffuse reflectance-infrared Fourier-transform spectra (DRIFTS) for light-driven CH4 conversion over Pd/WO3 sample. Upon light irradiation, apart from the peaks at 1305 and 3015 cm–1 corresponding to C–H deformation vibration of CH4, the peak at 1439 cm–1 for CH2/CH3 deformation vibration appears gradually, indicating the CH4 dissociation over the sample35,36. Moreover, the significant growth of vibrational peak at 1061 cm–1 and bands at 2927 and 2963 cm–1, corresponding to the methoxy and C–H stretching vibrations in CH3OH product, can be attributed to the CH4 activation in the presence of ·OH37. In sharp contrast, PdO/Pd–WO3 nanocomposites that can produce CH3COOH through light-driven CH4 conversion exhibit different behavior in DRIFTS (Fig. 3b). In addition to the vibration signals of CH3OH observed over Pd/WO3, the additional vibrational modes of C = O (1654 cm–1), C–O (979 cm–1), C–C (867 cm–1) and C–H (2858 cm–1) stretching vibrations can be monitored for the formation of CH3COOH over PdO/Pd–WO3-238,39. Notably, a broad peak at 2060 cm–1 in Fig. 3b is observed with the light irradiation proceeding, which can be assigned to the adsorbed *CO on Pd site (Supplementary Fig. 24)40. The vibration signals of C = O and *CO only appear with the existence of PdO species, implying that the synergistic effect of Pd/PdO heterostructure in the nanocomposite can facilitate the CH3COOH production with *CO as an intermediate.

a, b In situ DRIFT spectra for light-driven CH4 conversion over Pd/WO3 (a) and PdO/Pd–WO3-2 (b). c, d In situ NAP-XPS results of high-resolution C 1 s (c) and O 1 s (d) spectra over PdO/Pd–WO3-2 nanocomposite.
To further understand the process with elemental information, the surface carbon and oxygen species are also monitored by in situ near ambient pressure XPS (NAP-XPS) characterization. As shown in Fig. 3c, after introducing the reactant into NPA-XPS chamber, the peak of gas-phase CH4 (287.0 eV) is observed in the high-resolution C 1 s XPS spectrum (Supplementary Fig. 25). Upon light irradiation, three C 1 s peaks of surface ·CHx (285.5 eV), C–O (286.1 eV) and COO (289.1 eV) species appear and increase with the time evolution41,42,43. Meanwhile, the formation of oxygenates from CH4 oxidation is also verified by collecting the O 1 s spectra in NAP-XPS studies. Apart from the peak of lattice oxygen in sample (O1, 530 eV), the featured peaks of hydroxyl (O2, 531.1 eV), C–O (O3, 531.8 eV), adsorbed H2O (O4, 532.5 eV) and C = O (O5, 533.2 eV) species are resolved after light illumination (Fig. 3d)44,45,46. Of note, although the signals of *CO have been detected by in situ DRIFT and NAP-XPS measurements, gaseous CO is not observed as a product. Indeed, previous work has demonstrated that the adsorption of CO on PdO site is extremely strong so that *CO would be coupled with other intermediates before desorption47. Apparently, the surface ·CHx, C–O and C = O species are corroborated with the observation of in situ DRIFTS spectra. This indicates that the co-adsorption of CH4 and H2O over Pd/PdO heterostructure can produce various surface carbonaceous intermediates including methyl and carbonyl species and further generate liquid oxygenates.
Mechanistic study
As revealed by in situ characterizations, the carbonyl species is the key intermediate for the conversion of CH4 to CH3COOH, which is closely correlated with the presence of PdO in the prepared nanocomposite. The case of Pd/WO3 reveals that the generation of ·OH radicals alone cannot lead to the formation of C = O in the absence of PdO species (Fig. 3a), implying that the oxygen in C = O is most likely derived from PdO in the nanocomposite. To further trace the oxygen source of carbonyl intermediate in CH3COOH production, we prepare the 18O-labeled PdO-modified nanocomposite (denoted as Pd18O/Pd–WO3-2) by annealing pristine Pd/WO3 in 18O2 atmosphere. Subsequently, the light-driven CH4 oxidation is performed over Pd18O/Pd–WO3-2 and the products are analyzed by gas chromatography−mass spectrometry (GC−MS), in reference to Pd16O/Pd–WO3-2. In contrast to the case of Pd16O/Pd–WO3-2, the peaks at m/z = 45 and 47 by Pd18O/Pd–WO3-2 can be ascribed to CH3C18O+ and +C18OOH (Fig. 4a), indicating that the O atom in *CO is derived from the lattice oxygen of Pd18O in sample. As a result, the peak at m/z = 62 for CH3C18OOH can be detected.

a Mass spectra of CH3COOH product using Pd16O/Pd–WO3-2 and 18O-labeled Pd18O/Pd–WO3-2 nanocomposite. b The comparison of PdO contents in samples (50 mg) and the CH3COOH yields of the corresponding samples. c Schematic illustration for photochemical conversion of CH4 to CH3COOH over Pd/PdO heterointerface in the presence of ·OH radicals. The numbers represent the reaction steps.
To further understand the working mechanism of PdO in the production of CH3COOH, we quantitatively establish the relation between PdO consumption and CH3COOH production. With this purpose, the H2-temperature-programmed reduction (H2-TPR) is employed to determine the content of PdO in PdO/Pd–WO3-x nanocomposites (Supplementary Fig. 26). As revealed in Fig. 4b, the amounts of PdO in samples are very close to the CH3COOH yield at low PdO contents (i.e., PdO/Pd–WO3-1 and PdO/Pd–WO3-2). Given that the PdO is completely consumed after photochemical tests, this further confirms that the lattice oxygen of PdO solely contributes to the formation of C = O in CH3COOH during photochemical CH4 oxidation process. However, excessive PdO in nanocomposites will lower the content of metallic Pd to form PdO−WO3 interface, which suppresses charge separation and reduces performance48,49,50. In this case, the low CH4 photooxidation performance allows most lattice oxygen atoms in PdO to remain in the samples (i.e., PdO/Pd–WO3-3 to PdO/Pd–WO3-5 in Fig. 4b). It is worth pointing out that the formation of Pd–PdO interface in nanocomposite is critical for CH3COOH generation. Our control experiments indicate that CH3COOH cannot be produced through simply mixing Pd/WO3 with PdO, suggesting that Pd–PdO interface is a key factor for CH3COOH generation (Supplementary Fig. 27). Moreover, the CH3COOH production depends on the structural character of Pd/PdO heterointerface (Supplementary Figs. 28 and 29), corroborating the importance of Pd/PdO interface quality to CH3COOH synthesis.
Taken together, the experimental results have revealed the critical role of Pd/PdO heterointerface in CH4-to-CH3COOH conversion. Figure 4c illustrates the proposed reaction pathway. CH4 prefers to be activated at Pd site in the presence of ·OH and form Pd–CH3 intermediate (Supplementary Fig. 30). The methyl species can be gradually converted to Pd−CO intermediate through the combination with O atom from PdO and the dehydrogenation by ·OH. Subsequently, the C–C coupling between carbonyl and methyl species generates the Pd–COCH3 intermediate at Pd−PdO interface, and the further hydrolysis of Pd–COCH3 gives CH3COOH as a product51,52,53. Of note, W sites are also active for *CH3 generation by directly oxidizing CH4 on WO3. However, the *CH3 formed on WO3 can hardly approach the *CO on PdO so that the surplus *CH3 species would evolve into C1 oxygenates54,55.
From the working mechanism of Pd/PdO heterostructure, we can now understand that the complete consumption of lattice oxygen in PdO during light-driven CH3COOH production, in the case of PdO/Pd–WO3-2, will inevitably lead to the significant performance decay for CH3COOH production after the reaction (Supplementary Fig. 31). In comparison, the CH3OH production is not significantly affected by the oxygen loss in PdO. It is worth noting that the evolution of WO3 in photochemical CH4 conversion is also a factor for performance decay. Given that no H2 is detected in the photochemical process, the WO3 is inevitably reduced by photo-induced electrons, which is accompanied with gradual lattice oxygen loss, also causing performance decay (Supplementary Fig. 20). Nevertheless, the amount of lost oxygen atoms in WO3 is determined to be 1.28% through the calculation based on the demand of ·OH radical production, which is negligible as compared with the consumption of PdO (taking PdO/Pd–WO3-2 as example). Furthermore, the lost lattice oxygen in WO3 can be replenished together with that in PdO during the regeneration process, recovering photochemical activity.
Following the mechanistic studies, our investigation on photochemical CH4 conversion in gas-solid phase indicates that the generated *CH3 may undergo self-coupling to produce C2H6 as the primary product (Supplementary Fig. 32). For this reason, the controllable utilization of *CH3 in solution and gas phases is of great importance to further improve the production rate and selectivity for CH3COOH.
Photochemical flow synthesis of CH3COOH
The key to controllable *CH3 utilization is the efficient methyl−carbonyl coupling. Certainly, such an efficient coupling should be based on the supply of sufficient *CO species. Our control experiments show that the addition of CO to the reaction system using Pd/WO3 nanocomposite, in the absence of PdO, can deliver similar CH3COOH production (Supplementary Fig. 33). In comparison, the addition of methanol does not obviously promote the CH3COOH production over PdO/Pd–WO3-2 nanocomposite, suggesting that CH3COOH is not the product primarily from methanol carbonylation (Supplementary Fig. 34). The results provide us the clues for enhancing CH3COOH production—the cascade reaction between *CO and *CH3 on nanocomposite in continuous reaction channels that can promote the utilization of PdO and *CH3.
To this end, we design a photochemical flow reaction device with arc-shaped flow channels to further enhance the performance of CH3COOH production (Supplementary Fig. 35). In this design, the *CH3 species that have not coupled with *CO can migrate along the sample to further react with the adsorbed *CO or even evolve into *CO on the downstream PdO sites, promoting the conversion of CH4 to CH3COOH. Specifically, CH4 and H2O are premixed to form the monodispersed gas bubbles, which are then pumped into the flow reactor to generate gas-liquid-solid contact in channels (Fig. 5a). Benefitting from the flowing reactants and three-phase interface between CH4, H2O and sample (Fig. 5b, c), the generated *CH3 in solution from gas-solid phase CH4 oxidation can be rapidly captured by *CO on sample layer to realize continuous synthesis of CH3COOH. As such, the remarkable selectivity of 91.6% and production rate of 90.7 μmol g–1 h–1 are achieved for CH3COOH production over PdO/Pd–WO3-2 nanocomposite (Fig. 5d). As normalized to the Pd loading weight, the production rate reaches 1.5 mmol gPd–1, which exceeds the performance of existing photocatalysts for oxygenates production under mild condition (Supplementary Table 2). Furthermore, the integration of photochemical CH4 conversion with regeneration process also demonstrates the reproducibility and durability of the flow reaction device (Fig. 5e).

a Schematic illustration of photochemical flow reaction device, including reactants supplier, homemade reactor and products collector. b, c Side (b) and top (c) views of the arc-shaped flow channel in homemade reactor and the three-phase contact between CH4, H2O and sample. The purple, blue and brown colors represent CH4, H2O and nanocomposite, respectively. d Production rate and selectivity of light-driven CH4 conversion toward CH3COOH over PdO/Pd–WO3-2 nanocomposite using the flow reaction device or conventional device for the first 3 h. e Reaction-regeneration cycles on PdO/Pd–WO3-2 sample by employing the flow reaction device. The error bars represent the standard deviation of the experiments.
Discussion
We have demonstrated a direct light-driven synthesis of CH3COOH solely from CH4 on PdO/Pd–WO3 heterointerface nanocomposite, by controlling carbonyl intermediate formation and methyl−carbonyl coupling. As revealed by solid evidence from in situ characterizations, the PdO species can convert CH4 into carbonyl intermediate, holding the key to CH3COOH production. Our isotope labeling experiments indicate that the oxygen atom in carbonyl intermediate is derived from the lattice oxygen of PdO in nanocomposite, providing important information for establishing a conversion−regeneration process toward long-term recyclability. Leveraging our understanding on CH4-to-CH3COOH conversion pathway, we have designed a photochemical flow reaction device enabling cascade reactions to enhance the efficiency and selectivity of acetic acid production. As a result, the approach achieves the impressive production rate of 1.5 mmol gPd–1 h–1 and selectivity of 91.6% toward CH3COOH. This work highlights the importance of rational heterostructure engineering to controlling intermediates evolution, and provides new insights for selective C2+ oxygenates synthesis using methane as resource under mild conditions.
Methods
Chemicals
Sodium tungstate dehydrate (Na2WO4·2H2O, 99.5%), citric acid (CA, 99.5%) and palladium chloride (PdCl2, 98% metals basis) were purchased from Aladdin. Sodium borohydride (NaBH4, 98%) was obtained from Sigma-Aldrich. Hydrochloric acid (HCl, 36 ~ 38%) and ascorbic acid (AA, 99.7%) were purchased from Sinopharm Chemical Reagent Co., Ltd. The water used in all experiments was deionized. All of the chemical reagents were used as received without further purification.
Materials preparation
WO3 nanosheets were prepared by a two-step process. Typically, 1 mmol Na2WO4·2H2O and 1.5 mmol citric acid were dissolved in 30 mL H2O to form a transparent solution. Then 3 mL of HCl solution (6 M) was added into the solution with vigorous stirring for 30 min. The mixture was transferred into a 50 mL Teflon-lined autoclave and heated at 120 °C for 24 h. The resulting precursor of WO3·H2O nanosheets was centrifuged and washed with water several times, and dried in a vacuum oven. The WO3 nanosheets were obtained by calcinating the collected solid in air at 400 °C for 2 h. For the synthesis of Pd/WO3 nanocomposites, 60 mg WO3 nanosheets were dispersed in 30 mL water to form a homogeneous suspension. Then 6.5 mg of PdCl2 was dissolved in 0.5 mL HCl solution (10 mM), which was added into the WO3 suspension and further reacted with 5 mg of NaBH4. The slurry was washed with water three times and dried in a vacuum oven, producing Pd/WO3 sample. The PdO/Pd–WO3-1, PdO/Pd–WO3-2 and PdO/Pd–WO3-3 nanocomposites were obtained by calcinating the Pd/WO3 sample at 120 °C, 200 °C and 260 °C for 5 h, respectively, with a heating rate of 1 °C min–1 in air. The PdO/Pd–WO3-4 and PdO/Pd–WO3-5 nanocomposites were obtained by calcinating the Pd/WO3 sample at 350 °C and 450 °C for 3 h and 2 h, respectively, with a heating rate of 2 °C min–1 in air.
Characterization
Powder XRD patterns were measured by Philips X’Pert Pro Super X-ray diffractometer with Cu-Kα radiation (λ = 1.54178 Å). XPS characterizations of the prepared samples were carried out on JPS-9010MC (JEOL, Japan) with a hemispherical electron energy analyzer (1486 eV Al Kα radiation). TEM images were taken on a Hitachi Model H-7700 microscope at 100 kV. HRTEM images were taken on a JEOL JEM-2100 field-emission higher-resolution transmission electron microscope at 200 kV. The aberration-corrected HAADF-STEM images and EELS analysis were collected on the JEOL ARM-200F field-emission transmission electron microscope operated at 200 kV. EPR spectra for radical detection were obtained on the JEOL JES-FA200 spectrometer.
Photochemical CH4 conversion measurement
In a typical test, 10.0 mg of sample was dispersed in 10 mL water and added into a 30 mL custom-made quartz tube reactor. The light-driven CH4 conversion experiments were carried out in pure CH4 atmosphere (0.1 MPa) at room temperature. The reactor was irradiated by a 300 W xenon lamp (PLS-SXE300, Perfect light) with light intensity of 200 mW cm–1. The gas products were quantified by a gas chromatograph (GC, 7890B, Ar carrier, Agilent) equipped with thermal conductivity detector (TCD) and flame ionization detector (FID). Another GC (Techcomp GC-7900, China) equipped with a TDX-01 packed column was employed to measure the amounts of CO and CO2. The liquid products were quantified by 1H NMR (Bruker Avance, 600 MHz) with a water suppression pulse sequence. A certain concentration of dimethyl sulfoxide (DMSO) solution was used as external standard to calibrate the liquid products. The trapping experiments were performed by adding 1 mM K2Cr2O7, Na2C2O4 and salicylic acid into the reaction solution as photo-induced electron, hole and ·OH scavengers, respectively.
For using the designed photochemical flow device, 100 mg of sample was loaded on the channel of the homemade flow reactor. The reactor was clamped with mould and quartz plate. The reactants of CH4 and H2O were premixed by the microfluidic device to form the monodisperse gas and bubble, which were then pumped into the reactor for photochemical conversion under 300 mW cm–1 of light irradiation. The liquid products were received in bottle. For recovering the photochemical performance, the sample was calcinated at 230 °C for 3 h with a heating rate of 1 °C min–1 in air.
Isotope-labeling experiments
The isotope-labeling experiments were performed by using pure 13CH4 and 12CH4 as feeding gas. The liquid products were detected by 13C NMR. To trace the oxygen atom of CH3COOH, the PdO species in nanocomposite was generated by calcinating Pd/WO3 nanocomposite in 18O2 atmosphere at 200 °C for 8 h to label the oxygen atoms in PdO. The photochemical tests were performed in the homemade flow reaction device for maximizing CH3COOH yield. The CH3COOH product was concentrated and then analyzed by GC−MS (7890 A and 5975 C, He carrier, Agilent).
Photocurrent measurements
The photocurrent tests of the prepared samples were conducted on CHI 660D electrochemical workstation (CH Instruments) with three-electrode system under light or dark condition. Typically, 5.0 mg of material was dispersed in 500 μL of ethanol/water mixture (4:1, v/v) and then dropped onto a 1 × 3 cm fluorine-doped tin oxide (FTO)-coated glass for work electrode preparation. The Pt foil and saturated Ag/AgCl electrode were employed as counter and reference electrode, respectively. The measurements were performed using 0.5 M Na2SO4 aqueous solution as electrolyte. The photocurrent responses of the photoelectrodes (i.e., I–t curves) were collected by measuring the photocurrent densities under chopped light irradiation (light on/off cycles: 10 s) at a bias potential of 0.8 V vs. Ag/AgCl.
Detection of hydroxyl and methyl radicals
Briefly, the sample and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) were dispersed in ice-bath water. The mixture was vigorously shaken and irradiated by using a 500 W xenon lamp, and then analyzed by EPR spectroscopy. Methyl radical was trapped by the same procedure under pure CH4 in the reaction system.
In situ DRIFTS for photochemical CH4 conversion
In situ DRIFTS measurements were performed at BL01B in the NSRL in Hefei, China. The spectra were collected by using a Bruker IFS 66 v Fourier-transform spectrometer equipped with Harrick diffuse reflectance accessory with ZnSe and quartz window. Each spectrum was recorded by averaging 128 scans at a resolution of 2 cm–1. After sample loading, pure CH4 (99.999%) and water vapor were introduced into the chamber for background spectra collection. After that, the system was exposed to light irradiation and the spectra were collected when the irradiation times were 1, 5, 10, 20 and 30 min, respectively.
In situ NAP-XPS measurement for photochemical CH4 conversion
In situ NAP-XPS measurements were carried at the beamline BL02B1 of SSRF under light irradiation or dark condition. The sample was dropped onto a silicon wafer and subsequently cleaned by Ar plasmon for 10 min to remove the surface agent on sample. The prepared sample was stored in the vacuum before the measurement. The XPS spectra were recorded under dark condition firstly. After that, the reactant was sequentially introduced into the analysis chamber with the partial pressure up to 45 Pa. Subsequently, the in situ NAP-XPS spectra were collected under 365 nm LED light irradiation.
Data availability
The authors declare that all data supporting the findings of this study are available in the article and its Supplementary Information. Source data are provided with this paper. Figure 1g, Fig. 2a−c, Fig. 3a−d, Fig. 4a−b, Fig. 5d−e, Fig. S4, Fig. S6, Fig. S8, Fig. S9, Fig. S11, Fig. S17, Fig. S19, Fig. S22, Fig. S24. Additional data are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Schwach, P., Pan, X. & Bao, X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: challenges and prospects. Chem. Rev. 117, 8497–8520 (2017).
Saha, D., Grappe, H. A., Chakraborty, A. & Orkoulas, G. Postextraction separation, on-board storage, and catalytic conversion of methane in natural gas: a review. Chem. Rev. 116, 11436–11499 (2016).
Yuliati, L. & Yoshida, H. Photocatalytic conversion of methane. Chem. Soc. Rev. 37, 1592–1602 (2008).
Meng, X. et al. Direct methane conversion under mild condition by thermo-, electro-, or photocatalysis. Chem 5, 2296–2325 (2019).
Li, X., Wang, C. & Tang, J. Methane transformation by photocatalysis. Nat. Rev. Mater. 7, 617–632 (2022).
Choudhary, T. V. & Choudhary, V. R. Energy-efficient syngas production through catalytic oxy-methane reforming reactions. Angew. Chem. Int. Ed. 47, 1828–1847 (2008).
Zhou, L. et al. Light-driven methane dry reforming with single atomic site antenna-reactor plasmonic photocatalysts. Nat. Energy 5, 61–70 (2020).
Groothaert, M. H. et al. Selective oxidation of methane by the bis(μ-oxo)dicopper core stabilized on ZSM-5 and mordenite zeolites. J. Am. Chem. Soc. 127, 1394–1395 (2005).
Song, H. et al. Direct and selective photocatalytic oxidation of CH4 to oxygenates with O2 on cocatalysts/ZnO at room temperature in water. J. Am. Chem. Soc. 141, 20507–20515 (2019).
Lin, M. & Sen, A. Direct catalytic conversion of methane to acetic acid in an aqueous medium. Nature 368, 613–615 (1994).
Narsimhan, K. et al. Methane to acetic acid over Cu-exchanged zeolites: mechanistic insights from a site-specific carbonylation reaction. J. Am. Chem. Soc. 137, 1825–1832 (2015).
Lin, M., Hogan, T. E. & Sen, A. Catalytic carbon–carbon and carbon–hydrogen bond cleavage in lower alkanes. Low-temperature hydroxylations and hydroxycarbonylations with dioxygen as the oxidant. J. Am. Chem. Soc. 118, 4574–4580 (1996).
Zerella, M., Kahros, A. & Bell, A. T. Methane oxidation to acetic acid catalyzed by Pd2+ cations in the pesence of oxygen. J. Catal. 237, 111–117 (2006).
Zhu, W. et al. Facet-dependent enhancement in the activity of bismuth vanadate microcrystals for the photocatalytic conversion of methane to methanol. ACS Appl. Nano Mater 1, 6683–6691 (2018).
Dong, C. et al. Direct photocatalytic synthesis of acetic acid from methane and CO at ambient temperature using water as oxidant. J. Am. Chem. Soc. 145, 1185–1193 (2023).
Jiang, W. et al. Pd-modified ZnO–Au enabling alkoxy intermediates formation and dehydrogenation for photocatalytic conversion of methane to ethylene. J. Am. Chem. Soc. 143, 269–278 (2021).
Luo, L. et al. Binary Au–Cu reaction sites decorated ZnO for selective methane oxidation to C1 oxygenates with nearly 100% selectivity at room temperature. J. Am. Chem. Soc. 144, 740–750 (2022).
Tian, Y., Piao, L. & Chen, X. Research progress on the photocatalytic activation of methane to methanol. Green Chem 23, 3526–3541 (2021).
Luo, L. et al. Synergy of Pd atoms and oxygen vacancies on In2O3 for methane conversion under visible light. Nat. Commun. 13, 2930 (2022).
Nosaka, Y. & Nosaka, A. Y. Generation and detection of reactive oxygen species in photocatalysis. Chem. Rev. 117, 11302–11336 (2017).
Zhang, W. et al. High-performance photocatalytic nonoxidative conversion of methane to ethane andhydrogen by heteroatoms-engineered TiO2. Nat. Commun. 13, 2806 (2022).
Periana, R. A. et al. Catalytic, oxidative condensation of CH4 to CH3COOH in one step via CH activation. Science 301, 814–818 (2003).
Au-Yeung, J., Chen, K., Bell, A. T. & Iglesia, E. Isotopic studies of methane oxidation pathways on PdO catalysts. J. Catal. 188, 132–139 (1999).
Long, R. et al. Surface facet of palladium nanocrystals: a key parameter to the activation of molecular oxygen for organic catalysis and cancer treatment. J. Am. Chem. Soc. 135, 3200–3207 (2013).
Wang, T.-J. et al. Porous Pd-PdO nanotubes for methanol electrooxidation. Adv. Funct. Mater. 30, 2000534 (2020).
Gao, Z. et al. Pd-decorated PdO hollow shells: a H2-sensing system in which catalyst nanoparticle and semiconductor support are interconvertible. ACS Appl. Mater. Interfaces 12, 42971–42981 (2020).
Annanouch, F. E. et al. Aerosol-assisted CVD-grown PdO nanoparticle-decorated tungsten oxide nanoneedles extremely sensitive and selective to hydrogen. ACS Appl. Mater. Interfaces 8, 10413–10421 (2016).
Do, V.-H. et al. Pd–PdO nanodomains on amorphous Ru metallene oxide for high-performance multifunctional electrocatalysis. Adv. Mater. 35, 2208860 (2023).
Huang, C.-J. et al. Growth and field emission of reactive sputtered Pd–PdO core–shell nanoflakes on platinum. J. Electrochem. Soc. 156, J28 (2009).
Arjona, N. et al. Electrocatalytic activity of well-defined and homogeneous cubic-shaped Pd nanoparticles. J. Mater. Chem. A 1, 15524–15529 (2013).
Xiong, Y., Yang, Y., DiSalvo, F. J. & Abruña, H. D. Pt-decorated composition-tunable Pd–Fe@Pd/C core–shell nanoparticles with enhanced electrocatalytic activity toward the oxygen reduction reaction. J. Am. Chem. Soc. 140, 7248–7255 (2018).
Wang, D. et al. Electrodeposition of metallic nanowire thin films using mesoporous silica templates. Adv. Mater. 15, 130–133 (2003).
Steinhauer, S. et al. Thermal oxidation of size-selected Pd nanoparticles supported on CuO nanowires: the role of the CuO–Pd interface. Chem. Mater. 29, 6153–6160 (2017).
Xiao, Z. et al. Intermediate stabilization for tuning photocatalytic selective oxidation of CH4 to CH3OH over Co3O4/ZnO. J. Catal. 413, 20–30 (2022).
Li, C. & Xin, Q. FT-IR spectroscopic investigation of methane adsorption on cerium oxide. J. Phys. Chem. 96, 7714–7718 (1992).
Yu, X. et al. Selective photocatalytic conversion of methane into carbon monoxide over zinc-heteropolyacid-titania nanocomposites. Nat. Commun. 10, 700 (2019).
Wang, J. et al. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 3, e1701290 (2017).
Finnie, K. S. et al. Vibrational spectroscopy and EXAFS study of Ti(OC2H5)4 and alcohol exchange in Ti(iso-OC3H7)4. J. Mater. Chem. 10, 409–418 (2000).
Liu, Q. et al. Direct catalytic hydrogenation of CO2 to formate over a Schiff-base-mediated gold nanocatalyst. Nat. Commun. 8, 1407 (2017).
Wang, Y. et al. Exploring the ternary interactions in Cu–ZnO–ZrO2 catalysts for efficient CO2 hydrogenation to methanol. Nat. Commun. 10, 1166 (2019).
Tao, F. F. et al. Understanding complete oxidation of methane on spinel oxides at a molecular level. Nat. Commun. 6, 7798 (2015).
Liu, Z. et al. Water-promoted interfacial pathways in methane oxidation to methanol on a CeO2-Cu2O catalyst. Science 368, 513–517 (2020).
Mudiyanselage, K. et al. Importance of the metal–oxide interface in catalysis: in situ studies of the water–gas shift reaction by ambient-pressure X-ray photoelectron spectroscopy. Angew. Chem. Int. Ed. 52, 5101–5105 (2013).
Koushik, D. et al. Plasma-assisted atomic layer deposition of nickel oxide as hole transport layer for hybrid perovskite solar cells. J. Mater. Chem. C 7, 12532–12543 (2019).
Lu, X., Yim, W.-L., Suryanto, B. H. R. & Zhao, C. Electrocatalytic oxygen evolution at surface-oxidized multiwall carbon nanotubes. J. Am. Chem. Soc. 137, 2901–2907 (2015).
Chen, S., Duan, J., Jaroniec, M. & Qiao, S.-Z. Nitrogen and oxygen dual-doped carbon hydrogel film as a substrate-free electrode for highly efficient oxygen evolution reaction. Adv. Mater. 26, 2925–2930 (2014).
Hirvi, J. T. et al. CO oxidation on PdO surfaces. J. Chem. Phys. 133, 084704 (2010).
Bai, S., Jiang, J., Zhang, Q. & Xiong, Y. Steering charge kinetics in photocatalysis: intersection of materials syntheses, characterization techniques and theoretical simulations. Chem. Soc. Rev. 44, 2893–2939 (2015).
Zhang, P., Wang, T., Chang, X. & Gong, J. Effective charge carrier utilization in photocatalytic conversions. Acc. Chem. Res. 49, 911–921 (2016).
Kim, H. I. et al. Robust co-catalytic performance of nanodiamonds loaded on WO3 for the decomposition of volatile organic compounds under visible light. ACS Catal 6, 8350–8360 (2016).
Tang, Y. et al. Single rhodium atoms anchored in micropores for efficient transformation of mthane under mild conditions. Nat. Commun. 9, 1231 (2018).
Moteki, T., Tominaga, N. & Ogura, M. CO-assisted direct methane conversion into C1 and C2 oxygenates over ZSM-5 supported transition and platinum group metal catalysts using oxygen as an oxidant. ChemCatChem 12, 2957–2961 (2020).
Shan, J. et al. Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Nature 551, 605–608 (2017).
Qi, G. et al. Au-ZSM-5 catalyses the selective oxidation of CH4 to CH3OH and CH3COOH using O2. Nat. Catal 5, 45–54 (2022).
Mahlaba, S. V. L. et al. Platinum-catalysed selective aerobic oxidation of methane to formaldehyde in the presence of liquid water. Angew. Chem. Int. Ed 61, e202206841 (2022).
Acknowledgements
This work was financially supported by the National Key R&D Program of China (2020YFA0406103), NSFC (22122506, 22232003, 22075267, 21725102, 91961106), Strategic Priority Research Program of the CAS (XDPB14), Anhui Provincial Natural Science Foundation (2008085J05), Youth Innovation Promotion Association of CAS (2019444), Open Funding Project of National Key Laboratory of Human Factors Engineering (SYFD062010K), and Fundamental Research Funds for the Central Universities (KY2140000031, 20720220007, WK2060000039). The in situ DRIFTS measurements were performed at beamline BL01B in the NSRL. NAP-XPS measurements were performed at the beamline BL02B1 of SSRF supported by National Natural Science Foundation of China under contract no. 11227902. The authors thank the support from USTC Center for Micro- and Nanoscale Research and Fabrication.
Author information
Authors and Affiliations
Contributions
R.L. and Y.X. supervised the projects. W.Z., R.L. and Y.X. conceived the idea for this work. W.Z. prepared the photocatalysts, carried out catalytic measurements and in situ experiments. W.Z., D.X., Y.C., A.C. and Y.J. contributed to the characterization. W.Z., R.L. and Y.X. analyzed the data. W.Z., Z.Z., H.Z. and Z.L. contributed to the NAP-XPS measurements. W.Z. and H.L. contributed to the DRIFTS measurements. W.Z., R.L. and Y.X. wrote the manuscript. All the authors contributed to the interpretation of the data and preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wenzhong Wang, Vitaly Ordomsky and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, W., Xi, D., Chen, Y. et al. Light-driven flow synthesis of acetic acid from methane with chemical looping. Nat Commun 14, 3047 (2023). https://doi.org/10.1038/s41467-023-38731-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-38731-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
