
Abstract
The preparation of high value-added boronic acids from cheap and plentiful carboxylic acids is desirable. To date, the decarboxylative borylation of carboxylic acids is generally realized through the extra step synthesized redox-active ester intermediate or in situ generated carboxylic acid covalent derivatives above 150 °C reaction temperature. Here, we report a direct decarboxylative borylation method of carboxylic acids enabled by visible-light catalysis and that does not require any extra stoichiometric additives or synthesis steps. This operationally simple process produces CO2 and proceeds under mild reaction conditions, in terms of high step economy and good functional group compatibility. A guanidine-based biomimetic active decarboxylative mechanism is proposed and rationalized by mechanistic studies. The methodology reported herein should see broad application extending beyond borylation.
Similar content being viewed by others
Introduction
Chemistry of boronic acids has expanded far beyond the Suzuki reaction and is multiplying, and has become important in medicinal chemistry, analytical chemistry, chemical biology, and materials science. For example, the clinically approved antifungal drugs Tavaborole and eczema treatment drugs Eucrisa, a series of antibacterial or antitumor molecules under investigation with excellent biological activity, boronate-responsive probes or prodrugs, and even the borono-phenylalanine for Boron Neutron Capture Therapy1,2,3,4,5,6.
Given the importance of boronic acids, the development of methods that enables the borylation of all common carbon-heteroatom bonds, including C-O, C-N (ammonium salt and aryl diazonium), and C-X (Cl, Br, I and hypervalent iodine reagents), C-S (sulfonium salts) bonds is desirable7,8,9,10,11,12,13,14,15,16,17,18,19. Despite their synthetic versatility, the preparation of these compounds can be challenging, and these compounds can be unstable, with limited structure, or their synthesis may require forcing reaction conditions that somewhat limit their application. Among them, mild phenyl/aryl radical reactions are more suitable for the development of borylation20,21,22,23,24,25,26.
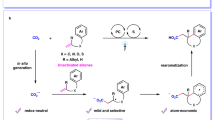
Stable and structurally diverse carboxylic acids exist in many natural products and are frequently accessible from commercial sources27,28,29. Carboxylic acids are well-established versatile building blocks for the construction of various chemical bonds, including C–H, C–O, C–N, C–S, and C–F bonds. However, it is curious that there were until recently no precedents for the construction of C-B bond from carboxylic acids30. Significant progress has been addressed indirectly through step-generated alkyl carboxylic acid N-Hydroxyphthalimide (NHPI) esters (redox-active esters) by the work of Baran, Aggarwal, etc.31,32,33. Glorius further completed the borylation of aryl carboxylic acid NHPI ester via a conceptually novel electron donor-acceptor complex34,35. Apart from the step synthesized redox-active ester strategy23,36,37,38,39,40,41,42,43, the group of Szostak realized the palladium-catalyzed decarbonylative borylation via in situ generated carboxylic acid anhydride intermediate in one-step synthesis44; Soon after, the group of Su developed the nickel-catalyzed decarbonylative borylation via in situ generated benzoic acid aryloxyboron intermediate under similar high-temperature condition45. Compared with metal-catalyzed borylation at high temperatures, the photocatalytic strategy with milder conditions has significant advantages in borylation reaction34,35 (Fig. 1a).

a Previous reported decarboxylative borylation. b Mechanism of Uroporphyrinogen decarboxylase (UroD). c This work on the direct borylation. Cat., catalyst.
More recently, oxidative radical decarboxylation with a higher step economy has been studied because of the lower activation energy for the decarboxylation (8–9 kcal mol−1). However, the rate of direct decarboxylation of benzoic acids (k ≈ 106 s−1) is not competitive with nondecarboxylative pathways, including the addition to arenes (k ≈ 108 M−1s−1), hydrogen atom transfer (HAT) (k ≈ 107 M−1 s−1), as well as back electron transfer (BET) between the generated aryl carboxyl radical and a reduced photocatalyst46,47,48,49. Moreover, even if the aryl radicals are formed, they may still be hard to be trapped by boronating reagents at rates that approach rates of its diffusion50,51,52,53, which further limits the direct decarboxylation borylation. Just recently, MacMillan published their latest work on the direct decarboxylative borylation without prefunctionalization of the native acid, which exploits UV photoinduced copper ligand-to-metal charge transfer (Cu-LMCT) to convert aryl acids into aryl radicals (Fig. 1a)54.
Inspired by the biomimetic approaches of enzymatic decarboxylation, scientists have been working on developing new decarboxylative functionalization strategies55,56. For example, based on the biosynthesis of polyketide, all kinds of decarboxylative reactions of malonic acid half thio/oxyester were developed by metal/organocatalysts57. Except for the biosynthesis of polyketide, uroporphyrinogen decarboxylase (UroD) may catalyze uroporphyrinogen decarboxylation for the biosynthesis of heme, chlorophyll, and the cytochromes in plants and animals58. It is worth noting that UroD can enhance the rate of substrate decarboxylation by a factor of 1.2 × 1017, which is one of the most significant rate enhancements and has been reported as an enzyme that acts without cofactors. The protonated basic residue (guanidino group of Arg-37) of UroD is thought to play a significant role in active carboxylic acid by furnishing a counterion, which assists the scissile carboxylate group of the substrate enter a relatively nonpolar environment and stabilizes the carbanion generated by the departure of CO2 (Fig. 1b). Inspired by this, we introduce a simplified guanidine-based molecule with both basic and nucleophilic properties to promote direct decarboxylative borylation. The guanidine can active the phenyl carboxyl acids, while promoting the generation and conversion of phenyl carboxyl radicals by balancing the capture rate and its diffusion rates. Herein we report a photocatalytic direct decarboxylative borylation method of aromatic carboxylic acids without extra stoichiometric additives or synthesis steps (Fig. 1c).
Results and discussion
Optimization of the reaction conditions
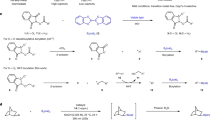
To test our hypothesis, we take 3-acetylbenzoic acid (1n) as a model compound to investigate the decarboxylation borylation. Due to the high oxidation potential (Eox = 1.4 V) from benzoic acid to phenyl radical, the metal-based photocatalysts [Ir(dF(CF3ppy)2)(5,5′-CF3-bpy)]PF6 [Ir-1] (IrIII*/II = 1.68 V versus SCE in MeCN)59,60,61 was chosen first in the presence of ethyl acetate solvent, boronating reagents B2pin2 and 440 nm blue light condition, the raw materials 1n does not have any conversion. (Table 1, entry 1) To our delight, when the guanidine-based reagents tetramethylguanidine (TMG) was added, the borylated product 2n was acquired in 15% yield, which is a preliminary confirmation of our biomimetic reaction design (entry 2). Besides, different transition-metal-catalysts were screened, and only when Co(dmgH)2pyCl (Co-1) was used in the state containing TMG, the yield of 2n was surprisingly raised from 15 to 56% (entry 4 and Supplementary Table 4). Based on the condition in entry 4, we further screened other TMG analogs with imine protecting group or conformationally constrained cyclic molecules (entry 5–8, also see Supplementary Table 2), but no better results were acquired. Also, no product was obtained when other photocatalysts with higher or similar oxidation potential were tested, such as Mes-AcrClO4 (E1/2 red = 2.06 V vs SCE), Ru(bpz)3PF6 (RuII*/I = 1.45 versus SCE in MeCN); then a series of screening on the Ir-based photocatalysts with different modification of the ligands was also conducted (entry 9–11, also see Supplementary Table 1), the change in yield is positively correlated with the oxidation potential of different photocatalysts. Interestingly, only the carboxylic acid ester solvents could promote the transformation, and the AcOtBu solvent gained the best result with a 70% yield (entry 12, and Supplementary Table 7). Compared with the argon protection conditions at less than 10% yield, the influence of the air atmosphere on the reaction is huge, indicating that the oxygen in the air participates in the catalytic cycle (see Supplementary Table 5). After a detailed optimization of photosensitizer, guanidine-based reagents, cobalt (III) reagents, and boronating reagents were conducted, a 78% yield was acquired as the optimal condition (entry 13), which was provided in the Supplementary Table 10.
Substrate scope
We further evaluated the substrate scope of the photocatalytic direct decarboxylative borylation using a series of carboxylic acids substrates (Fig. 2). Benzoic acids with electron-donating groups (e.g. alkyl and alkoxy) were completely suitable substrates (2a-2k). Various substitution patterns (o-, m-, and p-) were tolerated (2b-2e). Similarly, benzoic acids with electron-withdrawing groups (ethyl carboxylate, acetyl, trifluoromethyl) were converted to the corresponding boronic esters in low to moderate yields (2l-2r). The high functional group compatibility was shown by successful decarboxylative borylation with functional groups, including acid-base sensitive difluoro methyl (2r), oxidation-sensitive aldehydes (2 m), and thiophene ring (2ae and 2af), enolizable ketones (2o), especially aryl halides (F, Cl, Br, I) (2s-2ad), which are commonly used and readily accessible precursors to boronic acids. The synthetic utility was further demonstrated by the borylation of the drugs and bioactive molecules (2ai and 2aj). Borylation of activated carboxylic acids containing naphthalene and heteroaromatics, such as thiophene and furan, was also successful with a higher yield (2v-2z, 2aa-2ah). Benzoic acids bearing strong coordinating or oxidizable functional groups, including amines and phenol failed to transform.

Standard conditions: 1n (0.1 mmol), B2pin2 (1.5 eq), [Ir-1] (3 mol%), [Co-1] (15 mol%), TMG (50 mol%), tert-Butyl acetate (2 mL), 440 nm LED, 24 h. Isolated yield of products. TMG, N, N’-Tetramethylguanidine.
Mechanistic considerations
We next turned our attention to the mechanism of this decarboxylation process. Based on the above results and further control experiments, the biomimetic active process of TMG and [IrIII] catalyst with specific high oxidation potential were all essential for the success of this new decarboxylative borylation protocol (entry 4–5, Fig. 3a). The presence of Co catalyst increases the reaction driving force, enhancing the reaction yield by 30% (entry 1–2, Fig. 3a). After the reaction, CO2 was detected by gas chromatography-mass spectrometry (see Supplementary Fig. 2). According to a Stern–Volmer analysis (Fig. 3b and Supplementary Figs. 3–5), oxidative quenching of the photoexcited [IrIII]* catalyst by [CoIII] (kq(Co-1) = 1.2 × 1011 M−1s−1) is more likely than reductive quenching by carboxylate (kq (1n) = 2.5 × 109M−1s−1) in AcOtBu, which is consistent with oxidation to [IrIV], rather than the oxidation of the carboxylate by the photoexcited state of the photocatalyst (see below).

For detailed conditions, see Supplementary Information including a control experiment to determine the key factors affecting the occurrence of reactions. [Ir-1], [Ir(dF(CF3ppy)2)(5,5′-CF3-bpy)]PF6. AcOtBu, tert-butyl acetyl ester. TMG, N, N’-Tetramethylguanidine. n.d., not detected. a Control experiments for mechanistic study. b Fluorescence quenching studies of photocatalysts Ir-1 (the varied colors lines represent corresponding data at different quench concentrations). [Co], Co(dmgH)2pyCl. [S], 1n. c To test if the 1a’ is the key intermediate in the reaction. Standard conditions: 1n (0.1 mmol), B2pin2 (1.5 eq), [Ir-1] (3 mol%), [Co-1] (15 mol%), TMG (50 mol%), tert-Butyl acetate (2 mL), 440 nm LED, 24 h. RT, room temperature. d Radical trapping or inhibiting experiments. Yield was determined by HPLC with 1,3,5-trimethoxybenzene as an internal standard. TEMPO, 2,2,6,6-Tetramethyl-1-piperinedinyloxy.
Since cobalt phenyl complex 1a’ was not observed during the reaction, the cobalt phenyl complex 1a’ was prepared independently, and its reactivity has been investigated. The pinacol phenyl boronate product 2a cannot be formed in the reaction whether under the same light conditions or heating conditions (Fig. 3c, Supplementary Table 12). The above results indicate that the Co catalyst [Co-1] may not participate in the borylation process, but only take part in the oxidative activation of the [IrIII] photosensitizer (IrIII*/II = 1.68 V versus SCE in MeCN) to [IrIV] (IrIV/III = 1.94 V versus SCE in MeCN)60 with higher oxidation potential.
Furthermore, the radical trapping reagents TEMPO completely suppresses the borylation of 1n (Fig. 3d), which suggests the presence of free radicals in the reaction; Interestingly, only in the presence of TMG, the TEMPO-captured product 2n’ could be produced.
The in situ 1H nuclear magnetic resonance (NMR) experiments were then carried out to investigate the mechanism. The reactions were conducted in screw-cap NMR tubes with AcOtBu for convenient monitoring of the actual reaction progress. The mixing of substrate 1n and TMG forms the corresponding complex 1 based on the hydrogen bonding and makes the original H signal of 1n move to the high field and the H signal of TMG move to the low field (Fig. 4a, c, d). Under standard conditions, the reaction proceeds with 50% complex 1a and 50% product 2n after 5 h (Fig. 4a, e). This suggests that the complex 1a consisting of carboxylic acid substrate 1n and TMG is the crucial intermediate for decarboxylation of the carboxylic acid and turnover of the photocatalytic cycle. The presence of TMG resulted in a significant reduction in the 11B NMR signal of B2pin2, which showed the possible activation of TMG on B2pin2 to form complex 2 (Fig. 4b and Supplementary Fig. 7). Under the SET oxidation process of [IrIV] (Step D, Fig. 4b), the stabilized phenyl carboxyl ion free radical complex 1d was generated and transferred to borylated product 2 quickly via the interaction between phenyl free radical 1e and complex 2 (Step E and F, Fig. 4b).

a Condition of in situ 1HNMR analysis A: 1n (0.33 M) in AcOtBu (the date shown as the black line), B: TMG (0.016 M) in in AcOtBu (the date shown as the orange line), C: 1n (0.33 M), TMG (0.016 M) in AcOtBu (the date shown as the blue line), D: 1n (0.33 M), TMG (0.032 M) in AcOtBu (the date shown as the purple line), E: Standard condition in 5 h (the date shown as the pink line). [Ir-1], [Ir(dF(CF3ppy)2)(5,5′-CF3-bpy)]PF6. b Plausible mechanism.
According to Garda’s work62, benzoic acid and TMG forms stable complexes by hydrogen bond interaction, with the changing of chemical properties. We computed the redox potential from benzoic acid to phenyl carboxyl radical using quantum chemical methods of density functional theory, and the result is +2.51 V in ethyl-ethanoate solvent. Meanwhile, the IrIV/III redox potential of metal-based photocatalysts [Ir-1] was computed as +1.69 V (which was measured as +1.94 V versus SCE in MeCN experimentally60), which is quite lower than the redox potential just mentioned. Although the redox potential of PhCOO− is 0.54 V, it is hardly accessible in nonpolar organic solvent because of poor solubility, while for the whole sodium benzoate PhCOONa, the redox potential will increase to 1.82 V. When the TMG was added to PhCOOH (1) to form complex 1a, there are two allowed reaction paths: the one with oxidation followed by HAT (hydrogen atom transfer) (Oxidation-HAT), and the proton transfer followed by oxidation (PT-Oxidation), as shown in Fig. 5, the redox potential of benzoic acid drops significantly with the changed computation value to E = + 1.36 V in PT-Oxidation and +1.39 V in Oxidation-HAT, demonstrating the activation of carboxylic acids by TMG (see Supplementary Information for computational details).

The change of redox potential and the singly occupied molecular orbitals (SOMO) as the orbital where the redox process happens (see more details in Supplementary Table 18), tells the key roles of TMG in the oxidation process: First, by contributing the main component of the SOMO where the electron is transferred from as in complex 1a in Oxidation-HAT process, or being the proton acceptor as complex 1c in PT-Oxidation process, TMG has effectively lower the redox potential. Secondly, TMG and benzoic acids form hydrogen binding complex 1a/1c to promote the redox reaction by maintaining good solubility in organic solvents.
After forming the free radical of benzoic acid-TMG complex 1d’/1d, 1e is then generated with the spontaneous dissociation of TMGH+ (ΔG = −5.1/−8.5 kcal/mol) and departure of CO2 gas (ΔG‡ = 8.9 kcal/mol). In addition to the activation in decarboxylation, the role of TMG in forming complex 2 and then activating B2pin2 has also been computationally corroborated. Although it makes little change on the transition state energy barrier (ΔG‡ = 7.3 kcal/mol for 1e-TS’ vs 6.5 kcal/mol for 1e-TS), the final state energy of borylation with TMG (68.4 kcal/mol for 2 + 1 f’) is much lower than the reaction without TMG (110.2 kcal/mol for 2 + 1 f). We thus conclude that TMG could stabilize the cleavage product of B2pin2 during borylation process, and increase the thermodynamic driving force.
In conclusion, we have described an approach that enables direct decarboxylative borylation from renewable carboxylic acids that does not require extra stoichiometric additives, showing a path that does not involve the step synthesis of redox-active ester or in situ synthesis of carboxylic acid covalent derivatives in harsh conditions. The scope and functional group tolerance of this method were further demonstrated on several representative carbocyclic substrates and medicinally relevant compounds. A guanidine-based biomimetic active decarboxylative strategy was introduced successfully to improve decarboxylation kinetics of aromatic carboxylic acids and the borylation process, as confirmed by systematic computational studies. This work expands the types of biomimetic decarboxylation catalysis and improves the step economy of decarboxylation borylation reactions, which should see broad application extending beyond borylation.
Methods
General methods
For 1H and 13CNMR spectra of compounds in this manuscript and details of the synthetic procedures, see Supplementary Information.
General procedure for decarboxylative borylation
A 5 mL vial equipped with a magnetic stir bar was charged with 3- acetyl -carboxylic acid (16.5 mg, 0.1 mmol), [Ir(dF(CF3ppy)2)(5,5′-CF3-bpy)]PF6 (3.6 mg, 0.003 mmol), Co(dmgH)2pyCl (6 mg, 0.015 mmol), B2pin2 (39 mg, 0.15 mmol), AcOtBu (2 mL) was then into the vial. The reaction mixture was stirred without irradiation for 10 min at ambient temperature, and then TMG (7 μL, 0.05 mmol) was gradually added to the vial under stirring. The reaction mixture was stirred without irradiation for another 10 min at ambient temperature and then irradiated for 24 h while maintaining the temperature at ~35 °C through cooling with a fan. The solvent was removed on a rotary evaporator under reduced pressure, and the residue was purified by preparative thin-layer chromatography.
Computational study
All density functional theory (DFT) calculations were carried out with the Gaussian 16, Revision A.03. The B3LYP functional is mainly used in this research, and the 6–311 + g(d,p) basis set was used for organic molecules, For metal-containing molecules, light elements (C, H, N, O etc) were treated using the 6–31 G* basis set while metal atoms (Co and Ir) were treated using the LanL2DZ/SDD basis set. For all the calculations, Grimme-D3 correction was utilized for the Empirical Dispersion63, and the solvent effect of ethyl-ethanoate was included with Truhlar’s SMD model64. More details of the calculation were added to Supplementary Information.
Data availability
The authors declare that data relating to the characterization of materials and products, general methods, optimization studies, experimental procedures, mechanistic studies, HRMS data, and NMR spectra are available within the article and the Supplementary Information or from the corresponding author upon request.
References
Li, S., Hou, X. H., Ma, Y. F. & Wang, Z. Phenylboronic-acid-based functional chemical materials for fluorescence imaging and tumor therapy. ACS Omega 7, 2520–2532 (2022).
Hiller, N. D. et al. Arylboronic acids and their myriad of applications beyond organic synthesis. Eur. J. Org. Chem. 2020, 4841–4877 (2020).
Neochoritis, C. G. et al. Rapid approach to complex boronic acids. Sci. Adv. 5, 4607 (2019).
Akgun, B. & Hall, D. G. Boronic acids as bioorthogonal probes for site-selective labeling of proteins. Angew. Chem. Int. Ed. 57, 13028–13044 (2018).
Diaz, D. B. & Yudin, A. K. The versatility of boron in biological target engagement. Nat. Chem. 9, 731–742 (2017).
Lorbach, A., Hubner, A. & Wagner, M. Aryl(hydro)boranes: versatile building blocks for boron-doped pi-electron materials. Dalton Trans. 41, 6048–6063 (2012).
Wang, M. Y. & Shi, Z. Z. Methodologies and strategies for selective borylation of C-Het and C-C bonds. Chem. Rev. 120, 7348–7398 (2020).
Jin, S. F. et al. Visible light-induced borylation of C-O, C-N, and C-X bonds. J. Am. Chem. Soc. 142, 1603–1613 (2020).
Constantin, T., Juli, F., Sheikh, N. S. & Leonori, D. A case of chain propagation: alpha-aminoalkyl radicals as initiators for aryl radical chemistry. Chem. Sci. 11, 12822–12828 (2020).
Zhang, L. & Jiao, L. Visible-light-induced organocatalytic borylation of aryl chlorides. J. Am. Chem. Soc. 141, 9124–9128 (2019).
Nguyen, V. D., Nguyen, V. T., Jin, S. F., Dang, H. T. & Larionov, O. V. Organoboron chemistry comes to light: recent advances in photoinduced synthetic approaches to organoboron compounds. Tetrahedron 75, 584–602 (2019).
Tian, Y. M. et al. Selective photocatalytic c-f borylation of polyfluoroarenes by rh/ni dual catalysis providing valuable fluorinated arylboronate esters. J. Am. Chem. Soc. 140, 17612–17623 (2018).
Qiao, Y. S., Yang, Q. M. & Schelter, E. J. Photoinduced miyaura borylation by a rare-earth-metal photoreductant: the hexachlorocerate(III) anion. Angew. Chem. Int. Ed. 57, 10999–11003 (2018).
Zhou, J. et al. Preparing (multi)fluoroarenes as building blocks for synthesis: nickel-catalyzed borylation of polyfluoroarenes via C-F bond cleavage. J. Am. Chem. Soc. 138, 5250–5253 (2016).
Mfuh, A. M. et al. Additive- and metal-free, predictably 1,2-and 1,3-regioselective, photoinduced dual C-H/C-X borylation of haloarenes. J. Am. Chem. Soc. 138, 8408–8411 (2016).
Mfuh, A. M., Doyle, J. D., Chhetri, B., Arman, H. D. & Larionov, O. V. Scalable, metal- and additive-free, photoinduced borylation of haloarenes and quaternary arylammonium salts. J. Am. Chem. Soc. 138, 2985–2988 (2016).
Chen, K., Cheung, M. S., Lin, Z. Y. & Li, P. F. Metal-free borylation of electron-rich aryl (pseudo)halides under continuous-flow photolytic conditions. Org. Chem. Front. 3, 875–879 (2016).
Niwa, T., Ochiai, H., Watanabe, Y. & Hosoya, T. Ni/Cu-catalyzed defluoroborylation of fluoroarenes for diverse C-F bond functionalizations. J. Am. Chem. Soc. 137, 14313–14318 (2015).
Kawamorita, S., Ohmiya, H., Iwai, T. & Sawamura, M. Palladium-catalyzed borylation of sterically demanding aryl halides with a silica-supported compact phosphane ligand. Angew. Chem. Int. Ed. 50, 8363–8366 (2011).
Bose, S. K. et al. Zinc-catalyzed dual C-X and C-H borylation of aryl halides. Angew. Chem. Int. Ed. 54, 11843–11847 (2015).
Cheng, Y., Muck-Lichtenfeld, C. & Studer, A. Metal-free radical borylation of alkyl and aryl iodides. Angew. Chem. Int. Ed. 57, 16832–16836 (2018).
Wang, S., Wang, H. & König, B. Photo-induced thiolate catalytic activation of inert Caryl-hetero bonds for radical borylation. Chem 7, 1653–1665 (2021).
Hong, J. et al. Electrochemical radical borylation of aryl iodides. Chin. J. Chem. 37, 347–351 (2019).
Tian, Y. M. et al. Visible-light-induced Ni-catalyzed radical borylation of chloroarenes. J. Am. Chem. Soc. 142, 18231–18242 (2020).
Zhu, C. & Yamane, M. Transition-metal-free borylation of aryltriazene mediated by BF3.OEt2. Org. Lett. 14, 4560–4563 (2012).
Li, X., Wan, Z., Hu, X. & Zhang, H. Photoinduced aerobic C–S borylation of aryl sulfides. Org. Chem. Front. 9, 3034–3038 (2022).
Wei, Y., Hu, P., Zhang, M. & Su, W. P. Metal-catalyzed decarboxylative C-H functionalization. Chem. Rev. 117, 8864–8907 (2017).
Neeve, E. C., Geier, S. J., Mkhalid, I. A. I., Westcott, S. A. & Marder, T. B. Diboron(4) compounds: from structural curiosity to synthetic workhorse. Chem. Rev. 116, 9091–9161 (2016).
Xuan, J., Zhang, Z. G. & Xiao, W. J. Visible-light-induced decarboxylative functionalization of carboxylic acids and their derivatives. Angew. Chem. Int. Ed. 54, 15632–15641 (2015).
Xu, L. Decarboxylative borylation: new avenues for the preparation of organoboron compounds. Eur. J. Org. Chem. 2018, 3884–3890 (2018).
Li, C. et al. Decarboxylative borylation. Science 356, 7355 (2017).
Fawcett, A. et al. Photoinduced decarboxylative borylation of carboxylic acids. Science 357, 283–286 (2017).
Hu, D. W., Wang, L. H. & Li, P. F. Decarboxylative borylation of aliphatic esters under visible-light photoredox conditions. Org. Lett. 19, 2770–2773 (2017).
Candish, L., Teders, M. & Glorius, F. Transition-metal-free, visible-light-enabled decarboxylative borylation of aryl N-hydroxyphthalimide esters. J. Am. Chem. Soc. 139, 7440–7443 (2017).
Patra, T., Mukherjee, S., Ma, J. J., Strieth-Kalthoff, F. & Glorius, F. Visible-light-photosensitized aryl and alkyl decarboxylative functionalization reactions. Angew. Chem. Int. Ed. 58, 10514–10520 (2019).
Wang, J. et al. Cu-catalyzed decarboxylative borylation. ACS Catal. 8, 9537–9542 (2018).
Zeng, Z. Y., Feceu, A., Sivendran, N. & Goossen, L. J. Decarboxylation-initiated intermolecular carbon-heteroatom bond formation. Adv. Synth. Catal. 363, 2678–2722 (2021).
Zhang, Q. et al. Decarboxylative borylation of stabilized and activated carbon radicals. Angew. Chem. Int. Ed. 59, 21875–21879 (2020).
Zhu, S., Yan, J., Zhou, Y., Yang, K. & Song, Q. Pyridinium-catalyzed decarboxylative borylation of benzoyl peroxides. Green. Synth. Catal. 2, 299–302 (2021).
Li, J., Huang, C. Y., Ataya, M., Khaliullin, R. Z. & Li, C. J. Direct deoxygenative borylation of carboxylic acids. Nat. Commun. 12, 4970 (2021).
Wei, D., Liu, T. M., Zhou, B. & Han, B. Decarboxylative borylation of mCPBA-activated aliphatic acids. Org. Lett. 22, 234–238 (2020).
Barton, L. M., Chen, L. R., Blackmond, D. G. & Baran, P. S. Electrochemical borylation of carboxylic acids. Pro. Nat. Acad. Sci. USA 118, 1–7 (2021).
Dai, J.-J., Teng, X.-X., Fang, W., Xu, J. & Xu, H.-J. Electrochemically promoted decarboxylative borylation of alkyl N-hydroxyphthalimide esters. Chin. Chem. Lett. 33, 1555–1558 (2022).
Liu, C. W., Ji, C. L., Hong, X. & Szostak, M. Palladium-catalyzed decarbonylative borylation of carboxylic acids: tuning reaction selectivity by computation. Angew. Chem. Int. Ed. 57, 16721–16726 (2018).
Deng, X., Guo, J. D., Zhang, X. F., Wang, X. T. & Su, W. P. Activation of aryl carboxylic acids by diboron reagents towards nickel-catalyzed direct decarbonylative borylation. Angew. Chem. Int. Ed. 60, 24510–24518 (2021).
Xu, P., Lopez-Rojas, P. & Ritter, T. Radical decarboxylative carbometalation of benzoic acids: a solution to aromatic decarboxylative fluorination. J. Am. Chem. Soc. 143, 5349–5354 (2021).
Su, W. Q., Xu, P. & Ritter, T. Decarboxylative hydroxylation of benzoic acids. Angew. Chem. Int. Ed. 60, 24012–24017 (2021).
Hu, X. Q., Liu, Z. K., Hou, Y. X. & Gao, Y. Single electron activation of aryl carboxylic acids. Iscience 23, 101266 (2020).
Candish, L., Freitag, M., Gensch, T. & Glorius, F. Mild, visible light-mediated decarboxylation of aryl carboxylic acids to access aryl radicals. Chem. Sci. 8, 3618–3622 (2017).
Leifert, D. & Studer, A. The persistent radical effect in organic synthesis. Angew. Chem. Int. Ed. 59, 74–108 (2020).
Hartwig, J. F. & Larsen, M. A. Undirected, homogeneous C-H bond functionalization: challenges and opportunities. ACS Cent. Sci. 2, 281–292 (2016).
Lucarini, M., Pedulli, G. F. & Valgimigli, L. Reactions of substituted boryl radicals with nitroalkanes. EPR, kinetic, and product studies. J. Org. Chem. 61, 4309–4313 (1996).
Kryger, R. G., Lorand, J. P., Stevens, N. R. & Herron, N. R. Radicals and scavengers .7. Diffusion controlled scavenging of phenyl radicals and absolute rate constants of several phenyl radical reactions. J. Am. Chem. Soc. 99, 7589–7600 (1977).
Dow, N. W. et al. Decarboxylative borylation and cross-coupling of (hetero)aryl acids enabled by copper charge transfer catalysis. J. Am. Chem. Soc. 144, 6163–6172 (2022).
Mukherjee, G. et al. Inspiration from nature: influence of engineered ligand scaffolds and auxiliary factors on the reactivity of biomimetic oxidants. ACS Catal. 11, 9761–9797 (2021).
Zhao, H. M. Highlights of biocatalysis and biomimetic catalysis. ACS Catal. 1, 1119–1120 (2011).
Pan, Y. H. & Tan, C. H. Catalytic decarboxylative reactions: biomimetic approaches inspired by polyketide biosynthesis. Synthesis, 2044–2053 (2011).
Lewis, C. A. & Wolfenden, R. Uroporphyrinogen decarboxylation as a benchmark for the catalytic proficiency of enzymes. Proc. Natl Acad. Sci. USA 105, 17328–17333 (2008).
Lin, Y. X. et al. Photoredox organic synthesis employing heterogeneous photocatalysts with emphasis on halide perovskite. Chem. Eur. J. 26, 13118–13136 (2020).
Choi, G. J., Zhu, Q. L., Miller, D. C., Gu, C. J. & Knowles, R. R. Catalytic alkylation of remote C-H bonds enabled by proton-coupled electron transfer. Nature 539, 268–271 (2016).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363 (2013).
Ramkumar, V., Das, I. & Gardas, R. L. Structural arrangement and computational exploration of guanidinium-based ionic liquids with benzoic acid derivatives as anions. Cryst. Growth Des. 19, 2642–2657 (2019).
Grimme, S., Antony, J., Ehrlich, S., & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Marenich, A. V., Cramer, C. J., & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). 27–38.
Acknowledgements
We gratefully acknowledge the Beijing Municipal Natural Science Foundation (Grant No. Z200018), the National Nature Science Foundation of China (Grant no. U1867209), the Ministry of Science and Technology of the People’s Republic of China (Grant nos. 2021YFA1601400 and 2017YFA0506300), the Special Foundation of Beijing Municipal Education Commission (Grant no. 3500-12020123), Li Ge-Zhao Ning Life Science Youth Research Foundation (LGZNQN202004) to Z.L., the Central Guidance for Local Science and Technology Development Projects (no. 202138–03). We also acknowledge the facility support from the Analytical Instrumentation Center of Peking University.
Author information
Authors and Affiliations
Contributions
Q.W. and Z.B.L. conceived and designed the project. Q.W. conducted the experiments and analyzed the data. W.Q.L., X.L.C., Y.H.L., B.S.M., and X.-Y.C. W.S.W. and S.M.B. discussed the experimental results and commented on the paper. Y.H.L. and S.M.B performed DFT calculations, discussion, and revision. Q.W. prepared the manuscript with feedback from all authors. Z.B.L. conducted general guidance, project directing, and paper revisions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mingbin Yuan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, Q., Lee, Y., Liang, W. et al. Photocatalytic direct borylation of carboxylic acids. Nat Commun 13, 7112 (2022). https://doi.org/10.1038/s41467-022-34833-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-34833-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
