
Abstract
Osimertinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), potently and selectively inhibits EGFR-TKI-sensitizing and EGFR T790M resistance mutations. This analysis evaluates acquired resistance mechanisms to second-line osimertinib (n = 78) in patients with EGFR T790M advanced non-small cell lung cancer (NSCLC) from AURA3 (NCT02151981), a randomized phase 3 study comparing osimertinib with chemotherapy. Plasma samples collected at baseline and disease progression/treatment discontinuation are analyzed using next-generation sequencing. Half (50%) of patients have undetectable plasma EGFR T790M at disease progression and/or treatment discontinuation. Fifteen patients (19%) have >1 resistance-related genomic alteration; MET amplification (14/78, 18%) and EGFR C797X mutation (14/78, 18%).
Similar content being viewed by others


Introduction
Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) are effective treatments for patients with advanced non-small cell lung cancer (NSCLC) harboring an EGFR-TKI sensitizing mutation (EGFRm)1. Most patients who are treated with EGFR-TKIs develop resistance, with 50–60% of patients whose disease progresses whilst receiving first- and second-generation EGFR-TKIs harboring the EGFR T790M mutation2,3,4,5,6.
Osimertinib is a third-generation, irreversible, oral EGFR-TKI that potently and selectively inhibits both EGFRm and EGFR T790M resistance mutations7,8. It is approved as a first-line treatment for patients with EGFRm advanced NSCLC and for patients with EGFR T790M advanced NSCLC following progression on an EGFR-TKI, and has efficacy in patients with NSCLC central nervous system (CNS) metastases9,10,11. Approval for patients with EGFR T790M advanced NSCLC is based on the Phase III AURA3 study (NCT02151981). In this study, osimertinib significantly prolonged progression-free survival (PFS) (median 10.1 versus 4.4 months; hazard ratio (HR] 0.30 (95% confidence interval (CI) 0.23 to 0.41]; P < 0.001) and improved objective response rate (ORR; 71% versus 31%) versus platinum-doublet chemotherapy in patients with EGFR T790M advanced NSCLC, following disease progression on first-line EGFR-TKI therapy7. The final analysis for overall survival (OS) did not show a statistically significant benefit with osimertinib versus platinum-doublet chemotherapy (median 26.8 versus 22.5 months; HR 0.87 (95% CI 0.67 to 1.12; P = 0.277); however, there was a high crossover rate from chemotherapy to osimertinib12.
A number of small-scale studies have reported candidate resistance mechanisms to osimertinib at the point of disease progression when it is used as a second- or later-line treatment13,14,15,16, and more recently, when used in the first-line setting, including from the Phase III FLAURA study17,18. Functional studies for many pathways of acquired resistance to osimertinib have been reported previously19,20,21. However, understanding resistance mechanisms is important to help define appropriate combination therapies for patients with EGFRm advanced NSCLC following acquired resistance to EGFR-TKIs or to prevent the development of resistance. Tumor-specific molecular characteristics can be tested using circulating tumor DNA (ctDNA) isolated from the plasma of some cancer patients, thus providing a potentially valuable biomarker status that can be obtained in a minimally invasive manner22,23,24. The data available for resistance mechanisms to osimertinib are limited and collected from across different studies using diverse methodologies, with the majority of resistance studies focusing on ctDNA versus tissue analysis14,16,21. A heterogenous mixture of resistance mechanisms to osimertinib have been detected including EGFR mutations and MET amplification13,14,16. A loss of detectable T790M has also been reported in 42–68% of patients13,14,21. Here we report data on the plasma ctDNA genomic profile of patients with EGFR T790M advanced NSCLC, whose disease progressed on second-line osimertinib treatment during the Phase III AURA3 study.
Results
Demographics
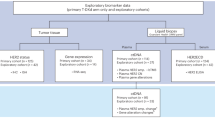
In AURA3, 279 patients were randomized to osimertinib and 140 to platinum-pemetrexed; 83 (30%) and 30 (21%), respectively, had paired (baseline sample and sample at disease progression and/or treatment discontinuation) plasma samples analyzed by NGS (Fig. 1). Among patients with treatment discontinuation samples, 54 (84%) in the osimertinib arm and 16 (94%) in the platinum-pemetrexed arm discontinued due to disease progression. Other reasons for treatment discontinuation included adverse event (n = 5 [8%]), subject decision (n = 2 [3%]), and other (n = 3 [5%]) in the osimertinib arm, and maximum cycle of chemotherapy reached (n = 1 [6%]) in the platinum-pemetrexed arm.

CONSORT flow diagram of patient disposition and eligibility in the analysis of mechanisms of acquired resistance in the AURA3 trial. *Plasma provided at baseline and at disease progression or treatment discontinuation. EGFR epidermal growth factor receptor, p.o, orally, qd once daily, TKI tyrosine kinase inhibitor.
Among patients with paired plasma samples, 103/113 (91%) had baseline detectable plasma EGFR mutations (Ex19del/L858R and/or T790M), and were included in the acquired resistance analysis subset: 78/83 (94%) in the osimertinib arm and 25/30 (83%) in the platinum-pemetrexed arm (Fig. 1). Within this subset, most patients (75/103, 73%) had plasma samples only available at treatment discontinuation, compared with patients with samples only available at disease progression (22/103, 21%). A small number of patients had samples available at both disease progression and treatment discontinuation (6/103; 6%); results from both time points were in the analysis for these patients.
Generally, baseline demographics and clinical characteristics for patients included in the resistance analysis subset were consistent with those reported for all patients randomized in AURA3; baseline demographics for the osimertinib arm are shown in Table 1. Slightly more patients in the osimertinib arm received prior treatment with erlotinib and slightly fewer received gefitinib compared with all patients randomized to osimertinib.
Acquired resistance mechanisms by treatment arm (plasma ctDNA analysis): Osimertinib arm
In the osimertinib arm acquired resistance analysis subset, 32/78 (41%) patients had at least one detectable acquired resistance mechanism; 46 (58%) had no detectable candidate mechanism of resistance (Fig. 2A). EGFR mutations and MET amplification were the most common acquired resistance mechanisms detected, occurring in 17 (22%) and 14 (18%) patients, respectively. Acquired HER2 amplification, MAPK/PI3K alterations and oncogenic fusions (FGFR3-TACC3, NTRK1-TMP3, RET-ERC1, and RET-CCDC6) were each detected in four patients (5%); PIK3CA amplification was detected in three patients (4%). Acquired EGFR mutations included C797X in 14 patients (18%), two patients with C797X co-occurring with L792X, and one patient each with G796S, L718Q, and exon 20 insertion. A total of 39 (50%) patients had a loss of detectable plasma EGFR T790M at progression and/or treatment discontinuation, of which 10/39 (26%) had acquired alterations (Fig. 2A). Ten patients (13%) had undetectable EGFR T790M at baseline. Among patients with MET amplification and detectable plasma EGFR T790M at baseline, 6/12 (50%) lost EGFR T790M detection at progression and/or treatment discontinuation (Fig. 2A). In contrast, none of the patients with acquired EGFR mutations and detectable baseline plasma EGFR T790M lost EGFR T790M detection at progression and/or treatment discontinuation.

Tile plots indicating A acquired alterations in osimertinib-treated patients (n = 78) and B acquired alterations in chemotherapy-treated patients (n = 25) from the AURA3 trial. Source data are provided in the Supplementary Data 1 file. *C797S or C797G; †L792F or L792H. EGFR epidermal growth factor receptor.
More than one acquired resistance mechanism was detected in 15 (19%) patients (Fig. 2A), meaning 47% of all 32 patients with an acquired resistance mechanism had multiple mechanisms detected. MET amplification co-occurred with EGFR C797X (C797S or C797G) in five patients, including two patients with cell cycle gene alterations, one of whom also had BRAF V600E, and one patient with KRAS G12D. Further co-occurrence with MET amplification was detected in three other patients with cell cycle gene alterations, detected in one patient with TPM3-NTRK1 fusion, and in one patient with EGFR G796S + HER2 amplification. Two further patients with HER2 amplification had co-occurring PIK3CA amplification and cell cycle gene alterations, including one patient with co-occurring EGFR C797G/L792H/L792F. One patient with FGFR3-TACC3 fusion had co-occurring EGFR C797S + BRAF V600E.
Non-genomic mechanisms of resistance, such as small cell transformations may have been present, but could not be detected using NGS; histological assessment of tumor tissue at progression/treatment discontinuation was not available to compare these resistance mechanisms.
Acquired resistance mechanisms by treatment arm (plasma ctDNA analysis): platinum-pemetrexed arm
In the platinum-pemetrexed arm acquired resistance analysis subset (n = 25), acquired alterations included loss of amplification in MET (n = 2), HER2 (n = 1), and PIK3CA (n = 1). One patient had a PTEN truncating mutation. There were no EGFR C797S mutations, no acquired mutations in H/N/KRAS, BRAF, FGFR1, PIK3CA, no acquired MET, HER2, or FGFR1 amplifications, and no oncogenic fusions. In total, 4/25 patients (16%) lost detectable EGFR T790M at progression/treatment discontinuation; five patients (20%) had undetectable EGFR T790M at baseline (Fig. 2B).
Osimertinib duration of treatment by candidate resistance mechanisms
Among patients in the osimertinib arm acquired resistance analysis subset (n = 78), duration of treatment was variable across patients with different acquired mechanisms of resistance and loss or retention of detectable EGFR T790M at progression (Fig. 3). Due to the heterogeneity of acquired resistance mechanisms, there was no clear correlation between type or co-occurrence of acquired mechanisms of resistance and duration of treatment.

Swimmer plot indicating the duration of treatment with osimertinib (months) by candidate mechanism and T790M status (n = 78). Source data are provided in the Supplementary Data 1 file. X time of death for patients who have died, O date last known alive for patients who have not died, D time of study discontinuation.
Discussion
The data presented in this exploratory analysis of the AURA3 study represent, to the best of our knowledge, the largest study of resistance mechanisms in a single cohort of patients with similar baseline characteristics from a randomized controlled study who received second-line osimertinib. Numerous acquired resistance mechanisms were detected with osimertinib, the most common being acquired EGFR mutations and MET amplification, occurring in 22% and 18% of patients, respectively. The most common EGFR mutation was C797S, reported in 14% of patients; less frequently reported EGFR mutations included, C797G, L792H/F, G796S, L718Q, and exon 20 insertion. Other resistance mechanisms observed with osimertinib included; HER2 amplification, PIK3CA amplification, cell cycle gene alterations, and oncogenic fusions FGFR3-TACC3, NTRK1-TPM3 RET-ERC1, and RET-CCDC6.
The acquired resistance mechanisms detected in this study are consistent with previous reports for osimertinib in later-line settings, where MET amplification and EGFR C797S have also been reported as among the most common resistance mechanisms14,16,17,21. EGFR C797S was detected in 22–29% of patients in these studies, higher than the frequency reported here, while higher frequencies of up to 50% have been reported for acquired MET amplification. It should be noted that there is no consensus on the criteria for defining MET amplification with NGS. Furthermore, MET polysomy cannot be effectively accounted for with NGS25. A similar pattern of acquired resistance mechanisms has been reported following first-line osimertinib treatment in the phase 3 FLAURA study18,26 and in a recent large study comparing acquired resistance between first- and second/later-line osimertinib17. There is a degree of overlap between the mechanisms of resistance reported here and after treatment with first- and second-generation EGFR-TKIs. Although EGFR T790M is the most common mechanism of resistance following first- or second-generation EGFR-TKIs, amplification of EGFR, MET, HER2, and PIK3CA mutations have also been identified3,27. Of note, no new resistance mechanisms were identified in this study with second-line osimertinib that could lead to a more aggressive disease.
Loss of detectable EGFR T790M was reported in approximately half of the AURA3 patients studied here and in these patients, no clear association with a shorter duration of treatment was observed compared with patients who retained EGFR T790M. Although the possibility that EGFR T790M was undetected due to the assay’s limit of detection cannot be discounted, the baseline EGFR-TKI sensitizing mutation was detectable in the majority of patients at progression, indicating sufficient levels of ctDNA. Approximately a third of those patients with a loss of detectable EGFR T790M had at least one detectable acquired resistance mechanism, including activation of pathways either downstream or parallel to EGFR, including MET, HER2, and PIK3CA amplifications or cell cycle gene alterations. In a previous analysis of patients with T790M NSCLC and acquired resistance to osimertinib, patients with loss of detectable EGFR T790M had a shorter median time to treatment discontinuation compared with patients who retained EGFR T790M (6.1 months versus 15.2 months)14.
Co-occurrence of acquired resistance mechanisms was common in this study and could have clinical implications when determining subsequent treatments, highlighting the need for combination therapies to overcome multiple resistance mechanisms; for example, MET amplication co-occurred with HER2 amplification and NTRK fusions. Data from the phase 1b TATTON study (NCT02143466) provided early evidence for the combination of osimertinib and savolitinib, a MET inhibitor, in patients with MET-amplified advanced NSCLC who progressed after receiving ≥1 first-, second-, or third-generation EGFR-TKIs (median duration of response 7.1 months, objective response rate (ORR] 52%)28,29, highlighting the importance of maintaining EGFR inhibition in subsequent lines of therapy. Another study addressing EGFR and MET inihibiton was the Phase Ib/II study of capmatinib plus gefitinib, which demonstrated preliminary clinical activity (ORR 47%) in patients with EGFR-mutated NSCLC and MET-amplified tumors after progression on EGFR-TKI therapy10.
To address the acquired EGFR C797S mutation, a potential therapeutic option could be to combine osimertinib with a first-generation EGFR-TKI which does not require EGFR C797 for activity. The ongoing ORCHARD platform study is investigating osimertinib plus gefitinib in patients with acquired EGFR C797X following progression on first-line osimertinib30. In addition, preclinical data have suggested that osimertinib combined with gefitinib, which is active against EGFR C797S, may delay the emergence of acquired resistance31. Preliminary clinical evidence also supports the potential for this combination, as demonstrated in an ongoing Phase I/II study where concurrent osimertinib plus gefitinib for the first-line treatment of EGFR-mutated NSCLC resulted in an ORR of 85% with rapid plasma clearance of the EGFR mutation32.
For other acquired mutations, interesting results have also been obtained in preclinical studies. For example, co-treating tumor cells that are EGFR T790M and BRAF V600E positive with osimertinib and the BRAF V600E inhibitor encorafenib increased tumor sensitivity compared with encorafenib treatment alone33. Further research is needed to elucidate the mechanisms of resistance to second-line osimertinib, and the therapeutic strategies to address them.
Caution should be taken when interpreting these data due to the exploratory nature of this analysis. Amplification events may be underestimated due to a high false-negative rate of plasma NGS for amplification compared with fluorescent in situ hybridization (FISH)25. As plasma NGS only detects genomic alterations in ctDNA, other non-genomic mechanisms of resistance including histological transformation (e.g., small cell lung cancer [SCLC]) were not evaluated, though it would be possible to study these potential mechanisms of resistance in the future trials that involve tissue samples17. Additionally, as no paired tissue biopsies were available for analysis, the plasma mutations could not be compared with tissue.
In conclusion, multiple mechanisms of resistance to second-line osimertinib were observed, similar to those observed in previous studies, with no predominating single mechanism identified. The most frequent resistance mechanisms were MET amplification and the EGFR C797S mutation and approximately half of the patients had a loss of detectable EGFR T790M. Importantly, no new mutations that lead to more aggressive cancer biology were detected. The results identify the need for tissue samples to be taken in order to further investigate non-genomic mechanisms of resistance including histological transformation and for continued investigation into combination therapy approaches to prevent or overcome emergent resistance.
Methods
Standard protocol approvals, registration, and patient consent
The study was approved by the institutional review board/independent ethics committee associated with each study center. This study was performed in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with International Conference on Harmonization/Good Clinical Practice and applicable regulatory requirements and the AstraZeneca policy on bioethics. Informed consent was obtained from all patients prior to enrollment into the study. Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data-sharing policy described at http://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. Full study protocol available at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/View?id=2318.
Study design and participants
Full details of phase 3 randomized, open-label, international AURA3 study have been published previously7. In brief, AURA3 assessed the efficacy and safety of osimertinib versus platinum-pemetrexed chemotherapy in patients with centrally confirmed, EGFR T790M advanced NSCLC whose disease had progressed on first-line EGFR-TKI therapy. Patients were stratified (Asian/non-Asian) and randomized 2:1 to receive oral osimertinib (80 mg once daily) or intravenous chemotherapy (pemetrexed, 500 mg/m2 body-surface area) plus either cisplatin (75 mg/m2) or carboplatin (target area under the free carboplatin plasma concentration versus time curve of 5) every 3 weeks for up to six cycles, followed by optional pemetrexed maintenance therapy.
The analysis presented here was an exploratory, retrospective analysis to investigate candidate mechanisms of acquired resistance to osimertinib in the second-line treatment setting in a subset of patients who progressed or discontinued treatment during AURA3. Provision of ctDNA samples was mandatory for all patients who gave informed consent; samples from patients who withdrew consent were excluded. Evaluable patients were required to have detectable plasma EGFRm (L858R/ex19del) and/or EGFR T790M at baseline and to have paired plasma samples from baseline (day 1 cycle 1) and at progression and/or treatment discontinuation. Patients with non-detectable plasma EGFRm and/or EGFR T790M were excluded from the analysis. Plasma samples were restricted to those that passed quality control checks. In addition, patients from China were excluded as plasma samples were unable to be exported for analysis. The data-cutoff for this analysis was 15 March 2019, the final data-cut for AURA3 when OS was reported. Progression events were not updated at this cutoff.
Compulsory blood samples for plasma ctDNA were collected during the screening period. Serial plasma samples were collected (predose) from patients treated with osimertinib and platinum-pemetrexed at screening and on days 1, 8, and 15 of cycle 1 and day 1 of cycles 2–6, and then every 6 weeks thereafter until disease progression, and/or treatment discontinuation. Treatment beyond progression was permitted, and plasma samples were taken at both disease progression and treatment discontinuation. To explore mechanisms of acquired resistance, ctDNA extracted from paired plasma samples was analyzed using a 74-gene NGS panel (Guardant Health, Guardant360® assay)34,35. The limit of variant allelic fraction detected was 0.04–0.06%. Genomic alterations were identified using Guardant Health’s proprietary bioinformatics pipeline34,35. Paired samples were defined as samples from the same patient obtained on day 1 of the first cycle (baseline) and at progression or treatment discontinuation; where samples were available at both progression and or treatment discontinuation, data are reported based on the discontinuation sample.
Assessments
Known and candidate-acquired resistance mechanisms were identified at progression and/or treatment discontinuation in both treatment arms, using the baseline plasma sample as a reference. Amplifications in MET, HER2, or PIK3CA were detected per GuardantHealth CLIA-validated protocols34.
PFS was assessed by the investigator according to response evaluation criteria in solid tumors (Response Evaluation Criteria in Solid Tumors [RECIST] 1.1). Tumor assessments were performed at baseline and every 6 weeks thereafter until objective disease progression. For the analysis reported here, duration of treatment, defined from the date of randomization to the end of osimertinib treatment, was determined according to candidate resistance mechanism in the osimertinib treatment arm.
Statistical methods
As a retrospective, exploratory analysis, data were summarized using descriptive statistics. Plasma samples at progression or treatment discontinuation included in the paired analysis were collected up until April 2018. Clinical data were analyzed from 15 April 2016, data cutoff, and no further disease progression was assessed by RECIST after this date.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The de-identified patient data generated in this study are provided in Supplementary Data 1. Specific consent for sequencing data deposition was not obtained from patients. Anonymized patient-level clinical data, aggregated clinical data, and/or anonymized clinical study documents underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data-sharing policy described at: http://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. Since at the time of this publication, the AURA3 trial is still ongoing, the study data will be accessible at https://vivli.org/ when the trial is completed. In the meantime, requests to access the data from the AURA3 trial described in the current manuscript can be submitted through: https://vivli.org/members/enquiries-about-studies-not-listed-on-the-vivli-platform/. Requested data are available from approval of the request typically for one year. Some patients/countries may need to be excluded based on the informed consent form or country‐level legislation. The use of data must comply with the requirements of the Human Genetics Resources Administration of China and patients who have withdrawn consent for data use will be removed from the shared dataset. Patient-level images or genetic data are not available for access.
References
Planchard, D. et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. ESMO Guidelines Committee Updated version published 15 September https://www.esmo.org/guidelines/lung-and-chest-tumours/clinical-practice-living-guidelines-metastatic-non-small-cell-lung-cancer (2020).
Oxnard, G. R. et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin. Cancer Res. 17, 1616–1622 (2011).
Yu, H. A. et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 19, 2240–2247 (2013).
Jänne, P. A. et al. Dacomitinib as first-line treatment in patients with clinically or molecularly selected advanced non-small-cell lung cancer: a multicentre, open-label, phase 2 trial. Lancet Oncol. 15, 1433–1441 (2014).
Wu, S. G. et al. The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget 7, 12404–12413 (2016).
Lin, C. C. et al. Outcomes in patients with non-small-cell lung cancer and acquired Thr790Met mutation treated with osimertinib: a genomic study. Lancet Respir. Med. 6, 107–116 (2018).
Mok, T. S. et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med. 376, 629–640 (2017).
Soria, J. C. et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 378, 113–125 (2018).
U.S. Food and Drug Administration. TAGRISSO® (osimertinib). Highlights of Prescribing Information. Revised 2018. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208065s006lbl.pdf. Last accessed November 2022. 29 June 2017.
Wu, Y. L. et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J. Clin. Oncol. 36, 3101 (2018).
Reungwetwattana, T. et al. CNS response to osimertinib versus standard epidermal growth factor receptor tyrosine kinase inhibitors in patients with untreated EGFR-mutated advanced non-small-cell lung cancer. J. Clin. Oncol. JCO2018783118 (2018).
Papadimitrakopoulou, V. A. et al. Osimertinib versus platinum-pemetrexed for patients with EGFR T790M advanced NSCLC and progression on a prior EGFR-tyrosine kinase inhibitor: AURA3 overall survival analysis. Ann. Oncol. 31, 1536–1544 (2020).
Le, X. et al. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC. Clin. Cancer Res. 24, 6195–6203 (2018).
Oxnard, G. R. et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 4, 1527–1534 (2018).
Planchard, D. et al. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 26, 2073–2078 (2015).
Piotrowska, Z. et al. MET amplification (amp) as a resistance mechanism to osimertinib. J. Clin. Oncol. 35, 9020 (2017).
Schoenfeld, A. J. et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin. Cancer Res. 26, 2654–2663 (2020).
Chmielecki, J., et al. Candidate mechanisms of acquired resistance to first-line osimertinib in EGFRmutated advanced non-small cell lung cancer Nat Commun, https://doi.org/10.1038/s41467-023-35961-y.
Leonetti, A. et al. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 121, 725–737 (2019).
Offin, M., et al. Acquired ALK and RET gene fusions as mechanisms of resistance to osimertinib in EGFR-mutant lung cancers. JCO Precis. Oncol 2, PO.18.00126 (2018).
Yang, Z. et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin. Cancer Res. 24, 3097–3107 (2018).
Diehl, F. et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 102, 16368–16373 (2005).
Douillard, J. Y. et al. Gefitinib treatment in EGFR mutated caucasian NSCLC: circulating-free tumor DNA as a surrogate for determination of EGFR status. J. Thorac. Oncol. 9, 1345–1353 (2014).
Leighl, N. B. et al. Clinical utility of comprehensive cell-free DNA analysis to identify genomic biomarkers in patients with newly diagnosed metastatic non-small cell lung cancer. Clin. Cancer Res. 25, 4691–4700 (2019).
Hartmaier, R. J. et al. Detection of MET-mediated EGFR tyrosine kinase inhibitor (TKI) resistance in advanced non-small cell lung cancer (NSCLC): biomarker analysis of the TATTON study. Cancer Res. 79, 4897 (2019). Abstract 4897 presented at the 110th Annual Meeting of the American Association for Cancer Research (AACR); 29 March-3 April 2019; Atlanta, GA, USA.
Ramalingam, S. S. et al. Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Ann. Oncol. 29, VIII740 (2018). Abstract LBA50 presented at the 43rd Congress of the European Society for Medical Oncology (ESMO); 19-23 October 2018; Munich, Germany.
Sequist, L. V. et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 3, 75ra26 (2011).
Sequist, V. L. et al. TATTON phase Ib expansion cohort: osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET-amplified NSCLC after progression on prior third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). Cancer Res 79, CT033 (2019). Abstract CT003 presented at the 110th Annual Meeting of the American Association for Cancer Research (AACR); 29 March-3 April 2019; Atlanta, GA, USA.
Yu, H. et al. TATTON phase Ib expansion cohort: osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET amplified NSCLC after progression on prior first/second-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). Cancer Res. 79, CT032 (2019). Abstract CT032 presentated at the 110th Annual Meeting of the American Association for Cancer Research (AACR); 29 March-3 April 2019; Atlanta, GA, USA.
Yu, H. A. et al. Biomarker-directed phase II platform study in patients with EGFR sensitizing mutation-positive advanced/metastatic non-small cell lung cancer whose disease has progressed on first-line osimertinib therapy (ORCHARD). Clin. Lung Cancer 22, 601–606 (2021).
Niederst, M. J. et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 21, 3924–3933 (2015).
Rotow, J. K. et al. Concurrent osimertinib plus gefitinib for first-line treatment of EGFR-mutated non-small cell lung cancer (NSCLC). J. Clin. Oncol. 38, 9507 (2020).
Ho, C. C. et al. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J. Thorac. Oncol. 12, 567–572 (2017).
Genetic Testing Registry (GTR). Guardant360. Available at: https://www.ncbi.nlm.nih.gov/gtr/tests/527948/. Last accessed 21 November 2022.
Odegaard, J. I. et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin. Cancer Res. 24, 3539–3549 (2018).
Acknowledgements
Thanks to all the patients and their families. The authors would also like to thank Aleksandra Markovets for her significant contributions to the work. The study (NCT02151981) was funded by AstraZeneca (Cambridge, UK), the manufacturer of osimertinib. The sponsor, AstraZeneca, was involved in the study design and analysis. The authors would like to acknowledge Robert Harrison, Ph.D., of Ashfield MedComms, an Inizio Company, for medical writing support that was funded by AstraZeneca, Cambridge, UK, in accordance with Good Publications Practice (GPP) guidelines (https://www.ismpp.org/gpp-2022).
Author information
Authors and Affiliations
Contributions
J.C. has made substantial contributions to the conception and design of the work, co-led the overall analysis, and contributed to and approved the manuscript. T.M. has made substantial contributions to the conception and design of the work, co-led the overall analysis, contributed to data interpretation, and contributed to and approved the manuscript. Y.-L.W. has made substantial contributions to the conception and design of the work, co-led the overall analysis, contributed to data collection and data interpretation, and contributed to and approved the manuscript. J.-Y.H. co-led the overall analysis and contributed to and approved the manuscript. M.-J.H. contributed to and approved the manuscript. S.S.R. has made substantial contributions to the conception and design of the work, co-led the overall analysis, and contributed to and approved the manuscript. T.J. co-led the overall analysis and contributed to and approved the manuscript. I.O. co-led the overall analysis and contributed to and approved the manuscript. J.C-H.Y. has made substantial contributions to the conception and design of the work, co-led the overall analysis, and contributed to and approved the manuscript. F.A.S. contributed to and approved the manuscript. K.C.B. contributed to data interpretation and contributed to and approved the manuscript. G.L. co-led the overall analysis and contributed to and approved the manuscript. B.C. co-led the overall analysis and contributed to and approved the manuscript. J.C.B. has made substantial contributions to the conception and design of the work, co-led the overall analysis, contributed to data interpretation, and contributed and approved the manuscript. R.J.H. co-led the overall analysis and contributed to and approved the manuscript. V.P. co-led the overall analysis and contributed to and approved the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: J.C. reports employment and stock ownership with AstraZeneca. T.M. reports employment with The Chinese University of Hong Kong; reports stock ownership with Aurora Tele-Oncology Ltd., Hutchison Chi-Med, and Sanomics Ltd.; has undertaken an advisory role for AbbVie Inc., ACEA Pharma, Amgen, AstraZeneca, Berry Oncology, Blueprint Medicines Corporation, Boehringer Ingelheim Pharmaceuticals Inc., Bristol-Myers Squibb Company, CStone Pharmaceuticals, Curio Science, Daiichi Sankyo Inc., Eisai, Fishawack Facilitate Ltd., G1 Therapeutics, Inc.,Gritstone Oncology, Inc., Guardant Health, geneDecode Co., Ltd., Hengrui Therapeutics Inc., Hutchison Chi-Med, Ignyta, Inc., Incyte Corporation, Inivata, IQVIA, Janssen, Lily, Loxo-Oncology Inc., Lunit, Inc., Merck Serono, Merck Sharp & Dohme, Mirati Therapeutics, Inc., Novartis, OrigiMed, Pfizer, Puma Biotechnology Inc., Roche/Genentech,Sanofi-Aventis R&D, SFJ Pharmaceutical, Takeda, Vertex Pharmaceuticals, Virtus Medical Group, and Yuhan Corporation; is on the board of directors for AstraZeneca and Hutchison Chi-Med; has received honoraria from Abbvie Inc., ACEA Pharma, Alpha Biopharma Co., Ltd., Amgen, Amoy Diagnostics Co., LTD., AstraZeneca, BeiGene, Berry Oncology, BI, Blueprint Medicines Corporation, BMS, CStone Pharmaceuticals, Curio Science, Daiichi Sankyo, Eisai, Fishawack Facilitate Ltd., Gritstone Oncology, Inc., Guardant Health, Hengrui Therapeutics Inc., Ignyta, Inc., Incyte Corporation, Inivata, IQVIA, Janssen, Lilly, Loxo-Oncology, Lunit, Inc., Merck Serono, MSD, Mirati Therapeutics Inc., MoreHealth, Novartis, OrigiMed, Pfizer, Puma Biotechnology Inc., Qiming Development (HK) Ltd., Roche Pharmaceuticals, Sanofi-Aventis, SFJ Pharmaceutical Ltd., Takeda Pharmaceuticals HK Ltd., Vertex Pharmaceuticals, and Yuhan Corporation; has received consulting fees from AbbVie Inc., ACEA Pharma, Amgen, AstraZeneca, Berry Oncology, Blueprint Medicines Corporation, Boehringer Ingelheim Pharmaceuticals Inc., Bristol-Myers Squibb Company, CStone Pharmaceuticals, Curio Science, Daiichi Sankyo Inc., Eisai, Fishawack Facilitate Ltd., G1 Therapeutics, Inc., Gritstone Oncology, Inc., Guardant Health, Hengrui Therapeutics Inc., Hutchison Chi-Med, Ignyta, Inc., Incyte Corporation, Inivata, IQVIA, Janssen, Lily, Loxo-Oncology Inc., Lunit, Inc., Merck Serono, Merck Sharp & Dohme, Mirati Therapeutics, Inc., Novartis, OrigiMed, Pfizer, Puma Biotechnology Inc., Roche/Genentech, Sanofi-Aventis R&D, SFJ Pharmaceutical, Takeda, Vertex Pharmaceuticals, Virtus Medical Group, and Yuhan Corporation; and has received grants or funds from AstraZeneca, Bristol-Myers Squibb, G1 Therapeutics, MSD, Merck Serono, Novartis, Pfizer, Roche, SFJ, Takeda, and XCovery. Y.-L.W. reports institutional funding from AstraZeneca, Bristol-Myers Squibb, Pfizer, and Roche; and has received speaker fees from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, MSD, Pfizer, Sanofi, and Roche. J-Y.H. has undertaken an advisory role for Novartis, MSD Oncology, AstraZeneca, Lilly, and Takeda; has received honoraria from Roche, AstraZeneca, and Takeda; and has received grants or funds from Roche, Pfizer, and ONO. M.-J.A. has undertaken an advisory role for AstraZeneca, Bristol-Myers Squibb, MSD, ONO Pharmaceutical, Roche, Takeda, and Alpha Pharmaceutical; and has received honoraria from AstraZeneca, Bristol-Myers Squibb, MSD, ONO Pharmaceutical, and Roche. S.S.R. has undertaken an advisory role for Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, Merck, Tesaro, Takeda, GlaxoSmithkline, Daichii Sankyo, and Eisai; has received consulting fees from Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, Merck, Tesaro, Takeda, Glaxo smithkline, Daichii Sankyo, and Eisai; and has received grants or funds from Amgen, Advaxis, AstraZeneca, BMS, Merck, Tesaro, Takeda, and Genmab. T.J. has undertaken an advisory role for, and received consulting fees from, Roche, Bristol-Myers Squibb, Merck, Ignyta, AstraZenca, Takeda, MSD, Specialized Therapeutics, and Pfizer. I.O. has undertaken an advisory role for AstraZeneca and Chugai Pharma AbbVie; has received honoraria from AstraZeneca, Taiho Pharmaceutical, ONO Pharmaceutical, MSD Oncology, Bristol-Myers Squibb, Chugai Pharma, and Pfizer; and has received grants or funds from Boehringer Ingelheim, AstraZeneca, Taiho Pharmaceutical, ONO Pharmaceutical, MSD Oncology, Astellas Pharma, Bristol-Myers Squibb, Chugai Pharma, and AbbVie. J.C-H.Y. reports employment with the National Taiwan University Cancer Center; has undertaken an advisory role for Bayer, Roche/Genentech, AstraZeneca, MSD, Merck Serono, Pfizer, Novartis, Yuhan Pharmaceuticals, Bristol-Myers Squibb, Daiichi Sankyo, Hansoh Pharmaceuticals, Takeda Pharmaceuticals, Jannsen, Boehringer Ingelheim, GSK, and IPSEN; is on the board of directors for the International Association for the Study of Lung Cancer; has received honoraria from Bayer, Roche/Genentech, Chugai, MSD, Pfizer, Novartis, Bristol-Myers Squibb, ONO Pharmaceuticals, and Boehringer Ingelheim; and has received consulting fees from Bayer, Roche/Genentech, Chugai, AstraZeneca, MSD, Merck Serono, Pfizer, Novartis, Yuhan Pharmaceuticals, Bristol-Myers Squibb, Daiichi Sankyo, Hansoh Pharmaceuticals, Takeda Pharmaceuticals, and Boehringer Ingelheim. F.A.S. reports stock ownership with AstraZeneca. K.C.B. reports employment and stock ownership with AstraZeneca. G.L. reports employment and stock ownership with AstraZeneca. B.C. is a contactor for AstraZeneca. J.C.B. reports employment and stock ownership with AstraZeneca. R.J.H. reports employment and stock ownership with AstraZeneca. V.P. has undertaken a scientific advisory role for AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Novartis, Merck, F. Hoffman-La Roche, Nektar Therapeutics, Janssen, AbbVie, Araxes, Arrys Therapeutics, Bolt Therapeutics, Clovis Oncology, Exelixis, G2 Innovation, Gritstone, Ideaya, Leeds Biolabs, Loxo-Oncology, Takeda, Tesaro, and TRM Oncology; has received honoraria from F. Hoffman-La Roche; and has received research funding from AstraZeneca, Eli Lilly, Novartis, Merck, F. Hoffman-La Roche, Nektar Therapeutics, Janssen, Bristol-Myers-Squibb, Checkmate, and Incyte.
Peer review
Peer review information
Nature Communications thanks Collin Blakely and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chmielecki, J., Mok, T., Wu, YL. et al. Analysis of acquired resistance mechanisms to osimertinib in patients with EGFR-mutated advanced non-small cell lung cancer from the AURA3 trial. Nat Commun 14, 1071 (2023). https://doi.org/10.1038/s41467-023-35962-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-35962-x
This article is cited by
-
Unraveling EGFR-TKI resistance in lung cancer with high PD-L1 or TMB in EGFR-sensitive mutations
Respiratory Research (2024)
-
Correlation of distribution characteristics and dynamic changes of gut microbiota with the efficacy of immunotherapy in EGFR-mutated non-small cell lung cancer
Journal of Translational Medicine (2024)
-
Candidate mechanisms of acquired resistance to first-line osimertinib in EGFR-mutated advanced non-small cell lung cancer
Nature Communications (2023)
-
Current and Emerging Treatment Options for Patients with Metastatic EGFR-Mutated Non-small Cell Lung Cancer After Progression on Osimertinib and Platinum-Based Chemotherapy: A Podcast Discussion
Advances in Therapy (2023)
-
Cerebrospinal fluid circulating tumour DNA genotyping and survival analysis in lung adenocarcinoma with leptomeningeal metastases
Journal of Neuro-Oncology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
