
Abstract
Polysulfide dissolution and slow electrochemical kinetics of conversion reactions lead to low utilization of sulfur cathodes that inhibits further development of room-temperature sodium-sulfur batteries. Here we report a multifunctional sulfur host, NiS2 nanocrystals implanted in nitrogen-doped porous carbon nanotubes, which is rationally designed to achieve high polysulfide immobilization and conversion. Attributable to the synergetic effect of physical confinement and chemical bonding, the high electronic conductivity of the matrix, closed porous structure, and polarized additives of the multifunctional sulfur host effectively immobilize polysulfides. Significantly, the electrocatalytic behaviors of the Lewis base matrix and the NiS2 component are clearly evidenced by operando synchrotron X-ray diffraction and density functional theory with strong adsorption of polysulfides and high conversion of soluble polysulfides into insoluble Na2S2/Na2S. Thus, the as-obtained sulfur cathodes exhibit excellent performance in room-temperature Na/S batteries.
Similar content being viewed by others
Introduction
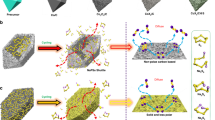
Low-cost sulfur-based sodium-ion storage has attracted tremendous interest for next-generation electric energy storage systems to meet increasing demands1,2,3. In the 1960s, high-temperature Na–S batteries were commercialized in smart grid stationary storage. Their operating temperature, however, around 300–350 °C, could potentially introduce severe safety issues and lead to Na2S3 as the final discharge product with low theoretical energy density of 760 W h kg−1 4,5. Consequently, room temperature sodium-sulfur (RT-Na/S) batteries are inspiring great interest, which could well address the safety hazard. They exhibit an increased energy density, up to 1274 W h kg−1, with Na2S as the final discharge product. This battery system suffers from rapid capacity fading and low reversible capacity, however, which can be mainly attributed to the sluggish reaction kinetics of sulfur and its Na2S product, along with serious polysulfide migration6,7,8,9,10. Significantly, various sulfur hosts have been developed for Li/S batteries to cope with the similar challenges, including a series of carbon matrices11,12,13,14,15,16,17,18, and polar sulfur hosts19,20,21,22,23,24,25,26. Nevertheless, sulfiphilic sulfur hosts have much lower conductivity than carbon materials, which inevitably compromise the rate capability and specific capacity of sulfur. To date, only a few sulfur hosts have been explored to enable RT-Na/S batteries4,5,27,28,29,30,31,32,33,34,35,36,37. By virtue of physical confinement, interconnected hollow mesoporous carbon can effectively encapsulate sulfur species inside of carbon shells during charge/discharge process27, although the low reversible capacity and insufficient lifespan of the cathode indicate that physical confinement alone is not sufficient to address the soluble polysulfide problem. Thus, constructing a multifunctional sulfur host by coupling a polar component with a functional carbon matrix is a promising way to achieve advancement on RT-Na/S batteries.
Herein, we present a multifunctional sulfur host with NiS2 nanocrystals implanted in nitrogen-doped porous carbon nanotubes (NiS2@NPCTs). First, the one-dimensional conductive NPCTs with a continuous carbon backbone inside can provide short ion diffusion paths and a fast transfer rate. Second, abundant cavities in each porous nanotube can serve as closed containers for suflur species, guarantying sufficient space for sulfur volumetric expansion and efficient polysulfide containment. Moreover, the implanted NiS2 nanocrystals have a polar feature that can bind strongly to sulfur species and spatially localize the deposition of the sulfide species. Significantly, N-doping sites and the NiS2 polar surface are capable of enhancing the adsorption energy of polysulfides, leading to strong catalytic activity towards polysulfide oxidation.
Results
Material characterization
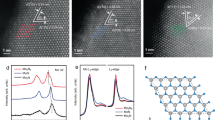
The NiS2@NPCTs/S nanocomposite with uniform one-dimensional (1D) morphology and nanocrystals encapsulated in a unique structure is prepared by a simplified synthesis strategy (Supplementary Figs. 1–3). As shown in Fig. 1a, the porous structures are well identified by scanning transmission electron microscopy (STEM); the corresponding energy dispersive spectroscopy (STEM-EDS) mapping images show the homogeneous distribution of N and S elements along C backbones. It is noticeable that the NiS2 nanocrystals (average size of about 8.3 nm) are well embedded into the carbon matrix and even the interior void space, which account for 10 wt.% in the composite (Supplementary Fig. 4a, b). To realize the mechanism of NiS2 grown within the carbon tubes, a capillary effect via vacuum treatment is introduced to drive the raw materials (nickel salt and thioacetamide) into the interior pores. For comparison, a control sample was prepared by conducting the same experiment but without vacuum treatment. As displayed in Supplementary Fig. 1f, most of the NiS2 compounds can be visually observed by SEM without vacuum treatment, indicating the NiS2 compounds were adsorbed on the exterior of NPCTs. However, no trace of NiS2 compounds is observed on the surface of the NiS2@NPCTs/S nanocomposite prepared by vacuum stirring, indicating the NiS2 nanocrystals grow within the carbon tubes. In addition, the following step of liquid nitrogen coupled with freeze-drying can further lock NiS2 within the carbon tubes, and the particle size can be effectively controlled by those pores and cavities at the same time. The EDS line scanning (Fig. 1b) of individual cavities clearly demonstrates that S is favorably dispersed on the surface of the NiS2 nanocrystals, indicating their sulfiphilic property. Fig. 1c contains a high-resolution transmission electron microscopy (HRTEM) image taken on NiS2@NPCTs/S composite shows that the interplanar distance between adjacent lattice planes is 0.279 nm, corresponding to (200) plane of NiS2. The inset 16 formula unit crystal structure model of pyrite NiS2 along [001] projected direction, which is highly consistent with the matched inverse fast Fourier transform (IFFT) pattern, indicating a high degree of crystallinity of the NiS2. In agreement with the X-ray diffraction (XRD) pattern (Fig. 1d), several intensive peaks are well indexed to pyrite NiS2 (JCPDS No. 89–1495). The low-intensity S peaks of well encapsulated sulfur can be attributed to the reduced size of the sulfur after sulfur loading process, indicate the successful encapsulation of sulfur. The loading mass of S in the NiS2@NPCTs/S composite was determined to be 56% (consistent with the Brunauer-Emmett-Teller (BET) analysis in Supplementary Fig. 4c) (Fig. 1e), which is 47% in NPCTs/S, further implying the high adsorption energy of S on NiS2. The slight weight loss of NiS2@NPCTs/S composite at high temperature is attributed to the decomposition of NiS236,37. The X-ray photoelectron spectroscopy (XPS) survey spectrum of the NiS2@NPCTs/S (Supplementary Fig. 5a) shows five characteristic peaks corresponding to S 2p, C 1s, N 1s, O 1s, and Ni 2p, respectively. The binding energy peaks observed in the Ni 2p spectrum (Fig. 1f) at 859 and 874 eV can be ascribed to the 2p3/2 and 2p1/2 of pyrite NiS238,39. Two peaks in the S 2p spectrum (Fig. 1g) at 162.9 and 164.0 eV are assigned to the 2p3/2 and 2p1/2 orbitals of S in NiS2, while the peaks at 163.4 and 164.7 eV are ascribed to the spin-orbit coupling of S 2p3/2 and S 2p1/2 in elemental S. The minor peak at 168.7 eV corresponds to C-SOx groups40. This result suggests the successful encapsulation of active S into the NiS2@NPCTs host. The N 1 s spectrum (Fig. 1h) shows the domination of pyridinic and pyrrolic nitrogen at 397.6–399.8 eV41. The N-doped carbon could serve as a conductive Lewis base matrix, which is expected to increase the adsorption energy of the polysulfides and promote the conversion kinetics42. In the C 1 s spectra in Fig. 1i, the three peaks at 288.5, 286.4, and 284.4 eV can be attributed to O–C=O, C–O, and C–C bonds, respectively, for both the NiS2@NPCTs/S and the NPCTs. The C–N bond energy in the NiS2@NPCTs/S (285.2 eV) is slightly lower than that in the NPCTs (285.7 eV), which is likely due to the interaction between C and the loaded S43,44. This observation is consistent with the Fourier transform infrared (FTIR) analysis (Supplementary Fig. 5b, c). Surprisingly, the sulfur-impregnated materials exhibit a higher D band to G band intensity ratio (ID/IG) than the NPCTs (Supplementary Fig. 5d), indicating high inclusion of defect sites on the surface45,46,47. These may provide more active sites for trapping polysulfides.

Characterizations of as-prepared sample. a STEM-EDS mapping images, b Colored STEM image coupled with EDS line scanning (inset) of a single cavity, and c HRTEM image with corresponding Fast Fourier Transform (FFT) pattern and molecular model matched IFFT image of the NiS2@NPCTs/S composite (insets). d XRD patterns of the NPCTs, NiS2@NPCTs/S, and sulfur. e Thermogravimetry (TG) and derivative thermogravimetry (DTG) curves of the NPCTs/S and NiS2@NPCTs/S. High-resolution XPS spectra of f Ni 2p, g S 2p, and h N 1s for NiS2@NPCTs/S composite. i Comparison of C 1s spectrum between NPCTs and NiS2@NPCTs/S composite
Electrochemical investigations of NiS2@NPCTs/S materials
It is expected that the well-designed nanostructures and critical functional components make NiS2@NPCTs/S a superior cathode for RT-Na/S batteries. It is impressive that NiS2@NPCTs/S delivers the high initial capacity of 960 mA h g−1 at 1 A g−1, and it maintains a stable capacity of 401 mA h g−1 for 750 cycles with distinct sodiation/desodiation plateaus (Fig. 2a, b), while NPCTs/S shows a large capacity loss of 55% within 100 cycles, highlighting the key role of the NiS2 component. The NiS2@NPCTs/S electrode also exhibits unprecedented rate performance, delivering capacity of 760, 691, 557, 457, 346, and 203 mA h g−1 at current density of 0.1, 0.2, 0.5, 1, 2, and 5 A g−1, respectively (Fig. 2c). Upon reverting back to 0.1 A g−1, the NiS2@NPCTs/S shows a fully restored capacity of 674 mA h g−1, which is in good agreement with the reversible capacity of 650 mA h g−1 over 200 cycles at 0.1 A g−1. Further electrochemical performances are presented in Supplementary Fig. 6. The discharge plateau shown in Fig. 2d can be clearly distinguished even at high rate, indicating the good confinement of sodium polysulfides and the fast reaction kinetics of the NiS2@NPCTs/S electrode. Remarkably, the NiS2@NPCTs/S composite delivered reversible capacity of 327 and 208 mA h g−1 for 1800 and 3500 cycles at 2 and 5 A g−1, respectively (Supplementary Fig. 7). It is notable that a large irreversible capacity loss is observed in the initial charge/discharge process for both samples, which can be attributed to the surface polysulfide dissolution and irreversible oxidization from polysulfide to sulfur27,48. Compared with previous reports, this is the best high-rate cycling stability result for a RT-Na/S battery with conventional current collector and carbonate-based electrolyte (Supplementary Table 1). In order to exclude the capacity contribution and highlight the advantages of the S host, the electrochemical performances of the NiS2@NPCTs and a commercial carbon nanotube/S mixture (CNTs-S) was compared. The CNTs-S mixture with high crystalline of S was found to be inactive (Supplementary Fig. 8 and 9). The Nyquist spectrum of CNTs-S after 10 cycles shows much higher charge transfer resistance (Rct) than that of NiS2@NPCTs/S electrode (Supplementary Fig. 9d), which is fitted to be 1628 and 207 Ω, respectively. When the cells are disassembled, the separator of CNTs-S is brown, which is ascribed to the side product of dissolved polysulfide out of CNTs framework. In contrast, no obvious change in the electrode and separator was observed in NiS2@NPCTs/S electrodes (Supplementary Fig. 10). Moreover, the SEM and cross-profile EDS mapping images of cycled CNTs-S electrodes show that thick film is formed on the electrode surface with dramatically reduced signal of sulfur. By contrast, uniform dispersion of S and Na is observed in NiS2@NPCTs/S. Therefore, the severe polysulfides dissolution and formation of thick passivation film for CNTs-S lead to its failure in Na–S system.

Room temperature sodium-sulfur battery test. a Cycling performance of NiS2@NPCTs/S (red) and NPCTs/S (black) at a current density of 1 A g−1. b The corresponding charge/discharge profiles of NiS2@NPCTs/S at different cycles. c Rate capability at 0.1, 0.2, 0.5, 1, 2, and 5 A g−1 for NiS2@NPCTs/S (red) and NPCTs/S (blue). d The corresponding charge/discharge profiles of NiS2@NPCTs/S at different current densities
Visible adsorbability of polysulfides
The strong polysulfide adsorption of the NiS2@NPCTs is evidenced by the UV–vis spectra (Fig. 3a). The Na2S6 solutions exposed to NiS2@NPCTs powder exhibit much weaker absorbance compared to the NPCTs, suggesting the effective adsorption capability of NiS2 nanocrystals towards polysulfides. It is evident that the yellow Na2S6 solution turns almost transparent when exposed to NiS2@NPCTs after 30 min (inset of Fig. 3a), although the color of the solution remains faint yellow for pristine NPCTs. Furthermore, optically transparent Na–S cells are shown in operation in Fig. 3b, c. After 4 h of discharging, a faint yellow color is observed in the transparent electrolyte for the NPCTs/S cell, which is due to the resultant polysulfide migration. In contrast, no obvious color change is observed for the NiS2@NPCTs/S electrode. The STEM-EDS mapping images of the NiS2@NPCTs/S electrode (Supplementary Fig. 11) in a sodiated state (open-circuit voltage around 0.8 V) show that the dispersion of elemental sodium and sulfur is highly overlapped, implying that all sulfur in this material is active for Na-ion storage. After 100 cycles in a desodiated state (open-circuit voltage around 2.8 V), the mapping images (Fig. 3d) show that the sulfur species have been well immobilized in the cavities and homogeneously dispersed along the carbon walls. It indicates that this hollow framework is capable of sulfur immobilization. The nitrogen-doped carbon shell with the fast electron diffusion ability and the electrocatalytic behaviors of the Lewis base matrix can provide more active sites for trapping polysulfides, which make the S species more favorable to reside in the shell of each pores during repeated charging/discharging processes. All of these observations indicate the efficient polysulfide trapping of the multifunctional NiS2@NPCTs host.

Visible adsorbability to polysulfides. a Ultraviolet/visible (UV–vis) spectra and corresponding photographs (inset) of pure Na2S6 solution and the solution after exposure to NiS2@NPCTs and NPCTs. Visual confirmation of polysulfide entrapment of b NPCTs/S and c NiS2@NPCTs/S at specific discharge depths. d STEM-EDS mapping images of NiS2@NPCTs/S composite after 100 cycles
Sodium-storage mechanism
High resolution in situ synchrotron XRD (λ = 0.6687 Å) was carried out in RT-Na/S batteries (Fig. 4a). A peak at 10.24° for the fresh cell can be indexed to the (222) planes of S8 (JCPDS No. 77–0145). Another two peaks located at 11.55° and 13.95° are attributed to the (111) and (200) planes of NiS2. During the initial discharge process, long-chain polysulfides (Na2Sx) appear with three new peaks at 10.47°, 11.87°, and 12.68° when discharged to 2.0 V, indicating the solid-liquid transition from S8 to long-chain polysulfides. To further understand the mechanism, S8 is removed by exposing NiS2@NPCTs/S composite in a 300 °C tube furnace under Ar flow for 10 mins. The XRD result (Supplementary Fig. 12a) shows only NiS2 remained in this composite. However, the TGA (Supplementary Fig. 12b) shows that about 32% sulfur still remained in this composite (NiS2@NPCTs/S32), indicating S8 has been removed and partial sulfur exists in an amorphous state in the carbon matrix. The tested coin cell with the NiS2@NPCTs/S32 composite shows that the short plateau around 2.2 V (formation of long-chain polysulfides) is no longer exist and only the plateau at 1.4 V (conversion of short-chain polysulfides) remained which resulted a high initial and reversible capacity than that of NiS2@NPCTs/S composite (Supplementary Fig. 12c, d). These results indicate the plateau around 2.2 V is highly related to the reduction of S8, and the amorphous sulfur remained in NiS2@NPCTs/S composite can be attributed to small sulfur molecules since the electrochemical reaction start from the conversion of short-chain polysulfides49. Once the voltage reached 1.5 V, the Na2Sx signals faded, and a new peak at 12.82° appeared, which can be indexed to the (213) planes of Na2S4 (JCPDS No. 71–0516). A further new peak at 17.1° that emerged when the cell reached 1.25 V corresponds to the (300) planes of Na2S2 (JCPDS No. 81–1771). The intermediate Na2S2 can be further reduced to Na2S from 1.1 to 0.8 V. Two new peaks at 10.3° and 16.3° can be attributed to the (111) and (220) planes of Na2S (JCPDS No. 77–2149). More intuitive information can be observed in the contour plot of XRD patterns. The signal of S8 disappeared during the charge process, indicating the irreversibility of S reduction. The signal of Na2S2 is also missing in the charge process, which might be attributed to the kinetically fast reaction. This redox mechanism illustrated by in situ synchrotron XRD is consistent with the cyclic voltammograms, as clearly detailed in Supplementary Fig. 13. In general, the reversible capacity of the RT-Na/S batteries based on the NiS2@NPCTs/S cathode comes from the reversibility of polysulfide conversion. The characteristic peak intensity of NiS2 decreases in the region between 1.1 and 0.8 V, and recovers in the charge process. This can be related to the accumulation of Na2S and partial Na+ intercalation into NiS2 based on the mechanism: NiS2 + xNa+ + xe− → NaxNiS2 (details in Supplementary Fig. 8). The electrocatalytic behaviors of the N-doped sites and the NiS2 component were further verified and highlighted via density functional theory (DFT) calculations. Fig 4b shows the adsorption conformations of Na2Sx on NiS2 nanocrystal. The chemical interactions are dominated by the bonds between the Na2Sx and the metal sulfide (Supplementary Table 2), although there is only physical adsorption dominated by van der Waals interactions for pure carbon, which are much weaker than chemical bonds. Thus, both N-doped carbon nanotube and NiS2 in our study can induce greater binding strength than pure carbon. As shown in Fig. 4c, the binding energies of Na2S6 on NiS2 and N-doped carbon nanotube are 0.79 and 0.57 eV, respectively, which are much higher than on the non-doped carbon nanotube (0.09 eV), indicating their high adsorption of soluble polysulfides. More importantly, the binding energy of Na2S on NiS2 is as high as 2.4 eV, which is more than triple that on N-doped carbon. This strong binding energy of Na2S illustrates the fast reaction mechanism transforming Na2S4 into Na2S. This electrocatalytic behavior can be explained by the rapid increase in binding energy via nitrogen dopant and NiS2 nanocrystal. It also suggests that the dual effect of chemical binding by the nitrogen dopant and NiS2 nanocrystal enables both strong entrapment of soluble polysulfides and preferential deposition of insoluble Na2S2/Na2S within the cathode during cycling.

Characterization of mechanism. a In situ synchrotron XRD patterns of the RT-Na/S battery containing a NiS2@NPCTs/S electrode with the corresponding galvanostatic charge/discharge curves at the current density of 200 mA g−1, and contour plot of XRD patterns in selected ranges of degrees. b Atomic conformations and binding energies for Na2Sx species adsorption on NiS2 (100) surface. c Comparison of the binding energies of various Na2Sx molecules bound to NiS2, N-doped carbon nanotube, and carbon nanotube, respectively, with atomic conformations of Na2S4 adsorption on N-doped carbon nanotube and carbon as insets
Discussion
Overall, we have developed an integrated structure to address the poor reaction kinetics of sulfur species and severe polysulfide migration. The physical confinement by the carbon shells and chemical bonding by doped nitrogen and NiS2 nanocrystals are of great benefit for polysulfide immobilization. Besides, both in situ synchrotron XRD and DFT results confirm that the doped nitrogen atoms coupled with the NiS2 nanocrystals serve as effective electrocatalytic sites, which significantly promote fast conversion from polysulfide to Na2S. Moreover, the possible side-reaction between the dissolved polysulfide and electrolyte can be prevented by the strong polysulfide immobilization of the multifunctional sulfur host as evidenced by EDS mapping. Consequently, the novel designed cathode can deliver a high reversible capacity of 650 mA h g−1 over 200 cycles at 0.1 A g−1 and excellent cycling stability for 3500 cycles. Our finding on electrocatalytic polysulfide immobilization and conversion may open up a new avenue for designing diverse S-based cathodes for superior RT-Na/S batteries.
Methods
Synthesis of polypyrrole nanotubes
To synthesize the polypyrrole nanotubes, Firstly, we prepared the methyl orange solution with 0.147 g methyl orange in 225 mL distilled water. After that, FeCl3 (2.95 g) was added in the solution, stirring until fully dissolved. Then, the distilled pyrrole monomer (5 × 10−3 m) was slowly dropped in the solution with continuous stirring for overnight under room temperature. Finally, the formed polypyrrole nanotubes in the above solution were washed with distilled water and ethanol for several times.
Synthesis of nitrogen-doped porous carbon nanotubes
The as-prepared polypyrrole nanotubes without any pre-treatment were slightly ground in an agate mortar, then calcined at 650 °C for 5 h in Ar atmosphere to obtain the desired structure of NPCTs.
Synthesis of the cathode composite
The NPCTs (50 mg) were added into 50 ml deionized water, followed by ultrasonication for 3 h to form a suspension. Meanwhile, Ni(NO)2*6H2O (15 mg) were dissolved in 50 mL thioacetamide deionized water solution. After stirring for 30 min, the above two solutions were mixed together, and then vigorously stirred at 50 °C. Once half deionized water evaporated, the mixed solution was stirred under vacuum at room temperature for 6 h. Then this solution was dropwise added into liquid nitrogen and freeze dried until all ice was removed. Then, the precursor was transferred to a quartz tube under Ar atmosphere and calcined at 450 °C for 5 h. The obtained composite was mixed with sulfur at weight ratio of 40/70 in a sealed quartz tube. The final NiS2@NPCTs/S composite was obtained by calcine the sealed quartz tube for 155 °C for 12 h first and then 300 °C for 1 h via the facile melt-diffusion strategy. The S incorporated NPCTs (NPCTs/S) and commercial carbon nanotubes (CNT-S) were fabricated with the same conditions. Besides, the NiS2@NPCTs were obtained by immersed NiS2@NPCTs/S in CS2 and washed for several times. Then, the material was transferred to a tube furnace under Ar atmosphere and calcined at 300 °C for 10 mins until the S has been evaporated.
Synthesis and preparation of Na2S6 solution
Eight milligram of these samples were separately immersed into 2.0 mL of 0.003 M Na2S6 solution in a mixed solvent of dimethoxyethane/tetraethylene glycol (DME/TEG) for 30 min.
Physical characterization
XRD patterns were employed with Cu Kα radiation in the 2θ range of 10°−70° (GBC MMA diffractometer, λ = 1.5406 Å, step size of 0.02° s−1). The morphology was detected via a field emission scanning electron microscope (FESEM, JEOL JSM-7500FA) equipped with energy-dispersive X-ray spectroscopy (EDS). A 200 kV scanning transmission electron microscope (STEM, JEM-ARM 200F) was equipped with a double aberration-corrector to achieve selected area electron diffraction (SAED) with a probe-forming, image-forming lens systems. The angular range of collected electrons for the high-angle annular dark-field (HAADF) images was around 70–250 mrad, while ABF-STEM images were recorded using a STEM-ABF detector simultaneously. The EDS mapping results were obtained via STEM using NSS software. Synchrotron powder diffraction data were collected at the Australian Synchrotron beamline with a wavelength (λ) of 0.6687 Å, calibrated with the standard reference material (National Institute of Standards and Technology (NIST) LaB6 660b). Schematic representations of the synchrotron XRD data were obtained by VESTA software. XPS with Al Kα radiation (hν = 1486.6 eV) was employed to detect the binding energies using a SPECSPHOIBOS 100 Analyser installed in a chamber in high-vacuum. The N2 absorption/desorption isotherms and pore size distribution were conducted by Micromeritics Tristar 3020 analyzer (USA). Raman spectra were collected using a 10 mW helium/neon laser at 632.8 nm excitation, which was filtered by a neutral density filter to reduce the laser intensity, and a charge-coupled detector (CCD). The thermal decomposition behavior of the products was monitored by using a Mettler Toledo TGA/SDTA851 analyzer from 50 to 900 °C in Ar with a heating rate of 5 °C min−1.
Electrochemical measurements
The cathode electrodes for Na–S cells which were assembled in an argon-filled glove box, were conducted by mixing 70 wt% active materials (NiS2@NPCTs/S, NPCTs/S, and CNTs-S), 20 wt% carbon black, and 10 wt% carboxymethyl cellulose (CMC) binder in distilled water. The formed slurry was then pasted on Al foil via a coater (Hohsen-MC20), which was followed by drying under vacuum at 60 °C overnight. The assembled Na–S coin cells were included the punched circular working electrodes with the average mass loading of 2.5 mg cm−2 for the active material and metallic sodium (reference and counter electrode) which were separated by glass fiber separator (Whatman GF/F). The 1 M NaClO4 electrolyte used in Na–S cells were prepared by ethylene carbonate (EC)/propylene carbonate (PC) in 1:1 volume ratio, with 3 wt% fluoroethylene carbonate as additives (EC/PC + 3 wt% FEC). The electrochemical data were collected by NEWARE coin cell tester and Biologic VMP-3 electrochemical workstation with a voltage window from 0.8 to 2.8 V (vs. Na/Na+).
Computational methods
Theoretical calculations were carried out based on the density functional theory and the plane-wave pseudopotential method50. The generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) exchange correlation function51 was adopted with the plane-wave cut-off energy set at 500 eV. All geometry optimizations and energy calculations were performed using the periodic boundary conditions. The distance between adjacent molecules and slabs was at least 15 Å. And only Γ point was used for the reciprocal space. The criterion of convergence was set that the residual forces are less than 0.01 eV Å−1 and the change of the total energy was <10–6 eV. The binding energy can be expressed as E(b) = E(Na2Sx) + E(slab) − E(Na2Sx@slab), where E(Na2Sx@slab), E(Na2Sx), and E(slab) are the total energies of the adsorbed system, the Na2Sx species, and the surface slab, respectively.
Data availability
Data supporting the findings of this study are available from the authors on reasonable request. See author contributions for specific data sets.
References
Liu, J.-C., Wang, Y.-G. & Li, J. Toward rational design of oxide-supported single-atom catalysts: atomic dispersion of gold on Ceria. J. Am. Chem. Soc. 139, 6190–6199 (2017).
Yan, Z. et al. A hydrostable cathode material based on the layered P2@P3 composite that shows redox behavior for copper in high-rate and long-cycling sodium-ion batteries. Angew. Chem. Int. Ed. 131, 1426–1430 (2019).
Fang, R. et al. More reliable lithium-sulfur batteries: status, solutions and prospects. Adv. Mater. 29, 1606823 (2017).
Carter, R. et al. A sugar-derived room-temperature sodium sulfur battery with long term cycling stability. Nano Lett. 17, 1863–1869 (2017).
Peng, H.-J. & Zhang, Q. Designing host materials for sulfur cathodes: from physical confinement to surface chemistry. Angew. Chem. Int. Ed. 54, 11018–11020 (2015).
Palomares, V. et al. Na-ion batteries, recent advances and present challenges to become low cost energy storage systems. Energy Environ. Sci. 5, 5884–5901 (2012).
Wang, Y.-X. et al. Room-temperature sodium-sulfur batteries: a comprehensive review on research progress and cell. Chem. Adv. Energy Mater. 7, 1602829 (2017).
Hueso, K. B., Armand, M. & Rojo, T. High temperature sodium batteries: status, challenges and future trends. Energy Environ. Sci. 6, 734–749 (2013).
Yin, Y. X., Xin, S., Guo, Y.-G. & Wan, L. J. Lithium–sulfur batteries: electrochemistry, materials, and prospects. Angew. Chem. Int. Ed. 52, 13186–13200 (2013).
Medenbach, L. et al. Sulfur spillover on carbon materials and possible impacts on metal–sulfur batteries. Angew. Chem. Int. Ed. 57, 13666–13670 (2018).
Li, Z. et al. Insight into the electrode mechanism in lithium-sulfur batteries with ordered microporous carbon confined sulfur as the cathode. Adv. Energy Mater. 4, 1301473 (2014).
Song, J. et al. Strong lithium polysulfide chemisorption on electroactive sites of nitrogen-doped carbon composites for high-performance lithium–sulfur battery cathodes. Angew. Chem. Int. Ed. 54, 4325–4329 (2015).
Hu, G. et al. 3D graphene-foam–reduced-graphene-oxide hybrid nested hierarchical networks for high-performance Li-S batteries. Adv. Mater. 28, 1603–1609 (2016).
Mao, Y. et al. Foldable interpenetrated metal-organic frameworks/carbon nanotubes thin film for lithium–sulfur batteries. Nat. Commun. 8, 14628 (2017).
Zhou, D. et al. Stable quasi-solid-state sodium-sulfur battery. Angew. Chem. Int. Ed. 57, 10168–10172 (2018).
Zhang, B.-W. et al. Long-life room-temperature sodium-sulfur batteries by virtue of transition-metal-nanocluster-sulfur interactions. Angew. Chem. Int. Ed. 58, 1484–1502 (2019).
Wu, T. et al. Controllable chain-length for covalent sulfur-carbon materials enabling stable and high-capacity sodium storage. Adv. Energy Mater. 9, 1803478 (2019).
Hou, H., Banks, C., Jing, M., Zhang, Y. & Ji, X. B. Carbon quantum dots and their derivative 3D porous carbon frameworks for sodium-ion batteries with ultralong cycle life. Adv. Mater. 27, 7861–7866 (2015).
Zhao, G. et al. Nickel chelate derived NiS2 decorated with bifunctional carbon: an efficient strategy to promote sodium storage performance. Adv. Funct. Mater. 28, 1803690 (2018).
Seh, Z.-W. et al. Sulphur–TiO2 yolk–shell nanoarchitecture with internal void space for long-cycle lithium–sulphur batteries. Nat. Commun. 4, 1331 (2013).
Cheng, M.-J., Clark, E.-L., Pham, H.-H., Bell, A. T. & Head-Gordon, M. Quantum mechanical screening of single-atom bimetallic alloys for the selective reduction of CO2 to C1 hydrocarbons. ACS Catal. 6, 7769–7777 (2016).
Kong, L. et al. A bifunctional perovskite promoter for polysulfide regulation toward stable lithium–sulfur batteries. Adv. Mater. 30, 1705219 (2018).
Song, J. et al. Nitrogen-doped mesoporous carbon promoted chemical adsorption of sulfur and fabrication of high-areal-capacity sulfur cathode with exceptional cycling stability for lithium-sulfur batteries. Adv. Funct. Mater. 24, 1243 (2014).
Qiang, Z. et al. Ultra-long cycle life, low-cost room temperature sodium-sulfur batteries enabled by highly doped (N,S) nanoporous carbons. Nano Energy 32, 59–66 (2017).
Lu, Y. et al. A simple melting-diffusing-reacting strategy to fabricate S/NiS2–C for lithium–sulfur batteries. Nanoscale 8, 17616 (2016).
Zhou, G. et al. Catalytic oxidation of Li2S on the surface of metal sulfides for Li−S batteries. Proc. Natl Acad. Sci. USA 114, 840–845 (2017).
Wang, Y. X. et al. Achieving high-performance room-temperature sodium–sulfur batteries with S@interconnected mesoporous carbon hollow nanospheres. J. Am. Chem. Soc. 138, 16576–16579 (2016).
Yu, X. & Manthiram, A. Highly reversible room-temperature sulfur/long-chain sodium polysulfide batteries. J. Phys. Chem. Lett. 5, 1943–1947 (2014).
Ma, D. et al. New strategy for polysulfide protection based on atomic layer deposition of TiO2 onto ferroelectric-encapsulated cathode: toward ultrastable free-standing room temperature sodium-sulfur batteries. Adv. Funct. Mater. 28, 1705537 (2018).
Chen, Y.-M. et al. A nitrogen doped carbonized metal–organic framework for high stability room temperature sodium–sulfur batteries. J. Mater. Chem. A 4, 12471–12478 (2016).
Hwang, T. H., Jung, D. S., Kim, J. S., Kim, B. G. & Choi, J. W. One-dimensional carbon–sulfur composite fibers for Na–S rechargeable batteries operating at room temperature. Nano Lett. 13, 4532–4538 (2013).
Xia, G. et al. Carbon hollow nanobubbles on porous carbon nanofibers: an ideal host for high-performance sodium-sulfur batteries and hydrogen storage. Energy Storage Mater. 14, 314–323 (2018).
Manthiram, A. & Yu, X. Ambient temperature sodium–sulfur batteries. Small 11, 2108–2114 (2015).
Wei, S. et al. A stable room-temperature sodium–sulfur battery. Nat. Commun. 7, 11722 (2016).
Zhang, B. W. et al. Atomic cobalt as an efficient electrocatalyst in sulfur cathodes for superior room-temperature sodium-sulfur batteries. Nat. Commun. 9, 4082 (2018).
Jin, C. et al. A hierarchical carbon modified nano-NiS2 cathode with high thermal stability for a high energy thermal battery. J. Mater. Chem. A 6, 7123–7132 (2018).
Chen, Q. et al. Yolk–shell NiS2 nanoparticle‐embedded carbon fibers for flexible fiber-shaped sodium battery. Adv. Energy Mater. 8, 1800057 (2018).
Zhao, F. et al. Stabilizing nickel sulfide nanoparticles with an ultrathin carbon layer for improved cycling performance in sodium ion batteries. Nano Res. 9, 3162–3170 (2016).
Yang, H. et al. Simultaneously dual modification of Ni-Rich layered oxide cathode for high-energy lithium-ion batteries. Adv. Funct. Mater. 29, 1808825 (2019).
An, T. et al. Interlaced NiS2–MoS2 nanoflake-nanowires as efficient hydrogen evolution electrocatalysts in basic solutions. J. Mater. Chem. A 4, 13439–13443 (2016).
Xu, Y. et al. Highly nitrogen doped carbon nanofibers with superior rate capability and cyclability for potassium ion batteries. Nat. Commun. 9, 1720 (2018).
Li, X. et al. Single-atom Pt as co-catalyst for enhanced photocatalytic H2 evolution. Adv. Mater. 28, 2427 (2016).
Wang, Z. et al. Enhancing lithium–sulphur battery performance by strongly binding the discharge products on amino-functionalized reduced graphene oxide. Nat. Commun. 5, 5002 (2014).
Wang, S. et al. Sodium ion batteries: free-standing nitrogen-doped carbon nanofiber films: integrated electrodes for sodium‐ion batteries with ultralong cycle life and superior rate capability. Adv. Energy Mater. 6, 1502217 (2016).
Li, W. et al. A high performance sulfur-doped disordered carbon anode for sodium ion batteries. Energy Environ. Sci. 8, 2916–2921 (2015).
Zhou, G. et al. A flexible nanostructured sulphur–carbon nanotube cathode with high rate performance for Li-S batteries. Energy Environ. Sci. 5, 8901 (2012).
Li, X. et al. Tailoring interactions of carbon and sulfur in Li–S battery cathodes: significant effects of carbon–heteroatom bonds. J. Mater. Chem. A 2, 12866–12872 (2014).
Ma, G. et al. A lithium anode protection guided highly-stable lithium–sulfur battery. Chem. Commun. 50, 14209 (2014).
Xin, S., Yin, Y., Guo, Y.-G. & Wan, L. J. A high-energy room-temperature sodium-sulfur battery. Adv. Mater. 28, 2427 (2016).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Revi. B 59, 1758 (1999).
Perdew, J., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Acknowledgements
The authors are grateful for financial support from an Australian Renewable Energy Agency (ARENA) Project (G00849), the Innovative Group of Guangdong Province (Grant No. 2014ZT05N013), the National Natural Science Foundation of China (Grant Nos. 11704114, 61427901), and the Australian Research Council (ARC) (DE170100928). Part of the experiments was carried out at the Powder Diffraction Beamline of the Australian Synchrotron. The authors would like to thank Guoqiang Zhao for support on TEM, Peng Li for the support on the contour plot of XRD patterns, Dr. Gilberto Casillas-Garcia for support on the STEM technique, and Dr. Tania Silver for critical reading of the paper.
Author information
Authors and Affiliations
Contributions
Z.Y. performed all synthetic experiments and prepared the manuscript. J.X. performed the density functional theory (DFT) calculations. L.W. and Hui Liu performed FTIR and UV–vis measurements and analyses. Z.Y. and F.G. conducted the TGA measurements. Z.Y., W.L., and Q.G. performed synchrotron X-ray diffraction measurements. Y.W. and S.-L.C. supervised the project. Z.Y., Y.W., S.-L.C., Hui Liu, Huakun Liu, and S.-X.D. analyzed the data and wrote the manuscript. All authors discussed the results and contributed to writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications would like to thank Kevin Huang and other, anonymous reviewers for their contributions to the peer review of this work. Peer review reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source Data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, Z., Xiao, J., Lai, W. et al. Nickel sulfide nanocrystals on nitrogen-doped porous carbon nanotubes with high-efficiency electrocatalysis for room-temperature sodium-sulfur batteries. Nat Commun 10, 4793 (2019). https://doi.org/10.1038/s41467-019-11600-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-11600-3
This article is cited by
-
Understanding the charge transfer effects of single atoms for boosting the performance of Na-S batteries
Nature Communications (2024)
-
Ultrasmall NiS2 Nanocrystals Embedded in Ordered Macroporous Graphenic Carbon Matrix for Efficiently Pseudocapacitive Sodium Storage
Transactions of Tianjin University (2023)
-
Single-Atom Catalysts: Advances and Challenges in Metal-Support Interactions for Enhanced Electrocatalysis
Electrochemical Energy Reviews (2022)
-
A Fe3N/carbon composite electrocatalyst for effective polysulfides regulation in room-temperature Na-S batteries
Nature Communications (2021)
-
Recent progresses of micro-nanostructured transition metal compound-based electrocatalysts for energy conversion technologies
Science China Materials (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
