
Abstract
Electrocatalytic recycling of waste nitrate (NO3−) to valuable ammonia (NH3) at ambient conditions is a green and appealing alternative to the Haber−Bosch process. However, the reaction requires multi-step electron and proton transfer, making it a grand challenge to drive high-rate NH3 synthesis in an energy-efficient way. Herein, we present a design concept of tandem catalysts, which involves coupling intermediate phases of different transition metals, existing at low applied overpotentials, as cooperative active sites that enable cascade NO3−-to-NH3 conversion, in turn avoiding the generally encountered scaling relations. We implement the concept by electrochemical transformation of Cu−Co binary sulfides into potential-dependent core−shell Cu/CuOx and Co/CoO phases. Electrochemical evaluation, kinetic studies, and in−situ Raman spectra reveal that the inner Cu/CuOx phases preferentially catalyze NO3− reduction to NO2−, which is rapidly reduced to NH3 at the nearby Co/CoO shell. This unique tandem catalyst system leads to a NO3−-to-NH3 Faradaic efficiency of 93.3 ± 2.1% in a wide range of NO3− concentrations at pH 13, a high NH3 yield rate of 1.17 mmol cm−2 h−1 in 0.1 M NO3− at −0.175 V vs. RHE, and a half-cell energy efficiency of ~36%, surpassing most previous reports.
Similar content being viewed by others


Introduction
Ammonia (NH3) is the critical feedstocks of artificial fertilizers and various chemicals and one of the most promising carbon-free energy carriers1,2,3. Currently, industrial NH3 synthesis heavily relies on the energy and carbon−emission intensive Haber−Bosch (H–B) process4,5,6,7. Alternatively, electrocatalytic N2-to-NH3 conversion (eN2–NH3), using water (H2O) as a proton source, has recently attracted significant research interests owing to its mild conditions and high compatibility with renewable electricity6,8,9,10,11,12,13,14,15. However, the inherent characters of N2, including high dissociation energy of the N≡N bond (945 kJ mol−1) and low water solubility, make the eN2–NH3 work at an insufficient selectivity and two orders of magnitude lower yield rate than that of H–B process9,10,11,16. To bridge the gap, the knowledge of the nitrogen cycle brings a renewed attention to the recycling of reactive N–containing species (e.g., NO and nitrate) to NH317,18,19,20. Among them, the nitrate (NO3−) anion is particularly attractive because it exhibits comparatively low dissociation energy of the N=O bond (204 kJ mol−1) and is widely abundant as pollution in agricultural and industrial wastewaters20,21,22,23,24,25,26. Furthermore, initial developments of plasma techniques promise to convert air to NO3− with low energy consumption27,28. Therefore, using NO3− as the precursor endows NH3 electrosynthesis with sustainable features and opens up an economical route to remedy environmental pollution.
NO3−-to-NH3 conversion in microorganisms is a tandem process; i.e., NO3− reduction to NO2− using nitrate reductase and subsequent NO2−-to-NH3 conversion employing nitrite reductase or nitrogenase, independently29,30,31. This enzyme-based tandem system allows efficient NH3 generation at ambient conditions in nature owing to its specific coordination binding with NO3− and NO2−, respectively29,31,32. However, NO3− typically shows low binding affinity to transition metals in aqueous electrolytes due to its symmetrical (D3h) resonant structure and strong hydrogen bonding to H2O31,33,34. Moreover, the NO3−-to-NH3 pathway involves a complex eight−electrons transfer and multiple intermediates17,35,36. As a result, there is a scaling relation between the binding strengths of NO3−, NO2− and other oxygen−containing intermediates (e.g., NO) on the transition metal surface37. Optimizing the adsorption of one species will typically take the others away from their optima38,39, making the simultaneous acceleration of sequential NO3−-to-NO2− and NO2−-to-NH3 reactions considerably challenging.
Copper (Cu)-based catalysts have been intensively investigated for the NO3− reduction reaction (NO3RR) due to its favorable ability to bind NO3− and catalyze NO3−-to-NO2− conversion40,41,42,43. However, pure Cu catalysts commonly suffer from rapid deactivation because of their strong adsorption of the NO3RR intermediates (e.g., NO2− and NO)42,43,44. Substantial efforts have recently been made to alleviate these limitations by regulating the proton- and/or electron-transfer, as well as the binding strengths of partially reduced intermediates adsorbed on Cu centres. This was performed either by alloying Cu with noble or other transition metals (e.g., Pt, Pd and Ni)40,45,46,47,48,49 or through the formation of hybrids with molecular solids or metal oxides (e.g., Cu2O)44,50,51. These strategies have increased the Faradaic efficiency (FE) of NH3 to an impressive 70–100% and the NH3 yield rate (YNH3) to a level of 30–200 μmol cm−2 h−1 44,45,51. However, restricted by the scaling relations, these advances require highly concentrated NO3− (e.g., 1 M) and/or relatively high overpotentials (<−0.4 V vs. RHE) to balance the rates of NO3−-to-NO2− and NO2−-to-NH3 reactions, resulting in an increased energy consumption44,45,51. To compete with the H–B process, further progress on YNH3 (>1 mmol cm−2 h−1)52 and on decreasing energy consumption are highly desirable.
Energy-efficient NO3RR points to low operating overpotentials (e.g., >−0.2 V vs. RHE), at which transition metal (e.g., Cu and Co)-based catalysts often suffer from potential−dependent phase evolution, leading to the coexistence of multiple phases, such as metallic, oxide and hydroxide phases53,54,55,56. In situ monitoring of the phase evolution of transition metals during the NO3RR, while correlating these intermediate phases with specific catalytic steps53,54,57,58, may not only guide the rational design of selective catalysts for NH3 but also provide insight into the NO3RR. Inspired by the tandem NO3−-to-NH3 conversion in nature29, we sought, therefore, to circumvent the scaling relations by combining two or more cooperative intermediate phases exhibiting complementary catalytic selectivity into one tandem system, intending to achieve NH3 synthesis at low overpotentials. Importantly, it has been clearly demonstrated that the reaction rate and selectivity of a tandem catalyst system, linked to the transport of key intermediates, could be optimized by judiciously tuning the proximity, hierarchy and content ratio of multiple active phases39,59,60,61,62. Accordingly, the YNH3 could be further improved, however, to the best of our knowledge, tandem catalysts based on earth−abundant elements have never been reported for electrocatalytic consecutive NO3–-to-NH3 conversion.
In this work, we introduce a facile electrochemically driven phase-separation strategy for a tandem catalyst design, which, different from previous approaches based on sequential assembling or deposition60,61, enables the in situ formation of multiple active intermediate phases and rich phase interfaces for rapid spillover and transport of reaction intermediates. As a proof-of-concept, we implemented this catalyst synthesis strategy by the electrochemical transformation of pre−synthesized Cu–Co binary metal sulfides into core–shell Cu/CuOx and Co/CoO phases on Cu foil. The employment of Co-based phases as a sub−component of the tandem catalysts was inspired by the previously reported high selectivity of Co-based materials and complexes for NH3 generation during the NO3RR and the specific NO2−-to-NH3 conversion63,64. Our electrocatalytic tests, kinetic studies, in situ scanning electrochemical microscopy (SECM) and in situ Raman spectra reveal that at low overpotentials, the inner Cu/CuOx phases preferentially catalyze NO3− reduction to NO2−, while the outer-layer Co/CoO phases selectively catalyze NO2− reduction to NH3, both of which can be combined for rapid “working-in-tandem” NH3 synthesis. As a result, we report NO3−-to-NH3 conversion with a FE of 93.3 ± 2.1% in a wide range of NO3− concentrations at −0.175 V vs. RHE, a high NH3 yield rate of 1.17 mmol cm−2 h−1 in 0.1 M NO3− at pH 13 and a half−cell energy efficiency of ~36%, which surpass most prevreports.
Results
Catalyst design and characterization
The synthesis of Cu/Co-based tandem catalysts is schematically illustrated in Fig. 1a. A nanorod array of metal-organic frameworks (MOFs) was first grown on Cu foil by optimizing the molar ratio of 2-methylimidazole and Co2+ to 20: 1 (hereafter named as ZIF-Co-R/Cu; Fig. 1b and Supplementary Fig. S1). The ZIF-Co-R/Cu was converted into Cu–Co binary metal sulfides following a previously reported electrochemically conversion of MOF (EC-MOF) strategy65,66,67. Upon adding 0.05 M Na2HPO4 as structure-tuning agents, the nanorod contour of ZIF–Co−R could be retained during the EC-MOF (Fig. 1c), which was otherwise changed into super-thin nanosheets (Supplementary Fig. S2). We denoted the PO43−-modified Cu–Co binary metal sulfides as CuCoSP_no and their products after further electrochemical redox activation as CuCoSP. This unique nanorod array configuration of CuCoSP can provide a more efficient pathway for mass and charge transport during the NO3RR, contributing to the best performance of CuCoSP for NO3RR as discussed below.

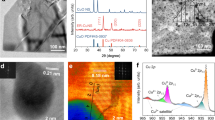
a Schematic illustration of the preparation of a Cu/Co-based binary ‘tandem catalyst’. SEM images of the ZIF-Co-R precursor (b), CuCoSP_no (c) and CuCoSP (d) on the Cu foil substrate. The inset figures are the SEM images at higher magnification. e, f Typical TEM image, EDX mappings, SAED patterns and HR-TEM image of CuCoSP_no (e) and CuCoSP (f). Unlabelled scale bars are 100 nm. The SAED patterns in (f) show the presence of CuO phases and Co-based oxides/hydroxides (CoOx(OH)y), where 1 = Co(OH)2 (100), 2 = Co(OH)2 (101), 3 = CuO (20\(\bar{2}\)), 4 = CoOOH (211) and 5 = CoO (220). The purple colours of the HR-TEM image in (f) are marked to guide the eye and correspond to the Co3O4 nanocrystals. g EDX line−scan of a selected area marked by the white arrows in (e) and (f). h XPS-determined atomic percentage-depth profile of CuCoSP_no and CuCoSP as a function of Ar+ etching time. 2-MI 2-methylimidazole; EC-MOF electrochemically conversion of metal-organic frameworks.
Low-resolution transmission electron microscopy (TEM) and high-resolution scanning electron microscopy (HR-SEM) images reveal that a curly nanosheet-assembled shell wraps the nanorod skeleton of CuCoSP_no (Fig. 1c, e and Supplementary Fig. S3a). The corresponding EDX mapping shows strong Co, Cu and S signals well-distributed in the whole nanorod and a weak signal of O due to surface oxidation. Remarkably, Cu is also spread over the nanorod, indicating that the Cu comprised in CuCoSP_no is from the dissolution of Cu foil followed by redeposition and inward diffusion of the released Cu ions during the EC-MOF. The enrichment of Cu on the CuCoSP_no surface is supported by the EDX-linear scan (Fig. 1g and Supplementary Fig. S3). Moreover, HR-TEM images, coupled with selected area electron diffraction (SAED), reveal that the curly nanosheets and skeleton of CuCoSP_no are mainly composed of ~3–5 nm Co9S8 and Cu2S nanocrystals, which are not detectable by X-ray diffraction (XRD) (Supplementary Fig. S4d)59,66.
After the electrochemical activation, the obtained CuCoSP preserves the contour of CuCoSP_no, except for the in situ formation of a hexagon-shaped nanosheet-assembled shell (Fig. 1d, f and Supplementary Fig. S5a). Compared to CuCoSP_no, the CuCoSP possesses more complex SAED patterns corresponding to multiple mixed phases (Fig. 1f), which include Co(OH)2, CoOOH, CoO and CuO EDX mappings and linear EDX scans (Fig. 1f and Supplementary Fig. S5f) show weaker S and enhanced O signals. These results suggest a significant transformation of Cu–Co binary sulfides into oxides and hydroxides, as corroborated by the XRD patterns (Supplementary Fig. S4e), as well as Raman and X-ray photoelectron spectroscopy (XPS). An HR-TEM image of a hexagon-shaped nanosheet on CuCoSP shows a typical (101) facet of Co(OH)2 and some Co3O4 nanocrystals at the edge. The EDX mapping of CuCoSP shows an intertwined distribution of Cu and Co on the nanorod (Fig. 1f), and the linear EDX scan reveals a higher content of Co in the shell region (Fig. 1g). Therefore, the shell assembled by xagon-shaped nanosheets is a Co element-rich phase, and the redox activation might induce the redistribution of Co-based and Cu-based phases.
To investigate the spatial arrangement of Cu-based and Co-based phases, we carried out XPS depth profiling. We calculated the atomic percentages of Cu, Co, O, S and P in both CuCoSP_no and CuCoSP as a function of Ar+ etching time (Fig. 1h and Supplementary Fig. S6). We found that the CuCoSP_no has a Cu-rich surface (Co: Cu = 0.862: 1) and Co-rich core (Co: Cu = 1.83: 1 at 40 min etching time), which is in line with the EDX-linear scan results. The surface of CuCoSP is enriched with Co with a Co/Cu ratio of 5.23: 1. After 5 min etching time, the Co/Cu ratio decreases to 3.07: 1 and slightly decreases in the following 35 min of Ar+ etching. The results reveal an electrochemical redox activation-induced outward diffusion of Co ions and a relative inward diffusion of Cu ions, contributing to the phase separation in CuCoSP. The outward diffusion of Co ions is attributed to the easier oxidation of cobalt sulfides than copper sulfides into corresponding oxides or hydroxides. EDX mapping images of CuCoSP_no after one-cycle redox activation provide further evidence (Supplementary Fig. S7), with the maps of Co and O overlapping on the outer-shell layer and those of Cu and residual S in the inner-core layer. This controllable phase separation in metal sulfide matrix driven by electrochemically redox activation may offer a strategy for a tandem catalyst design, which enables in situ formation of multiple active phases and rich phase interfaces for rapid spillover and transfer of reaction intermediates.
This core–shell arrangement of the Cu-based phases and Co-based phases, together with the rich phase interface between them, is the key to achieve a near-unity selectivity for subsequent NO3−-to-NH3 catalysis on CuCoSP. The Cu foil substrate of CuCoSP may play a similar role to the Cu-based phases (Fig. 1a). As a control, we used a Cu foil or ZIF-Co-R grown on carbon paper (CC) as precursors and treated them by the same EC-MOF and activation procedures (Supplementary Fig. S8). The related materials were denoted as CuSP and CoSP, respectively.
NO3RR performance
Linear sweep voltammetry (LSV) and one-hour electrolysis were performed for assessing the activity and selectivity of the as-synthesized catalysts in 0.01 M KNO3 and 0.1 M KOH (pH 13) (Fig. 2a–c). Unless otherwise noted, all potentials are corrected vs the reversible hydrogen electrode (RHE). Performing NO3RR in alkaline media was chosen due to the reported lower probability of the formation of toxic intermediates in solution (e.g., nitrogen oxides and NH2OH), as compared to those in acidic or neutral media, as well as the need of removing NO3− ions in alkaline nuclear wastewater21,42. We set the concentration of nitrate to be 0.01 M, which ensures the concentration of formed NH3 to be easily higher than that of environmental contaminants which in turn helps to compare the intrinsic activity of the catalysts via bypassing the positive effects of high-concentration NO3− (e.g., 1 M) with respect to facilitating the NO3RR52,68.

LSVs at a scan rate of 5 mV s−1 (a), Faradaic efficiencies (FE) for NH3 (b) and NO2− (c) on CuSP, CoSP and CuCoSP in 0.01 M NO3− and 0.1 M KOH (pH 13) at various applied potentials. d A comparison of half-cell energy efficiencies of NH3 (EENH3) and partial current densities of NH3 (jNH3) on CuSP, CoSP and CuCoSP catalysts at various potentials. e The FE of NO2− and NH3, the NH3 yield rate (YNH3), as well as the ratio of the formed NH3 concentration [NH3] to the converted NO3− concentration [NO3−] on the CuCoSP catalyst at −0.175 V vs. RHE at [NO3−] in the range of 1−100 mM at pH 13. f The ECSA-normalized YNH3 on CuSP, CoSP, CuCoSP and metallic CuCo hybrids in 0.01 M NO3− and 0.1 M KOH at various potentials. Error bars denote the standard deviations calculated from three independent measurements.
The LSV of CuSP in the presence of NO3− is similar to that of the NO3RR on the Cu (100) facet42, where a diffusion-limited maximum total current density (jtotal) of −55.3 mA/cm−2 is reached at around −0.3 V. At <−0.3 V, the surface of CuSP becomes blocked by the strongly adsorbed intermediates of the H2 evolution reaction (HER) and/or NO3RR until the surface is renewed because of the drastic HER at <−0.6 V42. CoSP requires a ~150 mV more negative potential to drive the NO3RR, but it shows a sharply increased jtotal for the HER at <−0.2 V, where metallic Co is supposedly formed (Supplementary Fig. S9). The CuCoSP has the catalytic features of both CuSP and CoSP, showing the highest NO3RR activity between −0.16 V and 0.085 V. Moreover, the Tafel slope of CuCoSP for the HER in 0.1 M KOH is 69 mV dec−1, and smaller than those of CuSP (125 mV dec−1) and CoSP (104 mV dec−1), suggesting faster kinetics for the HER (Supplementary Fig. S9). Thus, there is a strong synergy between the Cu-based and Co-based phases in CuCoSP for NO3RR at >−0.2 V and HER at <−0.2 V.
Determination of the product selectivity for CuSP, CoSP and CuCoSP shows a significant difference in the FE for NH3 and NO2− (Fig. 2b, c and Supplementary Figs. S10–13). At low over−potentials (>−0.2 V), CuSP exclusively catalyzes the formation of NO2−, while CoSP shows a high inherent NH3 selectivity without any interference from the underlying CC (Supplementary Fig. S14d). The CuCoSP inherits the advantages of CoSP and reaches a maximum FE of 95.9% for NH3 at −0.075 V, 14.5% higher than that of CoSP. This finding further suggests the synergy of the Cu-based and Co-based phases in CuCoSP for selective NO3−-to-NH3 conversion. Moreover, the CuCoSP shows a FE of 88.7% for NH3 and an NH3 partial current densities (jNH3) of −9.54 mA cm−2 at −0.025 V, a potential at which CuSP exhibits exclusive NO2− generation and CoSP shows a negligible NO3RR activity (Fig. 2a–d and Supplementary Figs. S11–13). CuCoSP retains this advantage even at 0.025 V (a FE of 66.4% for NH3 and a jNH3 of −3.84 mA cm−2). Similar to CuSP, the Cu foil and inner Cu-based phases of CuCoSP can catalyze the preferential formation of NO2−, which might then be further reduced to NH3 at the outer Co-based phases. This hypothesis, as further demonstrated below, may well account for the ~2-fold higher jNH3 for CuCoSP than that of CoSP at >−0.175 V (Fig. 2d).
At higher overpotentials (<−0.2 V), CuCoSP and CoSP show a sharp decline of the FE for NH3 but minor changes in their FE for NO2− (<1%) (Fig. 2b–d), indicating that the NO3RR on CuCoSP and CoSP is challenged by the drastically enhanced HER (Supplementary Fig. S9). The competing HER was previously suppressed by increasing the NO3− concentrations and/or the pH value of electrolytes45,52. As anticipated, jtotal of CuCoSP linearly increases with the NO3− concentrations (Supplementary Fig. S15), indicative of a first-order reaction kinetics. At −0.175 V, the CuCoSP catalyst reaches a nearly equal FE of 93.3 ± 2.1% for NH3 and a linearly increased NH3 yield rate (YNH3) with a value of 15.7, 33.4, 74.1, 155 and 327 μmol h−1 cm−2 in 1, 2, 5, 10 and 20 mM NO3−, respectively (Fig. 2e). When the NO3− concentration increases to 50 and 100 mM, the YNH3 of CuCoSP reaches 0.690 and 1.17 mmol h−1 cm−2, respectively. The latter is comparable to the highest reported value of 1.17 mmol h−1 cm−2 on strained Ru nanoclusters evaluated at −0.2 V in 1 M NO3− and 1 M KOH (pH 14)52. The turnover numbers (TON) of nitrate on CuCoSP, defined by the ratio of the yielded NH3 concentration [NH3] to the converted NO3− concentration [NO3−], are close to 1 at [NO3−] < 20 mM. This indicates that the formed NH3 is mainly derived from the NO3− electroreduction rather than any environmental contaminations (Fig. 2e and Supplementary Fig. S15d). However, the TON value decreases to ~0.8 in 100 mM nitrate electrolytes, suggesting that high [NO3−] might induce the formation of gaseous by-products (e.g., N2 and NOx)36. Finally, the CuCoSP achieves a half-cell energy efficiency of NH3 (EENH3) close to 40%. These results endow the proposed CuCoSP ranking among the best NH3 synthesizing electrocatalysts (Supplementary Table S1).
To assess the origin of the detected NH3 and correct the YNH3, we employed 1H NMR to detect the NH3 generation on CuCoSP in 0.1 M KOH containing 0.01 M 15N-labelled 15NO3− or 14NO3− (Supplementary Fig. S16). The 14NH3 yield quantified by 1H NMR is very close to that determined by colorimetric methods69, confirming the reliability of our results. The negligible NH3 generation in blank 0.1 M KOH and the typical 1H NMR double peaks of 15NH4+ after the electrolysis of 15NO3− suggest that the obtained NH3 indeed originates from the NO3RR25,28,44.
To derive the intrinsic activities of the catalysts, we normalize their YNH3 by the electrochemical active surface area (ECSA) (correlated with the double-layer capacity (Cdl)) (Fig. 2f and Supplementary Fig. S17). At low overpotentials, the YNH3 of CuCoSP can be ~45 times and two orders of magnitude higher than those of CoSP and CuSP, respectively, confirming the high intrinsic performance of CuCoSP. Remarkably, CuSP lost activity rapidly at >−0.525 V during the NO3− electrolysis (Supplementary Fig. S11), which occurred neither at CoSP nor at CuCoSP (Supplementary Fig. S12 and S13). This finding indicates that the Co-based phases existing in CoSP and CuCoSP may help to avoid similar poisoning effects. When the NO3RR was performed at −0.175 V for 10 h over the CuCoSP catalyst, no appreciable decay in activity and selectivity for NO3−-to-NH3 conversion was observed, and the structural features of CuCoSP were retained (Supplementary Fig. S18).
Understanding the high-rate NH3 generation on CuCoSP
Electrocatalytic NO3RR follows a consecutive pathway (Fig. 3a), where NO2− is generated as a stable intermediate17,36. A fast NO3−-to-NH3 conversion requires the simultaneous acceleration of the sequential NO3−-to-NO2− and NO2−-to-NH3 reactions35,36. To rationalize the high YNH3 on CuCoSP at low overpotentials, we firstly compared the potentials of the three catalysts required to reach −1 mA cm−2 (kinetic area with negligible mass transport limitation) in 0.01 M NO3− and NO2−, respectively (Supplementary Fig. S20). CuSP shows a 176 mV more positive potential than CoSP for NO3− reduction, while CoSP has a 251 mV more positive potential than CuSP for NO2− reduction. Therefore, CuSP and CoSP may play a complementary role for the consecutive NO3RR. Significantly, CuCoSP combines the positive properties of CuSP and CoSP for NO3− and NO2− reduction, respectively, implying that there are likely two types of active phases in CuCoSP: Cu-based phases which are similar to those in CuSP, and Co-based phases which are similar to those in CoSP. The two types of active phases in CuCoSP synergistically catalyze the tandem NO3RR.

a The LSV-derived potentials at a current density of −1 mA cm−2 and the calculated reaction constants (k, k1 for NO3−-to-NO2− and k2 for NO2−-to-NO conversion) for NO3− and NO2− reduction on CuSP, CoSP and CuCoSP catalysts. k was calculated based on the concentration evolution of NO3− or NO2− ions as a function of electrolysis time at −0.175 V vs. RHE in 0.01 M NO3− and NO2− at pH 13. Error bars denote the standard deviations of k calculated at different time points during 1 h electrolysis. b A comparison of the FE and the YNH3 on the control catalysts: CuCoSP_0.5, CuCoSP_4, CuCoSP_no, CuCoS and metallic CuCo hybrids. The LSV-derived Tafel slopes of CuSP, CoSP and CuCoSP in 0.01 M nitrate (c) and nitrite (d) at pH 13, respectively. The LSVs were recorded at a scan rate of 1 mV s−1. e Schematic representation of the SECM setup that is operated in a sample generation-tip collection (SG-TC) mode using a Pt-UME (WE 2) to detect NO2− and NH3 produced during NO3RR on Cu_Co(OH)2 model catalyst (WE 1). Inset: SEM image of Pt-UME tip; scale bar is 500 nm. f, g Current maps of NH3 oxidation (f) and NO2− reduction (g) recorded at 0.76 V and 0.06 V (vs. RHE) at the Pt-UME, respectively, when a potential of −0.12 V (vs. RHE) is applied to the Cu_Co(OH)2 model catalyst in 50 mM NO3− at pH 13.
To validate this hypothesis, we further evaluated the rate constants k1 and k2 of each catalyst for NO3−-to-NO2− and NO2−-to-NH3 conversion, respectively (Supplementary Fig. S21 and Table S2). We found that the rate constants of the three catalysts follow a similar trend as their potentials at −1 mA cm−2; a larger rate constant corresponding to a more positive potential for reaching −1 mA cm−2 (Fig. 3a). The largest k1 but the smallest k2 values of CuSP correspond to a fast reduction of NO3− to NO2− and subsequent desorption of NO2− to the electrolyte, resulting in the observed high FE of NO2− (Fig. 2c and Supplementary Fig. S11). On the CuCoSP surface, NO2− intermediates are preferentially formed on Cu-based phases and then spilt over to nearby Co-based phases. Compared to the k2/k1 ratio of CuSP (0.468), the larger k2/k1 ratios of CoSP (3.14) and CuCoSP (1.78) suggest a fast reduction of the intermediate NO2− to NH3 over their Co-based phases, thus well explaining their near-unity selectivity for NH3 generation. We used the k1 values of CoSP and CuCoSP to estimate their local surface NO2− concentration during the NO3RR and find a 1.54-fold higher value on CuCoSP than on CoSP (Supplementary Table S2), which is consistent with the observed 1.66-fold larger jNH3 of CuCoSP compared to CoSP at −0.175 V (Fig. 2d). This finding unveils the crucial role of Cu-based phases in CuCoSP for creating a high local NO2− concentration, which accelerates subsequent NH3 generation on its Co-based phases, ultimately contributing to the tandem catalysis of NO3RR.
Considering the lowest k1 value of CoSP, the smaller k1 value of CuCoSP than CuSP can be rationalized since the less active Co-based phases of CuCoSP may block its active Cu-based phases (especially the Cu foil substrate) for NO3− reduction. As such, we synthesized the CuCoSP_0.5 and CuCoSP_4 with 0.5 h and 4 h growth of ZIF-Co-R on Cu foil, respectively. The CuCoSP_0.5 shows catalytic features close to CuSP, thus giving a higher FE for NO2− than CuCoSP (Fig. 3b and Supplementary Fig. S22). This finding indicates that possible Co doping in Cu-based phases of CuCoSP_0.5 does not change their catalytic nature. Likewise, the CuCoSP _4 shows a similar catalytic feature to CoSP with a low sum FE for NO3RR. Therefore, the content ratio of Cu-based and Co-based phases in CuCoSP, rather than the Cu or Co doping, determines the rates of the NO3−-to-NO2− and NO2−-to-NH3 reactions. This is a typical feature of tandem catalysts39,59.
Electrokinetic analysis was conducted to determine the rate-determining step (RDS) of the NO3− and NO2− reduction catalyzed by the three catalysts. In 0.01 M NO3− (Fig. 3c), CuSP and CuCoSP show Tafel slopes of 94 and 112 mV dec−1, respectively, a little lower than 120 mV dec−1, suggesting that the RDS is the first one-electron transfer occurring during the NO3−-to-NO2− conversion34,48,70. The much higher Tafel slope of CoSP (167 mV dec−1) indicates that the NO3RR over CoSP is limited by the initial adsorption and activation of NO3− 48. In 0.01 M NO2− (Fig. 3d), CoSP and CuCoSP show Tafel slopes of 106 and 101 mV dec−1, respectively, suggesting that the RDS is the first one-electron transfer for the reduction of NO2− to NO34,48. CuSP has a Tafel slope of 72 mV dec−1, close to the critical value of 60 mV dec−1, implying that the RDS is a chemical step71,72. Based on the theoretical NO3RR pathways on Cu44, this RDS is likely the coupling of strongly adsorbed *NO and *H. Remarkably, the smaller Tafel slope of CuSP than those of CoSP and CuCoSP points to a faster NO2− reduction kinetics on CuSP, but CuSP suffers from a faster deactivation in 0.01 M NO2− than in 0.01 M NO3− (Supplementary Fig. S21), thus giving the smallest apparent k2 value. Accordingly, the observed poisoning of CuSP might be ascribed to the strongly adsorbed *NO species. Therefore, CuCoSP combines the nature of CuSP for NO3− reduction with that of CoSP for NO2− reduction.
To assess the possible roles of anionic ligands (PO43− and S2−) in our system, we tested the NO3RR performance of CuCoSP_no catalysts with the maximum PO43− and S2− ligands and CuCoS catalysts with only S2− ligand. The lower FE (for NH3) and YNH3 of CuCoSP_no compared with those of CuCoSP (Fig. 3b), in combination with the near-unity NH3 selectivity but low YNH3 of CuCoS (Fig. 3b and Supplementary Fig. S23), suggest that PO43− and S2− ligands most likely do not play a prominent role. We evaluated the final morphology of CuCoSP after repeating three electrolysis of one hour at −0.325 V and observed a separation of Cu-based phases (nanorods) and Co-based phases (hexagonal nanosheets) based on EDX mapping and HR-TEM images (Supplementary Fig. S24). This result rules out the formation of a bulk CuCo alloy, as corroborated by the XRD patterns (Supplementary Fig. S4f). To identify the impact of surface CuCo metallizing and/or alloying possibly during the NO3RR, we electrodeposited a hybrid of Cu–Co metals and alloy (Co: Cu = 2.82: 1, close to that on CuCoSP surface) on CC (Supplementary Fig. S25). The metallic CuCo hybrids show Tafel slopes of 61 mV dec−1 in 0.01 M NO3− and 79 mV dec−1 in 0.01 M NO2−, indicating a significantly different catalytic mechanism and kinetics than CuCoSP. Despite its higher apparent YNH3, the CuCo hybrids have a lower FE for NH3 and ECSA-normalized YNH3 than CuCoSP (Fig. 2f and 3b). The ECSA-normalized YNH3 of CuCo hybrids is about 85 and 67% of that formed on CuCoSP at −0.175 V and −0.025 V, respectively. As such, the excellent intrinsic performance of CuCoSP towards NH3 generation, especially at low overpotentials, is unlikely from surface Cu–Co metallizing or alloying. It can be concluded that the high-rate NO3−-to-NH3 conversion on CuCoSP is due to the coexistence of the complementary Cu-based and Co-based phases: the former catalyzes NO3−-to-NO2− reduction and the latter catalyzes the NO2−-to-NH3 conversion, contributing to the tandem catalysis of NO3RR.
To offer a direct evidence for this tandem catalysis of NO3RR, we performed SECM experiments in a surface-generation tip-collection mode, which involves using a positioned Pt ultramicroelectrode (Pt-UME) to detect the NO2− and NH3 formed on the surface of a Cu_Co(OH)2 model catalyst (Fig. 3e) during the NO3− electrolysis. This Cu_Co(OH)2 model catalyst has two adjacent layers to simulate the CuSP (Cu layer) and CoSP (Co(OH)2 layer) catalysts, while the border between the two layers may play a similar role of as the adjacent phases in CuCoSP (Supplementary Fig. S26). During the SECM measurements, the catalyst layer was polarized to −0.12 V (vs. RHE), while at the positioned Pt-UME cyclic voltammetry (CV) in the potential range between −0.12 V and 1.58 V (vs. RHE) at a rate of 200 mV s−1 was performed. We were applying −0.12 V at catalysts as we found that at this potential, CuSP and CoSP is relatively more stable and active compared to potentials of −0.075 V and −0.025 V, which is beneficial for in situ detection of the intermediately formed NO2− and NH3. The same reason is the use of a concentration of 50 mM NO3−. At −0.12 V, CuSP mainly produces NO2−, and CoSP exhibits a much lower NO3RR activity compared to CuCoSP, as indicated in Fig. 2a–c and Supplementary Fig. S11–13.
We firstly performed cyclic voltammograms at the Pt-UME separately in 10 mM NO3−, NO2− and NH4Cl at pH 13, which are compared with those in pure 0.1 M KOH (Supplementary Fig. S27a–c). The results indicate that the Pt-UME does not exhibit any activity for the NO3RR, but it can efficiently catalyze NO2− reduction and NH3 oxidation in alkaline media, which is well consistent with previous studies73,74,75. This enables selectively detecting the in situ generated NO2− (at 0.06 V) and NH3 (at 0.76 V) (Supplementary Fig. S27d–f), when the tip of Pt-UME approaches the surface of Cu_Co(OH)2 layer. Accordingly, a high current from NH3 oxidation and NO2− reduction corresponds to a high local concentration of NH3 and NO2−, respectively, above the surface of Cu_Co(OH)2 catalyst. The SECM array scans (Fig. 3f, g) reveal that the amount of generated NH3 is substantially increasing at the border (X ≈ 600 μm) between the Cu and Co(OH)2 layers, accompanying by a sharply decreased amount of generated NO2−. The SECM results unequivocally confirmed that the NO2− is preferentially formed on the Cu layer and then diffuses to the near Co(OH)2 layer, where the NO2− is reduced into NH3 according to a typical tandem catalysis process. Note that the formed NH3 at the border of the Cu_Co(OH)2 layers diffuses to both sides, leading to the formation of a concentration gradient around the border during the course of the SECM measurement.
Identification of the active phases for NO3RR
Ex situ XPS and Raman spectra were obtained to identify the surface phase compositions of the three catalysts (Fig. 4, Supplementary Fig. S28 and Fig. S29). We find that the redox activation induced the transformation of the initial Cu/Co-based sulfide phases into the corresponding oxides and hydroxides (see detailed assignments of the XPS and Raman peaks in Supplementary Notes 1 and 2). As a result, CuSP consists of Cu0, CuO, Cu2O and Cu(OH)253,54,76, whereas CoSP is composed of Co2+-dominated CoO and Co(OH)2, as well as Co3+-containing Co3O4 and CoOOH65,77,78,79. As anticipated, CuCoSP exists as a combination of CuSP and CoSP in phase compositions, except for the observed suppression of Co3+–CoOx phases and increase of Cu(OH)2 phases (Fig. 4a, b), which points to the synergy interaction between Cu-based and Co-based phases in CuCoSP. O 1s XPS spectra reveal a ~2-fold higher content of O vacancies on CuSP and CuCoSP than on CoSP (Fig. 4c and Supplementary Fig. S28c)80. Together with the observed smaller Tafel slopes of CuSP and CuCoSP compared to CoSP (Supplementary Fig. S28f), this result highlights a potential role of O vacancies in Cu-based phases for the initial adsorption and/or activation of free NO3− ions50, which is a key challenge for most of the reported NO3RR catalysts.

Cu LMM (a), Co 2p3/2 (b) and O 1s (c) XPS spectra. d Ex situ Raman spectra.
To derive the active phases for NO3RR experimentally, we used in situ Raman spectroscopy to monitor the phase evolution of the three catalysts at a series of applied potentials in 0.01 M KOH in the presence or absence of 0.01 M NO3− (Fig. 5 and Supplementary Fig. S30). NO3− ions exhibit a characteristic peak at ~1050 cm−1 81. K2SO4 was added to ensure sufficient ionic conductivity and provides SO42− ions as an external Raman reference with a typical signal at ~982 cm−1 53.

In situ Raman spectra of CuSP (a), CoSP (b) and CuCoSP (c) at different applied potentials in electrolytes containing 0.01 M NO3−, 0.04 M K2SO4 and 0.01 M KOH. d A proposed reaction mechanism of CuCoSP tandem catalysis of NO3RR at low overpotentials. In the middle of (d), the light blue region corresponds to the potential range for the reduction of NO3− to NO2 species at the Cu/CuOx phases, resulting in the etching of catalysts, while the light pink region shows the potential range for efficient tandem reduction of NO3− to NH3.
Figure 5a shows the Raman spectra of CuSP at reducing potentials related to NO3RR. The initial broad bands at 417, 523 and 628 cm−1, associated with Cu2O phases53,54, persist at as low as −0.525 V. Remarkably, at <−0.175 V, two sets of peaks emerge at 341, 407, 491 and 524 cm−1, previously assigned to Cu–O and Cu–OH modes, respectively53. The same but more notable signals are observed in the absence of NO3− (Supplementary Fig. S30a), partly due to the NO3RR delaying the surface phase evolution. It is worth noting that CuSP can prevent poisoning only at <−0.425 V, where the surface evolution of CuSP might promote stable NH3 generation. As a control, we further assessed the performance of a metallic Cu foil for NO3RR (Supplementary Fig. S14). The results show that at potentials >−0.425 V, Cu foil suffered from deactivation in a much higher rate, compared to CuSP containing mixed Cu/CuOx phases. Moreover, at low overpotentials, the Cu foil mainly electrochemically catalyzes the reduction of NO3− to highly oxidative NO2 and acts as reducing reagents for the subsequent chemical reduction of NO2 to NO2−, which is supported by the over 100% FE for NO3RR at −0.025 and −0.125 V (vs. RHE) (Supplementary Fig. S14c). This chemical step will lead to the partial oxidation of metallic Cu to CuOx in alkaline electrolytes. According to recent DFT calculations on Cu and mixed Cu/Cu2O phase, the latter was suggested to be more active for NO3− to NO2− conversion44. Therefore, at low overpotentials, the catalytic nature of CuSP for NO3RR is related to the Cu/CuOx phases, considering the XPS-evidenced presence of Cu0 phase.
On the CoSP catalysts, the characteristic Raman peaks of multiple Co3+-based phases and Co(OH)2 are fast attenuated with decreasing potentials from 0.025 V to −0.325 V (Supplementary Fig. S30b)77,78,79. This indicates the gradual conversion of Co3+-based phases and Co(OH)2 into CoO and metallic Co in the absence of NO3− 77. In 0.01 M NO3− the attenuation rate of these Raman peaks was much slower (Fig. 5b). Remarkably, the Raman signals associated with CoOOH and Co3O4 phases increase at 0.025 and −0.025 V, suggesting the partial oxidation of Co2+ in CoSP to Co3+77,78. Because of this, the electrolysis product of NO3− at 0.025 and −0.025 V should be only the highly oxidative NO2 species in Ar-saturated alkaline electrolytes49, and their formation slows down the phase conversion. In this context, the Co2+-based phases act as chemical reducing reagents and offer additional electrons for reduction of NO2 to NO2−, after which the formed NO2− will be electrochemically reduced to NH3 as evidenced by the low overpotential of CoSP for the NO2− reduction (Fig. 3a), finally contributing to the apparently highest FE for NH3 at low overpotentials (Fig. 2b). At <−0.025 V, the Raman signals of Co3+-based phases and Co(OH)2 start to weaken. This result, together with the potential at −1 mA cm−2 of CoSP catalysts for NO3− reduction (−0.071 V), suggests that the fast reduction of NO3− into NO2− on CoSP requires the in−situ formation of metallic Co. By contrast, we found that the Co2+ in CoSP was oxidized to Co3+ already at >0.025 V77,78,79. Since the potential at −1 mA cm−2 of CoSP for NO2− reduction is limited to 0.176 V (Fig. 3a), it can be concluded that the sharply increased Co3+-based phases at potentials >0.025 V are inactive for NO2− reduction. Thus, the active phase of CoSP for reducing NO3− to NO2− is related to metallic Co, while that for NO2−-to-NH3 conversion is a Co2+-dominated CoOx phase.
For CuCoSP, the Raman peaks associated with Co-based phases were quickly attenuated with decreasing potentials, while a phase assigned to CuOx emerges at 338 and 400 cm−1 (Fig. 5c and Supplementary Fig. S30c)54. These results suggest an electrochemically driven phase separation in CuCoSP, leading to the formation of hybrid of Cu/CuOx and Co/CoO phases. The CuOx persists to potentials as low as −0.325 V, further decreasing the probability of in situ surface CuCo alloying, especially at potentials >−0.2 V. This is further supported by the XRD patterns, HR-TEM and EDX-mapping images of CuCoSP after repeating three electrolysis cycles of one hour at −0.325 V (Supplementary Fig. S4f and Fig. S24). The rate at which these Raman peaks are evolving is almost not impacted by the NO3RR (Fig. 5c and Supplementary Fig. S30c). This might be attributed to the high rate of NO3RR on CuCoSP, which establishes a depletion layer of NO3− explaining the minor effects of the NO3RR on the phase-evolving rate. In stark contrast to CoSP (Fig. 5b), the Raman signals of Co3+-based phases in CuCoSP were not enhanced at both 0.025 and −0.025 V in 0.01 M NO3−. This finding indicates fast reduction of NO3− to NO2− rather than to oxidative NO2 species on the Cu-based phases of CuCoSP. Accordingly, the active Co2+-based phases of CuCoSP are stabilized by the Cu/CuOx phases, both of which are combined to form a tandem system for cascade NO3−-to-NH3 conversion at low overpotentials (Fig. 5d).
Discussion
In summary, we present a concept for designing efficient tandem catalysts, which involves the coupling of potential-dependent intermediate phases of transition metals to act as cooperative catalytic sites for cascade NO3−-to-NH3 conversion. This concept was verified using Cu/CuOx–Co/CoO hybrids with a well-defined spatial arrangement that is achieved by electrochemical redox activation-induced phase reconstruction of Cu/Co-based binary metal sulfides. In this tandem catalysis system, NO3− ions are reduced to NO2− preferentially on Cu/CuOx phases, while the NO2− intermediates are then transferred and selectively converted to NH3 on Co/CoO phases. The sequential NO3− and NO2− reduction on two different adjacent metal/metal oxide phases enables a high-rate NH3 generation at low overpotentials. At −0.175 V vs. RHE, the designed CuCoSP catalysts show an excellent FE for NH3 (90.6%) and super−high YNH3 of 1.17 mmol cm−2 h−1 in 0.1 M NO3− at pH 13, outperforming most of the NO3RR catalysts at the same conditions. Although tandem catalysis was widely employed in heterogeneous systems, this study provides a direct demonstration of efficiently using distinct potential-dependent intermediate phases as tandem catalytic sites. This concept of splicing active phases of transition metals represents a powerful strategy towards designing high-performance, multi-functional electrocatalysts for multi-step chemical reactions, such as e.g. urea electrosynthesis by integrating NO3RR with CO2 reduction.
Methods
Chemicals
Na2HPO4 (≥99.0 %) and NaClO solution (17 %) were purchased from VWR. Maleic acid (≥99.0 %) was obtained from Riedel-de Haën. All other chemicals and Cu foil (99.98 %) were from Sigma-Aldrich. All chemicals were used without further purification. Carbon cloth was provided by PHYCHEMI.
Growth of ZIF-Co-R nanorods on Cu foil and carbon cloth
The Cu foil (\(3\times 0.5\) cm2) was washed with acetone, ethanol and distilled water, finally dried by blotting paper before use. The CC was initially treated with 1 M HCl (≥37%) for 12 h, washed with deionized water and dried in a 70 °C oven. Then, the CC was soaked in an aqueous solution of 2-methylimidazole (0.4 M, 99% purity) for 12 h and dried by blotting paper before use. For the growth Co-ZIF-R nanorods, the Cu foil or CC was immersed into a 14 ml growth solution, which was prepared by adding 10 ml of 2-methylimidazole (0.4 M) into 4 ml aqueous solution of Co(NO3)2 6H2O (50 mM, ≥98%). The Co−ZIF-R nanorods were grown under static conditions at 25 °C for 105 min. The purple Co-ZIF-R nanorods on Cu foil or CC were washed with deionized water and dried in a 70 °C oven.
EC-MOF for synthesis of CuCoSP_no
In a typical three-electrode system, the Co-ZIF-R on Cu foil was used as the working electrode, and Ag/AgCl (sat. KCl) and an FTO slide were employed as the reference and counter electrodes, respectively. In Ar-saturated electrolytes (aqueous solution of 0.5 M thiourea (≥99.0%), 0.25 M KCl (≥99.0%) and 0.05 M Na2HPO4), the ZIF-Co-R was electrochemically converted via continuous CV scanning between −1.76 V and −0.15 V at a series of scan rates for different CV cycles (at 0.2 V s−1 for 1200 CV cycles, at 0.1 V s−1 for 300 CV cycles, at 0.05 V s−1 for 150 CV cycles and finally at 0.02 V s−1 for 100 CV cycles) using an Autolab potentiostat. The black products on Cu foil were taken out, rinsed with water and acetone, blow-dried by Ar gas and stored at −21 °C for further characterization. Using the same method, the CuSP_no was prepared from Cu foil directly; the CoSP_no on CC was synthesized using ZIF-Co-R nanorods on CC as precursors; the CuCoS_no on Cu foil was obtained in an electrolyte with no Na2HPO4. As a control, metallic CuCo hybrids were prepared via electrodeposition of CuCo metals and alloy on CC in 0.05 M H2SO4 (98%) solution containing 4 mM CuSO4 (≥99%) and 16 mM CoSO4 (≥99%) at −1.75 V vs. Ag/AgCl for 400 s.
Electrochemical redox activation of the samples
Using a Gamry interface 1000 workstation, the samples were activated in 0.1 M KOH (≥85%) and 0.01 M potassium nitrate (≥99%) at 60 mA cm−2 for 45 s. They then underwent continuous LSV sweeping at a rate of 20 mV s−1 from −0.8 V to −1.65 V (vs. Ag/AgCl) until the polarization curves reached a steady state. This activation process was repeated two or three times. Then, the samples were gently washed with water and acetone, and dried under Ar flow for further tests or characterizations. After the redox activation, the samples of CuCoSP_no, CuSP_no, CoSP_no and CuCoS_no were named as CuCoSP, CuSP, CoSP and CuCoS, respectively.
Material characterization
SEM was performed using a Quanta 3D FEG scanning electron microscope. TEM images, high-angle annular dark-field TEM images, SAED patterns, element mappings and EDX line-scan were carried out on a JEOL-2800 TEM/STEM system using gold grids. XPS was recorded using an AXIS Nova spectrometer (Kratos Analytical) equipped with a monochromatic Al Kα X-ray source (1487 eV, 15 mA emission current) and an inert ion gas gun for depth-profiling composition analysis as a function of etching time. For the core-level spectra, the binding energies were calibrated based on the C 1s feature located at 284.8 eV. The nuclear magnetic resonance (NMR) spectroscopy was performed on a Bruker 400 MHz NMR spectrometer. XRD were obtained using a Bruker D8 Discover X-ray diffractometer with Cu Kα radiation (\(\lambda =1.5418{\AA }\)).
Electrochemical NO3RR tests
The electrochemical tests were performed using a three-electrode system connected to the Gamry workstation in a typical H-type cell. The H-type cell was separated by a Nafion 117 membrane (Dupont) that was pretreated following reported procedures82. The catalysts were used as the working electrode, while Ag/AgCl (3 M KCl) and platinum mesh were used as the reference and counter electrodes, respectively. The electrolytes were Ar-saturated 0.1 M KOH (pH 13) containing different concentrations of NO3−. The electrochemical cell was maintained in an Ar atmosphere during experiments. The LSV curves were collected at a scan rate of 5 mV s−1. All potentials were calibrated to the RHE reference scale using \({E}_{{{{{{\rm{RHE}}}}}}}={E}_{{{{{{\rm{Ag}}}}}}/{{{{{\rm{AgCl}}}}}}}+0.207{{{{{\rm{V}}}}}}+0.0591\times {{{{{\rm{pH}}}}}}\). The current density was normalized to the geometric electrode area (~0.5 cm2). Note that the electrode area was 0.2 cm2 for tests in 0.05 and 0.1 M nitrate. The solution resistance (RS) was measured using potentiostatic electrochemical impedance spectroscopy with a frequency range of 0.1 Hz to 200 kHz and an amplitude of 10 mVpp. The potentials were compensated by iRS-drop from the electrolyte resistance. Potentiostatic measurements were performed for 1 h in 30 ml cathode electrolyte with a stirring rate of 300 rpm, and then the electrolyte was stored at 4 °C (no more than 2 days) before analysis. To assess the performance change of CuCoSP during ten cycles of one-hour electrolysis at −0.175 V (vs. RHE), the electrolyte (30 ml, 0.1 M KOH and 0.01 M nitrate) was collected after each one-hour electrolysis for product analysis and a fresh electrolyte was used for the next cycle of one-hour electrolysis. Note that NH3 volatilization in the electrolytes (pH 13) is negligible during the one-hour electrolysis (Supplementary Fig. S19). The Cdl was determined by CV scanning in a non-faradaic potential window at different scan rates (10–120 mV s−1). The plot of capacitive anode and cathode current differences \([({j}_{{{{{{\rm{a}}}}}}}-{j}_{{{{{{\rm{c}}}}}}})/2]\) at a set potential against the CV scan rates shows a linear relationship, and the slope is Cdl.
Kinetic evaluation
The LSVs of the catalysts were recorded at a scan rate of 1 mV s−1 in 0.1 M KOH (pH 13) containing 0.01 M NO3− or 0.01 M NO2− (KNO2, ≥96%). To obtain the rate constant, the electrolysis at −0.175 V (vs. RHE) were performed for 1 h in 22 ml electrolyte (0.1 M KOH) containing 0.01 M NO3− or 0.01 M NO2− in the cathode chamber. The reaction constant (k1 for NO3− reduction and k2 for NO2− reduction) was obtained by monitoring the concentration evolution of NO3− or NO2− ions as a function of electrolysis time, assuming that their concentrations decayed exponentially as per first-order rate, that is, \({C}_{{{{{{\rm{t}}}}}}}={C}_{0}{\exp }(-k\times t)\), where, C0 is the initial molar concentration of reactant (NO3− or NO2−) and Ct is the molar concentration of reactant at time t.
Determination of ion concentrations
NH4 + quantification
The produced NH3 was quantitatively determined using the indophenol blue method10,69. Typically, a certain amount of electrolyte was taken out from the reaction cell and diluted to 2 ml. Then, 2 ml of 1 M NaOH (≥98%) solution containing sodium citrate (≥99%) and salicylic acid (≥99%) (stored at 4 °C) and 1 ml of freshly prepared 0.05 M NaClO was added. The mixed solution was shaken for few seconds. Finally, 0.2 ml of 1 wt.% sodium nitroferricyanide (≥99%) solution (stored at 4 °C) were added for the colour reaction. After keeping at room temperature for 1 h, the resulting solution was measured using an ultraviolet–visible (UV–Vis) spectrophotometer. The absorbance at ~655 nm was used to determine the concentration of NH3. In order to quantify the amount of NH3, a calibration curve was built using standard NH4Cl (≥99.5%) solution in 0.1 M KOH.
NO2 − quantification44
A specific colour reagent for NO2− quantification was prepared by mixing 0.20 g of N-(1-naphthyl) ethylenediamine dihydrochloride (≥98%), 4.0 g of sulfonamide (≥99%) and 10 ml of phosphoric acid (85 wt.% in H2O) (\({{{{{\rm{\rho }}}}}}=1.7{{{{{\rm{g}}}}}}/{{{{{\rm{ml}}}}}}\)) with 50 ml of deionized water. In a typical colourimetric test, 1 ml HCl (1 M) was firstly added into the 5 ml of diluted post-electrolysis electrolytes, and then 0.1 ml of colour reagent was added and shaken to obtain a uniform solution. The UV–Vis absorbance at 540 nm was recorded after 20 min at room temperature. The amount of NO2− was determined using a calibration curve of NaNO2 (≥96%) solutions. N2H4 and NH2OH were probably produced during the electroreduction of nitrate. However, their concentrations are expected to be very low and only measurable at intermediate times, owing to their high reactivity in basic media42,43. Thus, we mainly focused on analyzing the yields of NH3 and NO2−.
NO3 − quantification44,83
A certain amount of post-electrolysis electrolytes was diluted to 4 ml. Then, 1 ml of 1 M HCl and 0.1 ml sulfamic acid (98%) solution (0.8 wt.%) were added, and the final mixed solution was shaken to obtain a uniform solution. UV–Vis spectrophotometer was used to record the absorption intensities at wavelengths of 220 nm and 275 nm. The calculated absorbance value A (\(A={A}_{220{{{{{\rm{nm}}}}}}}-2\times {A}_{275{{{{{\rm{nm}}}}}}}\)) is linearly related to the NO3− concentrations. In order to quantify the amount of NO3−, a calibration curve was obtained using KNO3 standard solution.
Calculation of the FE, YNH3 and j NH3
The FE was defined as the charge consumed for the formation of a specific product (e.g., NH3) divided by the total charge passing through the electrodes (Q) during electrolysis. Given that eight electrons are consumed to produce one NH3 molecule, the FE of NH3 (FENH3), YNH3 and jNH3 can be calculated as follows: \({{{{{{\rm{FE}}}}}}}_{{{{{{\rm{NH}}}}}}3}=(8\times {{{{{\rm{F}}}}}}\times {C}_{{{{{{\rm{NH}}}}}}3}\times V)/Q\), \({{{{{{\rm{Y}}}}}}}_{{{{{{\rm{NH}}}}}}3}=({C}_{{{{{{\rm{NH}}}}}}3}\times V)/(A\times t)\), and \({j}_{{{{{{\rm{NH}}}}}}3}=(Q\times {{FE}}_{{{{{{\rm{NH}}}}}}3})/(A\times t)\), where F is the Faraday constant, CNH3 is the molar concentration of detected NH3, V is the volume of the electrolytes, A is the electrode geometric area, and t is the reaction time. Given that two electrons are consumed to produce one NO2− molecule, the FE of NO2− can be calculated as follows:\(\,{{{{{\rm{FE}}}}}}({{{{{{\rm{NO}}}}}}}_{2}^{-})=(2\times {{{{{\rm{F}}}}}}\times C({{{{{{\rm{NO}}}}}}}_{2}^{-})\times V)/Q\), where C(NO2−) is the molar concentration of detected NO2−.
15NO3 − Isotope labelling experiments and 14NH3 quantification by 1H NMR
To quantify the 14NH4+ yield after electrolysis of 0.01 M K14NO3 at −0.175 V (vs. RHE) for 1 h, a calibration curve of 1H NMR (400 MHz) measurements was constructed using a series of 14NH4Cl solutions with defined concentrations (1, 2, 3, 4 and 5 mM) as standards. In a typical procedure84, 125 μl of the standard solution/electrolytes was mixed with 125 μl of 15 mM maleic acid in DMSO-D6 (99.9 atom% D), 50 μl of 4 M H2SO4 in DMSO-D6 and 750 μl of DMSO-D6. The peak area integral ratio of 14NH4+ to maleic acid is positively correlated with the concentrations of 14NH4+. To confirm the source of NH3 qualitatively, 0.01 M Na15NO3 (>98 atom%15N, ≥99% purity) and 0.1 M KOH were used as the feeding electrolytes for 1 h electrolysis at −0.175 V (vs. RHE) and 15NH4+ in the electrolyte was detected using 1H NMR84.
Operando SECM test
To prepare the model catalyst, a CuSP slid (\(0.5\times 3\) cm2) was cleaned using 0.1 M HCl under ultrasonication for 20 min, washed by deionized water, and dried by blotting paper. Then, half of the CuSP slid was immersed in a MOFs growth solution (a mixture of 1.5 ml of 2-methylimidazole (0.4 M) and 1.5 ml aqueous solution of Co(NO3)2 6H2O (50 mM)) in Ar-atmosphere for 6 h. The formed Cu_ZIF-Co hybrid layers were washed with deionized water, dried with blotting paper, and further immersed in 1 M KOH solution bubbled with Ar gas for 1.5 h in a gas-tight cell, during which the ZIF-Co film was completely converted into a uniform and compacted Co(OH)2 layer, due to the poor stability of ZIF-Co MOFs in water85. After washed with water and ethanol and drying at 25 °C, half of the CuSP slide was exposed in Air and the other half was covered by Co(OH)2 nanosheets, both of which constitute the Cu (CuOx)_Co(OH)2 model catalyst.
The SECM setup with shear-force-based distance control is located in a faraday cage to allow for shielding of electrical noise, with the exception of the lock-in amplifier and the potentiostat86,87. During the SECM measurements, the Cu_Co(OH)2 catalyst were used as the sample (working electrode 1; WE 1), while a Pt-UME with a diameter of ~1 μm (working electrode 2; WE 2), Ag/AgCl/3 M KCl (reference electrode) and a Pt-mesh (counter electrode separated by a Zirfon membrane) were assembled to a four-electrode system. The sample was polarized to −0.12 V (vs. RHE) for triggering the NO3RR, while at the Pt-UME cyclic voltammograms were performed in a potential range between −0.12 V and 1.58 V (vs. RHE) at a scan rate of 200 mV s−1 to identify intermediately formed NO2− (at 0.06 V vs. RHE) and NH3 (at 0.76 V vs. RHE). Each SECM array scan was recorded from the Cu/CuOx layer to the Co(OH)2 layer with an overall x-displacement of 1200 μm, while the border between the two layers is at ~600 μm.
In situ Raman spectroscopy
Raman spectroscopy was performed with a Lab−RAM HR Raman microscopy system (Horiba Jobin Yvon, HR550) equipped with a 532 nm laser as the excitation source, a water immersion objective (Olympus LUMFL, 60×, numerical aperture = 1.10), a monochromator (1800 grooves/mm grating) and a Synapse CCD detector. Each spectrum is an average of five continuously acquired spectra with a collection time of 50 s each. A three-electrode electrochemical cell was used for in situ Raman tests. Pt wires and Ag/AgCl (3 M KCl) were used as counter and reference electrodes, respectively. To protect the objective from the corrosive 0.1 M KOH electrolyte, 0.01 M KOH (pH 12) was used instead. K2SO4 (≥99.0%) was added to ensure sufficient ionic conductivity (keeping the total concentration of K+ to be 0.1 M) and provides SO42− ions as an external Raman reference. Typically, in the presence of 0.01 M KNO3, the supporting electrolytes were 0.01 M KOH and 0.04 M K2SO4. In the absence of KNO3, the electrolytes were 0.01 M KOH and 0.045 M K2SO4.
Data availability
The data generated and analyzed during this study are provided in the main text and Supplementary information file or can be obtained from the corresponding authors on reasonable request.
References
Foster, S. L. et al. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 1, 490–500 (2018).
Ashida, Y., Arashiba, K., Nakajima, K. & Nishibayashi, Y. Molybdenum-catalysed ammonia production with samarium diiodide and alcohols or water. Nature 568, 536–540 (2019).
Tang, C. & Qiao, S. -Z. How to explore ambient electrocatalytic nitrogen reduction reliably and insightfully. Chem. Soc. Rev. 48, 3166–3180 (2019).
Kitano, M. et al. Ammonia synthesis using a stable electride as an electron donor and reversible hydrogen store. Nat. Chem. 4, 934–940 (2012).
Martín, A. J., Shinagawa, T. & Pérez-Ramírez, J. Electrocatalytic reduction of nitrogen: From Haber−Bosch to ammonia artificial leaf. Chem 5, 263–283 (2019).
Guo, C., Ran, J., Vasileff, A. & Qiao, S. -Z. Rational design of electrocatalysts and photo(electro)catalysts for nitrogen reduction to ammonia (NH3) under ambient conditions. Energy Environ. Sci. 11, 45–56 (2018).
Soloveichik, G. Electrochemical synthesis of ammonia as a potential alternative to the Haber–Bosch process. Nat. Catal. 2, 377–380 (2019).
Wan, Y., Xu, J. & Lv, R. Heterogeneous electrocatalysts design for nitrogen reduction reaction under ambient conditions. Mater. Today 27, 69–90 (2019).
Cui, X., Tang, C. & Zhang, Q. A review of electrocatalytic reduction of dinitrogen to ammonia under ambient conditions. Adv. Energy Mater. 8, 1800369 (2018).
Wang, J. et al. Ambient ammonia synthesis via palladium-catalyzed electrohydrogenation of dinitrogen at low overpotential. Nat. Commun. 9, 1795 (2018).
Chen, P. et al. Interfacial engineering of cobalt sulfide/graphene hybrids for highly efficient ammonia electrosynthesis. Proc. Natl Acad. Sci. USA 116, 6635–6640 (2019).
Song, Y. et al. A physical catalyst for the electrolysis of nitrogen to ammonia. Sci. Adv. 4, e1700336 (2018).
Lv, C. et al. An amorphous noble-metal-free electrocatalyst that enables nitrogen fixation under ambient conditions. Angew. Chem. Int. Ed. 57, 6073–6076 (2018).
Li, S. -J. et al. Amorphizing of Au Nanoparticles by CeOx-RGO hybrid support towards highly efficient electrocatalyst for N2 reduction under ambient conditions. Adv. Mater. 29, 1700001 (2017).
Geng, Z. et al. Achieving a record-high yield rate of 120.9 µgNH3 mgcat.−1 h−1 for N2 electrochemical reduction over Ru single-atom catalysts. Adv. Mater. 30, 1803498 (2018).
Chen, G. −F. et al. Ammonia electrosynthesis with high selectivity under ambient conditions via a Li+ incorporation strategy. J. Am. Chem. Soc. 139, 9771–9774 (2017).
Rosca, V., Duca, M., de Groot, M. T. & Koper, M. T. M. Nitrogen cycle electrocatalysis. Chem. Rev. 109, 2209–2244 (2009).
Chen, J. G. et al. Beyond fossil fuel-driven nitrogen transformations. Science 360, eaar6611 (2018).
Long, J. et al. Direct electrochemical ammonia synthesis from nitric oxide. Angew. Chem. Int. Ed. 59, 9711–9718 (2020).
Wang, Y., Yu, Y., Jia, R., Zhang, C. & Zhang, B. Electrochemical synthesis of nitric acid from air and ammonia through waste utilization. Natl Sci. Rev. 6, 730–738 (2019).
van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290–294 (2021).
McEnaney, J. M. et al. Electrolyte engineering for efficient electrochemical nitrate reduction to ammonia on a titanium electrode. ACS Sustain. Chem. Eng. 8, 2672–2681 (2020).
Hao, D. et al. Emerging alternative for artificial ammonia synthesis through catalytic nitrate reduction. J. Mater. Res. Technol. 77, 163–168 (2021).
Katsounaros, I. On the assessment of electrocatalysts for nitrate reduction. Curr. Opin. Electrochem. 28, 100721 (2021).
Wu, Z. -Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Yu, Y., Wang, C., Yu, Y., Wang, Y. & Zhang, B. Promoting selective electroreduction of nitrates to ammonia over electron-deficient Co modulated by rectifying Schottky contacts. Sci. China Chem. 63, 1469–1476 (2020).
Sun, J. et al. A hybrid plasma electrocatalytic process for sustainable ammonia production. Energy Environ. Sci. 14, 865–872 (2021).
Li, L. et al. Efficient nitrogen fixation to ammonia through integration of plasma oxidation with electrocatalytic reduction. Angew. Chem. Int. Ed. 60, 14131–14137 (2021).
Milton, R. D. & Minteer, S. D. Enzymatic bioelectrosynthetic ammonia production: recent electrochemistry of nitrogenase, nitrate reductase, and nitrite reductase. ChemPlusChem 82, 513–521 (2017).
Duca, M., Weeks, J. R., Fedor, J. G., Weiner, J. H. & Vincent, K. A. Combining noble metals and enzymes for relay cascade electrocatalysis of nitrate reduction to ammonia at neutral pH. ChemElectroChem 2, 1086–1089 (2015).
Ford, C. L., Park, Y. J., Matson, E. M., Gordon, Z. & Fout, A. R. A bioinspired iron catalyst for nitrate and perchlorate reduction. Science 354, 741–743 (2016).
Coelho, C. & Romão, M. J. Structural and mechanistic insights on nitrate reductases. Protein Sci. 24, 1901–1911 (2015).
Harris, L. E. The lower electronic states of nitrite and nitrate ion, nitromethane, nitramide, nitric acid, and nitrate esters. J. Chem. Phys. 58, 5615–5626 (1973).
Li, Y. et al. Enzyme mimetic active intermediates for nitrate reduction in neutral aqueous media. Angew. Chem. Int. Ed. 59, 9744–9750 (2020).
Wang, Z., Richards, D. & Singh, N. Recent discoveries in the reaction mechanism of heterogeneous electrocatalytic nitrate reduction. Catal. Sci. Technol. 11, 705–725 (2021).
Duca, M. & Koper, M. T. M. Powering denitrification: the perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 5, 9726–9742 (2012).
Liu, J. -X., Richards, D., Singh, N. & Goldsmith, B. R. Activity and selectivity trends in electrocatalytic nitrate reduction on transition metals. ACS Catal. 9, 7052–7064 (2019).
Wang, S. et al. Universal transition state scaling relations for (de)hydrogenation over transition metals. Phys. Chem. Chem. Phys. 13, 20760–20765 (2011).
Li, F. et al. Cooperative CO2-to-ethanol conversion via enriched intermediates at molecule–metal catalyst interfaces. Nat. Catal. 3, 75–82 (2020).
Martinez, J., Ortiz, A. & Ortiz, I. State-of-the-art and perspectives of the catalytic and electrocatalytic reduction of aqueous nitrates. Appl. Catal. B: Environ. 207, 42–59 (2017).
Zhang, X. et al. Recent advances in non-noble metal electrocatalysts for nitrate reduction. Chem. Eng. J. 403, 126269 (2021).
Pérez-Gallent, E., Figueiredo, M. C., Katsounaros, I. & Koper, M. T. M. Electrocatalytic reduction of nitrate on copper single crystals in acidic and alkaline solutions. Electrochim. Acta 227, 77–84 (2017).
Reyter, D., Bélanger, D. & Roué, L. Study of the electroreduction of nitrate on copper in alkaline solution. Electrochim. Acta 53, 5977–5984 (2008).
Wang, Y., Zhou, W., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350–5354 (2020).
Wang, Y. et al. Enhanced nitrate-to-ammonia activity on copper–nickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702–5708 (2020).
Lu, C., Lu, S., Qiu, W. & Liu, Q. Electroreduction of nitrate to ammonia in alkaline solutions using hydrogen storage alloy cathodes. Electrochim. Acta 44, 2193–2197 (1999).
Durivault, L. et al. Cu–Ni materials prepared by mechanical milling: their properties and electrocatalytic activity towards nitrate reduction in alkaline medium. J. Alloy. Compd. 432, 323–332 (2007).
Dima, G. E., de Vooys, A. C. A. & Koper, M. T. M. Electrocatalytic reduction of nitrate at low concentration on coinage and transition−metal electrodes in acid solutions. J. Electroanal. Chem. 554–555, 15–23 (2003).
Su, J. F., Ruzybayev, I., Shah, I. & Huang, C. P. The electrochemical reduction of nitrate over micro-architectured metal electrodes with stainless steel scaffold. Appl. Catal. B: Environ. 180, 199–209 (2016).
Reyter, D., Chamoulaud, G., Bélanger, D. & Roué, L. Electrocatalytic reduction of nitrate on copper electrodes prepared by high-energy ball milling. J. Electroanal. Chem. 596, 13–24 (2006).
Chen, G. -F. et al. Electrochemical reduction of nitrate to ammonia via direct eight−electron transfer using a copper-molecular solid catalyst. Nat. Energy 5, 605–613 (2020).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036–7046 (2020).
Zhao, Y. et al. Speciation of Cu surfaces during the electrochemical CO reduction reaction. J. Am. Chem. Soc. 142, 9735–9743 (2020).
Deng, Y., Handoko, A. D., Du, Y., Xi, S. & Yeo, B. S. In situ Raman spectroscopy of copper and copper oxide surfaces during electrochemical oxygen evolution reaction: identification of CuIII oxides as catalytically active species. ACS Catal. 6, 2473–2481 (2016).
Wang, L. et al. Migration of cobalt species within mixed platinum−cobalt oxide bifunctional electrocatalysts in alkaline electrolytes. J. Electrochem. Soc. 166, F3093–F3097 (2019).
Zheng, W., Liu, M. & Lee, L. Y. S. Electrochemical instability of metal-organic frameworks: In situ spectroelectrochemical investigation of the real active Sites. ACS Catal. 10, 81–92 (2020).
Mefford, J. T. et al. Correlative operando microscopy of oxygen evolution electrocatalysts. Nature 593, 67–73 (2021).
Wang, J., Tan, H. -Y., Zhu, Y., Chu, H. & Chen, H. M. Linking the dynamic chemical state of catalysts with the product profile of electrocatalytic CO2 reduction. Angew. Chem. Int. Ed. 60, 17254–17267 (2021).
Morales−Guio, C. G. et al. Improved CO2 reduction activity towards C2+ alcohols on a tandem gold on copper electrocatalyst. Nat. Catal. 1, 764–771 (2018).
Yan, H. et al. Tandem In2O3−Pt/Al2O3 catalyst for coupling of propane dehydrogenation to selective H2 combustion. Science 371, 1257–1260 (2021).
Yamada, Y. et al. Nanocrystal bilayer for tandem catalysis. Nat. Chem. 3, 372–376 (2011).
O’Mara, P. B. et al. Cascade reactions in nanozymes: spatially separated active sites inside Ag-core-porous-Cu-shell nanoparticles for multistep carbon dioxide reduction to higher organic molecules. J. Am. Chem. Soc. 141, 14093–14097 (2019).
Fajardo, A. S., Westerhoff, P., Sanchez-Sanchez, C. M. & Garcia−Segura, S. Earth−abundant elements a sustainable solution for electrocatalytic reduction of nitrate. Appl. Catal. B: Environ. 281, 119465 (2021).
Guo, Y., Stroka, J. R., Kandemir, B., Dickerson, C. E. & Bren, K. L. Cobalt metallopeptide electrocatalyst for the selective reduction of nitrite to ammonium. J. Am. Chem. Soc. 140, 16888–16892 (2018).
He, W., Ifraemov, R., Raslin, A. & Hod, I. Room-temperature electrochemical conversion of metal-organic frameworks into porous amorphous metal sulfides with tailored composition and hydrogen evolution activity. Adv. Funct. Mater. 28, 1707244 (2018).
He, W., Liberman, I., Rozenberg, I., Ifraemov, R. & Hod, I. electrochemically driven cation exchange enables the rational design of active CO2 reduction electrocatalysts. Angew. Chem. Int. Ed. 59, 8262–8269 (2020).
Liberman, I., He, W., Shimoni, R., Ifraemov, R. & Hod, I. Spatially confined electrochemical conversion of metal-organic frameworks into metal-sulfides and their in situ electrocatalytic investigation via scanning electrochemical microscopy. Chem. Sci. 11, 180–185 (2020).
Choi, J. et al. Identification and elimination of false positives in electrochemical nitrogen reduction studies. Nat. Commun. 11, 5546 (2020).
Zhao, Y. et al. Ammonia detection methods in photocatalytic and electrocatalytic experiments: How to improve the reliability of NH3 production rates? Adv. Sci. 6, 1802109 (2019).
Lefebvre, M. C. In Modern Aspects of Electrochemistry (eds Conway, B. E. et al.) 249−300 (Springer USA, 2002).
Fang, Y. -H. & Liu, Z. -P. Tafel kinetics of electrocatalytic reactions: from experiment to first-principles. ACS Catal. 4, 4364–4376 (2014).
Petrii, O. A., Nazmutdinov, R. R., Bronshtein, M. D. & Tsirlina, G. A. Life of the tafel equation: Current understanding and prospects for the second century. Electrochim. Acta 52, 3493–3504 (2007).
Kim, H. et al. Operando stability of platinum electrocatalysts in ammonia oxidation reactions. ACS Catal. 10, 11674–11684 (2020).
Duca, M. et al. New insights into the mechanism of nitrite reduction on a platinum electrode. J. Electroanal. Chem. 649, 59–68 (2010).
Yang, J., Sebastian, P., Duca, M., Hoogenboom, T. & Koper, M. T. M. pH dependence of the electroreduction of nitrate on Rh and Pt polycrystalline electrodes. Chem. Commun. 50, 2148–2151 (2014).
Krylova, V. & Andrulevičius, M. Optical, XPS and XRD studies of semiconducting copper sulfide layers on a polyamide film. Int. J. Photoenergy 2009, 304308 (2009).
Rivas-Murias, B. & Salgueiriño, V. Thermodynamic CoO-Co3O4 crossover using Raman spectroscopy in magnetic octahedron−shaped nanocrystals. J. Raman Spectrosc. 48, 837–841 (2017).
Yang, J., Liu, H., Martens, W. N. & Frost, R. L. Synthesis and characterization of cobalt hydroxide, cobalt oxyhydroxide, and cobalt oxide nanodiscs. J. Phys. Chem. C. 114, 111–119 (2010).
Liu, Y. -C., Koza, J. A. & Switzer, J. A. Conversion of electrodeposited Co(OH)2 to CoOOH and Co3O4, and comparison of their catalytic activity for the oxygen evolution reaction. Electrochim. Acta 140, 359–365 (2014).
Dupin, J. -C., Gonbeau, D., Vinatier, P. & Levasseur, A. Systematic XPS studies of metal oxides, hydroxides and peroxides. Phys. Chem. Chem. Phys. 2, 1319–1324 (2000).
Xu, M., Larentzos, J. P., Roshdy, M., Criscenti, L. J. & Allen, H. C. Aqueous divalent metal–nitrate interactions: hydration versus ion pairing. Phys. Chem. Chem. Phys. 10, 4793–4801 (2008).
Hanifpour, F., Sveinbjörnsson, A., Canales, C. P., Skúlason, E. & Flosadóttir, H. D. Preparation of Nafion membranes for reproducible ammonia quantification in nitrogen reduction reaction experiments. Angew. Chem. Int. Ed. 59, 22938–22942 (2020).
Bastian, R., Weberling, R. & Palilla, F. Ultraviolet spectrophotometric determination of nitrate: application to analysis of alkaline carbonates. Anal. Chem. 29, 1795–1797 (1957).
Hodgetts, R. Y. et al. Refining universal procedures for ammonium quantification via rapid 1H NMR analysis for dinitrogen reduction studies. ACS Energy Lett. 5, 736–741 (2020).
Zhou, K. et al. Characterization and properties of Zn/Co zeolitic imidazolate frameworks vs. ZIF-8 and ZIF-67. J. Mater. Chem. A 5, 952–957 (2017).
Dieckhöfer, S. et al. Probing the local reaction environment during high turnover carbon dioxide reduction with Ag-based gas diffusion electrodes. Chem. Eur. J. 27, 5906–5912 (2021).
Monteiro, M. C. O. et al. Probing the local activity of CO2 reduction on gold gas diffusion electrodes: effect of the catalyst loading and CO2 pressure. Chem. Sci. 12, 15682–15690 (2021).
Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement CasCat [833408]) and in the framework of the Marie Skłodowska−Curie MSCA-ITN “Implantsens” [813006] as well as from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy-EXC 2033-390677874-RESOLV and the “Center for Solvation Science ZEMOS” funded by the German Federal Ministry of Education and Research BMBF and by the Ministry of Culture and Research of Nord Rhine−Westphalia is acknowledged.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
W.S. supervised this project. W.S. and W.H. conceived the project. W.H conducted the experiments. J.Z. performed the TEM measurements. S.D. performed the XPS and SECM measurements. S.V. performed the SEM and EDS measurements. W.H. and A.C.B. performed the Raman measurements. J.R.C.J. and A.L. performed the 1H NMR measurements. S.S. carried out the XRD measurements. All authors contributed to the data analysis. W.S. and W.H. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hoang-Long Du, Seung Woo Lee, Sergi Garcia-Segura, Zhong-Qun Tian and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, W., Zhang, J., Dieckhöfer, S. et al. Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia. Nat Commun 13, 1129 (2022). https://doi.org/10.1038/s41467-022-28728-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28728-4
This article is cited by
-
Defect-induced triple synergistic modulation in copper for superior electrochemical ammonia production across broad nitrate concentrations
Nature Communications (2024)
-
Efficient nitrate electroreduction over Mn-doped Cu catalyst via regulating N-containing intermediates adsorption configuration
Science China Chemistry (2024)
-
Recent Development of Catalysts for the Nitrate Reduction Reaction: Electrochemical Solution to Produce Ammonia
Topics in Catalysis (2024)
-
Mechanochemical route to fabricate an efficient nitrate reduction electrocatalyst
Nano Research (2024)
-
Boosting ammonia production in electrocatalytic NOx reduction on a robust Fe/FeMoO4 catalyst
Nano Research (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
