
Abstract
Cerebrovascular spasm is a life-threatening event in salt-sensitive hypertension. The relationship between store-operated calcium entry (SOCE) and vasoconstriction in hypertension has not been fully clarified. This study investigated the changes in cerebrovascular contractile responses in high salt intake-induced hypertension and the functional roles of the main components of SOCE, namely, polycystin-2 (TRPP2), stromal interaction molecule 1 (STIM1), and Orai3. Polycystic kidney disease 2 (which encodes TRPP2) knockout mice displayed decreased cerebrovascular SOCE-induced contraction. The blood pressure of age-matched rats fed a normal or high-salt diet for 4 weeks was monitored weekly using noninvasive tail-cuff plethysmography. The systolic blood pressure of the rats fed a high-salt diet was significantly higher than that of controls. Western blotting and immunohistochemical results showed that these hypertensive rats expressed higher levels of cerebrovascular TRPP2, STIM1, and Orai3 than controls. Cerebrovascular tension measurements of the basilar artery indicated that SOCE-mediated contraction was significantly increased in hypertensive rats compared with control rats. In addition, SOCE-mediated contraction was decreased in the basilar arteries of rats pretreated with the SOCE inhibitor BTP-2 (10 μM) or transfected with TRPP2-specific or STIM1-specific small interfering RNA. Staining with 2,3,5-triphenyltetrazolium chloride (TTC) was used to quantify the infarcted brain area 24 h after middle cerebral artery occlusion, a model of ischemic stroke, in rodents. The infarcted brain area was significantly greater in hypertensive rats and significantly lower in BTP-2-treated rats than in controls. Taken together, these findings indicate that SOCE-induced contraction may be overactive in the basilar arteries of salt-sensitive hypertensive rats, suggesting the dysregulation of TRPP2 and SOCE and its other components.
Similar content being viewed by others

Introduction
The intracellular calcium concentration ([Ca2+]i) initiates or modulates a variety of physiological functions, including apoptosis, proliferation, and migration, in various cell types. Cytosolic Ca2+ generally consists of Ca2+ released from the endoplasmic reticulum (ER) and Ca2+ that has entered the cell from the extracellular space via voltage- or receptor-gated Ca2+ channels [1, 2]. Thus, the maintenance of ER Ca2+ stores and the activity of plasma membrane Ca2+ channels are essential for cytosolic Ca2+ homeostasis. After exposure to a stimulus that increases the [Ca2+]i, transient Ca2+ release from the ER may induce additional Ca2+ influx to replenish the ER Ca2+ store. This process is called store-operated calcium entry (SOCE), and its main components have been identified as stromal interaction molecules (STIMs), Orai proteins, and transient receptor potential channels [3, 4]. Polycystin-2 (TRPP2), encoded by the polycystic kidney disease 2 (PKD2) gene, is a Ca2+-permeable channel expressed on the ER and plasma membranes and may mediate SOCE [5, 6]. STIM1, a single transmembrane protein predominantly found on the ER membrane, serves as a Ca2+ sensor [2]. Expressed on the plasma membrane, Orai has three isoforms, namely, Orai1, Orai2, and Orai3, and they form Ca2+ channels that mediate Ca2+ entry [7]. The interactions between STIM1/Orai proteins and TRPP2/inositol trisphosphate receptors increase the [Ca2+]i, thus contributing to the contraction of vascular smooth muscle cells (VSMCs) [6].
High salt intake is one of the most important environmental risk factors for hypertension [8], which has been reported to cause more than half of the deaths from cardiovascular and cerebrovascular events in China [9]. Cerebrovascular spasm is a life-threatening event in salt-sensitive hypertension. In the vascular system, TRPP2 is widely expressed in the endothelium and smooth muscle of the cerebral arteries [4], whereas STIM proteins are found in vascular and airway smooth muscle [10]. Accumulating evidence has suggested that altered expression and dysfunction of STIM proteins are associated with vascular pathologies, including restenosis and hypertension [11]. STIM1 is upregulated in the tunica media of the aorta in spontaneously hypertensive rats [12]. In addition, STIM/Orai-mediated Ca2+ signaling pathways activate downstream effector proteins and transcription factors that promote smooth muscle cell proliferation and migration [13]. Previous studies have also indicated that increased TRPP2 plays a pivotal role in potentiating vasoconstriction in high salt intake-induced hypertensive rats, whereas knockout of the PKD2 gene exerts the opposite effect [14].
In the present study, to further explore the effect of salt-induced hypertension on TRPP2-regulated SOCE in the VSMCs of the cerebral vasculature, we investigated SOCE-induced cerebral vasoconstriction and ischemia-reperfusion lesions in salt-induced hypertensive rats and determined the expression level changes in TRPP2, STIM1, and Orai3 as well as their involvement in SOCE-induced vascular tension.
Materials and methods
Reagents
Endothelin 1 (ET-1) was obtained from Calbiochem (La Jolla, CA, USA). BTP-2 (sc-221441), anti-Orai3 primary antibody (sc-292104), anti-TRPP2 primary antibody (sc-25749), and anti-STIM1 primary antibody (sc-68897) were obtained from Santa Cruz (Dallas, Texas, USA), and GAPDH primary antibody (D110016) was obtained from Sangon Biotech (Shanghai City, China). TRPP2- and STIM1-specific small interfering RNAs (siRNAs) for rats and lipofectamine RNAiMAX transfection reagent were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The sequences for these siRNAs were as follows: TRPP2-specific siRNA, AACCUGUUCUGUGUGGUCAGGUUAU (sense strand) and AUAACCUGACCACACAGAACAGGUU (antisense strand); STIM1-specific siRNA, UAAGGGAAGACCUCAAUUA (sense strand) and UAAUUGAGGUCUUCCCUUA (antisense strand). siRNA transfection was achieved with lipofectamine RNAiMAX transfection reagent according to the manufacturer’s instructions. Acetylcholine, 2,3,5-triphenyltetrazolium chloride, U46619, and verapamil were purchased from Sigma (St. Louis, MO, USA).
Animals
All animal experiments were conducted according to the National Institutes of Health guidelines (publication no. 8523) and were approved by the Animal Experimentation Ethics Committee of Anhui Medical University. Mice on a C57BL/6J background with the smooth muscle PKD2 gene conditionally knocked out and their wild-type littermates (6–8 months of age; ∼30 g body weight) were bred in-house, and there were approximately equal numbers of males and females in each group. In addition, 6-week-old male Sprague-Dawley rats (150–200 g) randomly assigned to the high-salt diet and regular-salt diet groups (as previously described [14]) were housed in a temperature-controlled room with a 12-h light-dark cycle. The high-salt diet consisted of 4% (w/w) NaCl, and the regular-salt diet consisted of 0.4% NaCl; the diets were provided for 4 weeks.
Noninvasive blood pressure measurement
Systolic blood pressure was determined by a noninvasive procedure using tail-cuff plethysmography as previously described [15]. Briefly, conscious rats were placed in a restrainer in a prewarmed chamber for 15–20 min before blood pressure examination. A pneumatic pulse transducer was placed on the tails of the rats, and the signals from it were collected and analyzed automatically using a data acquisition and analysis system (BL-420E+, Chengdu Technology & Market Corp, China). Each rat underwent three successive rounds of measurements, and the mean of these values was recorded.
Basilar artery tension measurement
Basilar artery tension measurements were performed as described in other relevant studies [1, 16]. Control and PKD2-Cre mice and rats treated with or without a high-salt diet were humanely killed by CO2 inhalation. The brains were removed, dissected, and placed in an oxygenated ice-cold Krebs solution that contained (in mM) 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 KH2PO4, 1.2 MgSO4 (7 H2O), 25.2 NaHCO3, and 11.1 glucose. Subsequently, the basilar artery was rapidly isolated from the surrounding connective tissue and sliced into 2-mm-long rings under a dissecting microscope. In the endothelium-denuded group, the arteries were mechanically removed by repeatedly using wires. Segments of the rat basilar artery were placed in myograph organ baths (DMT 610M, Danish Myo Technology, Aarhus, Denmark) with 5 mL of Krebs solution at 37 °C and aerated with 95% O2 and 5% CO2 to maintain a pH of 7.4. All experiments were conducted under conditions of isometric contraction.
After a 20-min equilibration period of baseline force (2 mN for rat tissue; 1 mN for mouse tissue), the contractile function of the cerebral basilar artery was determined using a 60 mM K+ solution [16]. The vessels were thoroughly cleaned and exposed to 100 nM U46619 and 10 μM acetylcholine to examine endothelial integrity [17]. Only those vessels, in which the acetylcholine diastolic effect was below 10% of the maximum contraction caused by U46619 were cleaned of endothelium and used in experiments. In the SOCE-induced contraction experiments, the basilar arteries were first pretreated with Ca2+-free gramfloxacin, 100 nM ET-1, and 1 μM verapamil for 10 min, and then 2.5 mM CaCl2 was added. In some vessels, SOCE-induced contractions were examined before treatment with 10 μM BTP-2 [18] for 10 min. Vessel segments from control rats were randomly divided into three groups, the TRPP2 group, the STIM1 group, and the control group, and incubated with TRPP2 siRNA, STIM1 siRNA, or scrambled control siRNA, respectively, for 20 h.
Western blot analysis
The proteins were extracted from rat cerebral basilar arteries with detergent extraction buffer, which contained 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM disodium salt of ethylenediaminetetraacetic acid (Na2EDTA), 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, and 2.5 mM sodium pyrophosphate plus protease inhibitor cocktail tablets. For immunoblots, the extracted proteins were separated on a 10% SDS-PAGE gel and then transferred to polyvinylidene difluoride membranes by using a semidry transfer system (Bio-Rad). After blocking with PBST buffer containing 0.1% Tween 20 and 5% nonfat dry milk for 2 h, the membranes were incubated with an anti-TRPP2 (1:250), anti-STIM1 (1:250), anti-Orai3 (1:250), or anti-GAPDH (1:1000) primary antibody at 4 °C overnight. Immunodetection was performed with horseradish peroxidase-conjugated secondary antibodies, followed by the application of an enhanced chemiluminescence detection system. The optical densities of the protein bands were analyzed by Image J software. All protein bands on the membrane were normalized to GAPDH in the same lane and are expressed as the relative optical density.
Immunohistochemical staining analysis
The basilar arteries of the rats were quickly removed and rinsed with physiological saline. The vessels were fixed in 4% paraformaldehyde for 24 h and then cut into 5-μm-thick tissue sections after being dehydrated and embedded. Graded alcohol solutions and tap water were used for the dewaxing and washing of the vessel sections. Endogenous peroxidase enzyme activity was blocked with 3% (v/v) H2O2, and antigen retrieval was performed with 0.01 M citrate buffer heated in a microwave. After being washed with phosphate-buffered saline (PBS), the sections were incubated with a rabbit anti-TRPP2 antibody, a rabbit anti-STIM1 antibody, or an anti-Orai3 antibody (all diluted 1:100) at 4 °C overnight. The sections were washed again with PBS, and a Power Vision two-step histochemical staining reagent (PV-9000, ZSGB-BIO, Beijing, China) was applied for detection. All sections were stained with diaminobenzidine and counterstained with hematoxylin. In the negative control group, the primary antibody was omitted. All images were obtained using a light microscope, and the integrated optical density of the images was calculated by Image Pro Plus 5.1 (Media Cybernetics, USA) software.
Middle cerebral artery occlusion (MCAO) and TTC staining
High salt intake-induced hypertensive rats and normotensive control rats were subjected to MCAO by using previously reported methods with slight modifications [19,20,21]. The rectal temperature of the rats was maintained at 37 ± 0.5 °C with a homeothermic blanket. After being deeply anaesthetized with an intraperitoneal injection of pentobarbital sodium (45 mg/kg), the rats were placed on an operating table in the supine position. Following disinfection of the skin near the relevant area, a midline incision was made in the neck to expose the right common carotid artery, external carotid artery, and internal carotid artery. The common carotid artery was occluded with a bulldog clamp, and the external carotid artery branch was ligated distally. A 4–0 heparinized nylon suture with a round tip was inserted from the external carotid artery to the lumen of the internal carotid artery until it blocked the origin of the middle cerebral artery. Subsequently, the incision was sutured layer by layer. Reperfusion was established by gently pulling out the nylon suture 2 h later. In some rats, SOCE-induced contraction was examined before treatment with 10 μM BTP-2 for 30 min.
Twenty-four hours after cerebral ischemia-reperfusion injury, the neurological functions of the rats that underwent MCAO were assessed. Neurological deficit scores were assigned based on previously published criteria using the Bederson scale, with scores ranging from 0 to 3 [22]. Bederson scores were as follows: 0, no apparent deficits; 1, unable to extend the contralateral forelimb when suspended by the tail; 2, torso turning to the contralateral side with moderate forelimb weakness when held by the tail; and 3, spontaneous contralateral circling. Rats without the expected postoperative effects at the time of observation were removed from the study and replaced with other rats that underwent MCAO whenever necessary to ensure that each group had three rats.
Twenty-four hours after the MCAO operation, the rats were anesthetized and decapitated, and the brains were removed. Each brain was frozen at −20 °C for 20 min and then cut into five serial 2-mm-thick coronal slices, which were stained with 2% TTC at 37 °C and then fixed in a 4% paraformaldehyde fixation solution [23]. As a result, normal tissues were stained red, while the infarcted tissue appeared white. The images were photographed and recorded. The infarcted areas and the area of each hemisphere in each of three slices were quantified using an image analysis system (Image-Pro Plus version 6.0, Rockville, MD, USA).
Statistical analysis
The collected data are presented as the means ± SE. The Mann–Whitney U test (two-tailed) or two-way analysis of variance followed by the Games-Howell post hoc test when more than two treatments were compared were used to compare results across the different groups (Statistical Product and Service Solutions, version 16.0, SPSS, Inc., Chicago, IL, USA). Differences between groups were considered statistically significant at a value of P < 0.05.
Results
Changes in vessel contractile response in PKD2-Cre mice
Studies have reported that the decreased expression of TRPP2 reduces intracellular and ER Ca2+ levels [24, 25]. In addition, Narayanan et al. found a significant decrease in pressure-induced cerebral artery constriction in mice with PKD2 knockdown [26]. We investigated basilar artery tension to determine whether there were changes in SOCE-induced cerebral vasoconstriction in PKD2-Cre mice. We found that contractions induced by ET-1, a potent vasoconstrictor, were decreased in PKD2-Cre mice (Fig. 1a, b), but 60 mM high K+-induced contractions were unchanged between control and PKD2-Cre mice (Fig. 1c, d). This effect may be associated with the TRPP2 deficit in the cerebrovascular smooth muscle in PKD2-Cre mice.

Vasocontraction induced by store-operated calcium entry (SOCE) or 60 mM high K+ in denuded cerebral basilar arteries derived from control or PKD2-Cre mice. Representative traces (a) and summary data (b) of SOCE-induced cerebral basilar artery ring contractions. Cerebral basilar artery rings were pretreated with 100 nM endothelin-1 and 1 μM verapamil in a Ca2+-free solution for 10 min. Representative traces (c) and summary data (d) of cerebral basilar artery contractile changes in response to 60 mM high K+. The values are the means ± SE; n = 5 samples. *P < 0.05 vs. control (Mann–Whitney U test (two-tailed))
Changes in systolic blood pressure
The results of our previous study on TRPP2 and vasoconstriction suggested that increased TRPP2 exerts pivotal effects on enhancing vessel contraction in high salt intake-induced hypertensive rats [14]. The present study further investigated the functional roles of SOCE and its downstream molecules in cerebrovascular contraction and salt-sensitive hypertensive rats with cerebral ischemia-reperfusion injury. Systolic blood pressure in the control and high-salt diet rats was monitored weekly using a noninvasive tail-cuff method. Our results indicated that, from the second through the 4th week, the systolic blood pressure of the rats fed the high-salt diet was significantly higher than that of the age-matched normal rats fed the control diet (Fig. 2). The systolic blood pressure of all hypertensive rats was confirmed before all subsequent experiments.

Changes in systolic blood pressure in age-matched control and high-salt diet-fed rats. The values are the means ± SE; n = 12 samples. *P < 0.05 vs. control (regular diet) (two-way ANOVA)
Roles of SOCE in enhanced cerebrovascular contraction in hypertensive rats
Many studies have demonstrated that TRPP2, STIM1, and Orai3 proteins are involved in SOCE-induced vasocontraction [24, 27]. To examine the mechanism underlying SOCE-induced vasoconstriction in hypertensive rats, we determined the expression levels of TRPP2, STIM1, and Orai3 in the basilar artery VSMCs of rats fed a high-salt diet and control rats. The results of our western blotting data showed that TRPP2, STIM1, and Orai3 expression levels were significantly higher in basilar arteries derived from the hypertensive rats than those derived from the control rats (Fig. 3a–c).

Expression levels of TRPP2, STIM1, and Orai3 in denuded cerebral basilar arteries. a, b, and c western blot images of TRPP2, STIM1, and Orai3 expression levels in cerebral basilar arteries derived from high salt intake-induced hypertensive rats and age-matched control rats (three rats in each group). d TRPP2, STIM1, and Orai3 immunostaining results in segments of cerebral basilar arteries. d (a–d) are segments derived from rats fed a normal diet (control). d (e–h) are segments from high salt intake-induced hypertensive rats. Background indicates immunostaining controls with no primary antibody present. Magnification: 400×. e–g Summary of immunohistochemical staining data for TRPP2, STIM1, and Orai3 in segments of cerebral basilar arteries. The values are the means ± SE. n = 3–4 rats. *P < 0.05 vs. control (Mann–Whitney U test (two-tailed))
To support this finding, we also used immunohistochemical staining analyses. As shown in Fig. 3d–g, the expression levels of TRPP2, STIM1, and Orai3 were significantly higher in basilar artery VSMCs from hypertensive rats than in those from control rats. Together, these findings suggest that the upregulation of TRPP2, STIM1, and Orai3 may occur through a single mechanism by which the SOCE-induced contraction of VSMCs is increased.
We measured basilar artery tension to explore the differences in SOCE-induced and ET-1-induced cerebral vasoconstriction between high salt intake-induced hypertensive rats and rats fed a normal diet. The SOCE-induced and ET-1-induced contractile responses of the basilar artery were significantly increased in hypertensive rats compared with control rats (Fig. 4a–d), whereas 60 mM high K+-induced contractions were unchanged in hypertensive rats (Fig. 4e, f).

Vasocontraction induced by store-operated calcium entry (SOCE), endothelin-1 (ET-1), or 60 mM high K+ in denuded cerebral basilar arteries derived from rats fed a normal diet (control) or a high-salt diet. Representative traces (a) and summary data (b) of SOCE-induced cerebral basilar artery ring contraction. Cerebral basilar artery rings were pretreated with 100 nM endothelin-1 and 1 μM verapamil in a Ca2+-free solution for 10 min. Representative traces (c) and summary data (d) of ET-1-induced cerebral basilar artery ring contraction. Representative traces (e) and summary data (f) of contraction changes in cerebral basilar arteries in response to 60 mM high K+. The values are the means ± SE; n = 6–3 samples. *P < 0.05 vs. control (Mann–Whitney U test (two-tailed))
To investigate the functional roles of TRPP2 and STIM1 in SOCE-induced vasoconstriction, we used an siRNA specific for TRPP2 or STIM1 to knock down the expression of TRPP2 and STIM1 proteins in rats. Our western blotting results showed that, compared with scrambled control siRNA, TRPP2-specific siRNA and STIM1-specific siRNA effectively suppressed the expression of TRPP2 and STIM1, respectively, in the rat basilar artery (Fig. 5a, b). After treatment with an siRNA specific for TRPP2 or STIM1 or with scrambled siRNA for 20 h, we tested the SOCE-induced vasocontraction of the basilar artery. Treatment with either TRPP2 siRNA or STIM1 siRNA, compared with treatment with scrambled siRNA, significantly reduced SOCE-induced contractions in basilar artery rings (Fig. 5c, d). Cerebral vessels derived from rats fed a normal diet and from rats fed a high-salt diet and treated with BTP-2, an SOCE inhibitor, showed no difference in SOCE-induced contraction (Fig. 5e), providing further evidence that TRPP2, STIM1, and Orai3 may play important roles in the SOCE-induced contraction of VSMCs.

Functional roles of TRPP2, STIM1, and Orai3 in the store-operated calcium entry (SOCE)-induced contraction of rat cerebral basilar arteries. Representative images of TRPP2 and STIM1 expression in cerebral basilar arteries pretreated with scrambled siRNA (control), TRPP2-specific siRNA (a), or STIM1-specific siRNA (b). Summary data showing the effects of pretreatment with TRPP2-specific siRNA (c), STIM1-specific siRNA (d), or 10 μM BTP-2 (e) on SOCE-induced cerebral basilar artery ring contraction. Cerebral basilar artery rings were pretreated with 100 nM endothelin-1 and 1 μM verapamil in a Ca2+-free solution for 10 min. The values are the means ± SE; n = 4–7 samples. *P < 0.05 vs. scrambled siRNA in c and d, and vs. control in e; #P < 0.05 vs. hypertension in e (Mann–Whitney U test (two-tailed))
Functional roles of enhanced cerebral vascular SOCE in cerebral ischemia-reperfusion
We used the MCAO rodent model of ischemic stroke to identify the role of SOCE in cerebral ischemia-reperfusion injury. Our TTC staining results showed that, compared with rats fed a normal diet, hypertensive rats had an increased area of infarction, whereas normotensive rats treated with the SOCE inhibitor BTP-2 had a reduced area of infarction (Fig. 6). In addition, sham-operated rats did not exhibit an infarcted area. These results indicate that cerebral ischemic injury was most severe in high salt intake-induced hypertensive rats and suggest that this injury might be related to the abnormal expression of TRPP2, STIM1, and Orai3 in the hypertensive cerebrovascular system.

Percentage of cerebral infarct area relative to the area of the whole brain 24 h after ischemia-reperfusion caused by middle cerebral artery occlusion in normotensive and hypertensive rats. a Coronal brain slices stained with 2% 2,3,5-triphenyltetrazolium chloride. The white region is infarcted tissue, whereas the red region is noninfarcted tissue. b Quantitative analysis of cerebral ischemia. The middle cerebral arteries of rats in the sham group were not blocked by nylon sutures, and no infarcted area was found in the sham group. Control rats were fed a normal diet and were subjected to ischemia-reperfusion injury. The values are the means ± SE. n = 6–8 rats. **P < 0.01 vs. control (Mann–Whitney U test (two-tailed))
Discussion
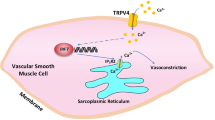
The present study examined the effects of TRPP2 and other SOCE constituents on vasoconstriction and how that is altered in high salt intake-induced hypertensive rats. Our results showed that SOCE-induced vasoconstriction in the cerebral arteries of PKD2-Cre mice (which are deficient in TRPP2) was decreased. We also found that, compared with rats fed a normal diet, age-matched rats fed a high-salt diet had increased systolic blood pressure, substantially increased expression levels of TRPP2, STIM1, and Orai3 in VSMCs, and enhanced SOCE-induced cerebral artery vasoconstriction (Scheme 1). The transfection of a TRPP2- or STIM1-specific siRNA decreased the expression levels of the corresponding proteins and attenuated ET-1-induced SOCE-mediated cerebral vasoconstriction. In addition, after treatment with the SOCE inhibitor BTP-2, SOCE-induced cerebrovascular contraction in high salt intake-induced hypertensive rats was significantly weaker than that in rats fed a normal diet, whereas the high K+-induced cerebrovascular contractions in these two groups were not different. Moreover, following MCAO, the infarcted area in the brains of hypertensive rats was significantly greater than that in control rats, whereas the infarcted area was significantly lower in normotensive rats treated with BTP-2 than that in control rats.

TRPP2-mediated Ca2+ signal pathway in vascular smooth muscle cells
In VSMCs, the depletion of Ca2+ stores can activate STIM1/Orai after initial Ca2+ release. Subsequently, activated Orai mediates SOCE, allowing the sustained influx of Ca2+ and the contraction of VSMCs [28,29,30]. Owing to the importance of Ca2+ in VSMCs, disturbances in Ca2+ homeostasis may induce abnormal VSMC contraction and thus be a mechanism that contributes to the etiology of hypertension. Studies on PKD2-Cre mice have indicated that their intracellular and ER Ca2+ levels are reduced [26]. Our present finding that cerebrovascular contraction was decreased in PKD2-Cre mice is consistent with that of Narayanan et al., who found a significant decrease in pressure-induced cerebral artery constriction in mice with PKD2 gene knockdown [26]. These findings indicate that TRPP2 plays a critical role in vasoconstriction in vivo.
TRPP2 acts as a Ca2+ channel that can mediate the secondary release of Ca2+ and partly contribute to SOCE [31,32,33]. Giachini et al. found that enhanced STIM1/Orai1 activity leads to an impairment in [Ca2+]i control along with vascular function in hypertensive animals [12]. The dysfunction or aberrant expression of TRPV4 (a Ca2+ channel) in the endothelium of resistant arteries in high-salt diet-fed animals causes salt-sensitive hypertension [34, 35]. Studies have also shown that TRPP2 is a major TRPP subtype in the cerebral vasculature that is mainly distributed in the plasma membrane of cerebral artery VSMCs [26]. Our present western blotting and immunohistochemical results indicated that TRPP2 expression was markedly increased in the VSMCs of cerebral vessels in hypertensive rats compared with normotensive control rats. This finding is similar to the finding of Zhao et al. that the expression of TRPP2 is increased in VSMCs of high salt intake-induced hypertensive rats [14].
To further investigate the role of TRPP2 in vasocontraction, we used TRPP2-specific siRNA to knock down TRPP2 expression in cerebrovascular VSMCs. We found that TRPP2-specific siRNA suppressed TRPP2 expression in the basilar artery and decreased ET-1-induced SOCE-mediated vasoconstriction. This finding supports the previous discovery that resting [Ca2+]i and SOCE activity are decreased in VSMCs in PKD2-Cre mice [36, 37] and suggests a potential direction for the future development of hypertension treatment. Our study also found that the SOCE-mediated contractile response induced by the potent vasoconstrictor ET-1 in denuded basilar arteries was increased in rats fed a high-salt diet. These results suggest that, like TRPP2, SOCE might exert unique effects on cerebrovascular contraction.
As a significant participant in VSMC Ca2+ signaling, SOCE is thought to have pivotal effects on vasoconstriction and is regulated by interactions between STIM and Orai [38,39,40,41,42]. STIM1 is a single-pass transmembrane protein diffusely distributed in the ER membrane. It functions as a Ca2+ sensor by interacting with Orai and translocating itself to the ER-plasma membrane junction upon Ca2+ store depletion, thus triggering SOCE [43,44,45,46]. Several studies have shown that STIM1 regulates SOCE in VSMCs [40, 42, 47] and is associated with blood pressure regulation [48, 49]. However, the role of STIM1 in hypertensive cerebrovascular contraction has not been previously clarified. Our present study found that ET-1–evoked SOCE was significantly enhanced in the cerebral vasculature of high salt diet-fed rats compared with rats fed a normal diet. Our western blotting and immunohistochemical results indicated that the expression of STIM1 in the cerebral vessels of hypertensive rats was significantly higher than that in age-matched control rats. In contrast, knocking down STIM1 protein levels with STIM1-specific siRNA transfection reduced SOCE-induced contraction in the basilar artery.
Orai3, which belongs to the Orai family, is a major component of the SOCE pathway. Studies have reported that the knockdown of Orai1 suppresses angiotensin-II–mediated Ca2+ entry [24] and that Orai3 in combination with STIM1 mediates the majority of SOCE in astrocytes [50]. However, few studies have examined the function of Orai3 in cerebrovascular Ca2+ homeostasis. Our data from western blotting and immunohistochemical assays showed overexpression of the Orai3 protein in the cerebral vessels of hypertensive rats compared with those of normotensive rats, which was likely related to enhanced vasoconstriction. However, the contractile response of the cerebral vasculature was weakened by BTP-2. Spirli et al. reported that TRPP2 affects coupling between STIM1 and Orai channels [24]. Our results may indicate that the increased expression of TRPP2 enhances the interaction of STIM1 and Orai3, while the increased expression of STIM1 and Orai3 further enhances SOCE (Scheme 1). We also found that, 24 h after cerebral ischemia-reperfusion, the area of infarction in the brains of high salt intake-induced hypertensive rats was greater than that in rats fed a normal diet, but the infarcted area was decreased in normotensive rats treated with BTP-2. This finding may be related to the enhancement of ET-1-induced SOCE-mediated vasoconstriction in the cerebral vasculature of hypertensive rats, but this postulation needs further confirmation.
In conclusion, our study results showed that increased expression levels of TRPP2, STIM1, and Orai3 were associated with increased cerebrovascular contraction in high salt intake-induced hypertension in rats. Our results suggest that TRPP2 and other SOCE components may be potential targets for drug intervention for hypertensive cerebrovascular disease. Given that SOCE and TRPP2 have complicated interactions with each other, the entire mechanism underlying Ca2+ modulation in hypertensive cerebrovascular disease pathology is yet to be elucidated.
References
Streb HIR, Berridge MJ, Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. 1983;306:3.
Rosado J. Calcium entry pathways in non-excitable cells. Adv Exp Med Biol. 2016;898:1–471.
Hogan PG, Rao A. Store-operated calcium entry: mechanisms and modulation. Biochem Biophys Res Commun. 2015;460:40–9.
Du J, Fu J, Xia XM, Shen B. The functions of TRPP2 in the vascular system. Acta Pharm Sin. 2016;37:13–8.
Papanikolaou M, Lewis A, Butt AM. Store-operated calcium entry is essential for glial calcium signalling in CNS white matter. Brain Struct Funct. 2017;222:2993–3005.
Zhong X, Fu J, Song K, Xue N, Gong R, Sun D, et al. The role of TRPP2 in agonist-induced gallbladder smooth muscle contraction. Sci China Life Sci. 2015;59:409–16.
Jing W, Jie F, Jing L, Yang W, Long T, Su-Wen B, et al. Enhanced store-operated Ca2+ entry in high glucose-cultured neonatal and adult diabetic rat cardiomyocytes. Int J Clin Exp Pathol. 2017;10:877–89.
Liu Z, Qi H, Liu B, Liu K, Wu J, Cao H, et al. Genetic susceptibility to salt-sensitive hypertension in a Han Chinese population: a validation study of candidate genes. Hypertens Res. 2017;40:876–84.
Sun SW, Lu FH, Sun Y, Zhao YX, Liu ZD, Wang SJ. 2010 Chinese guidelines for the management of hypertension. Am J Hypertens. 2012;25:271.
Kip SN, Hunter LW, Ren Q, Harris PC, Somlo S, Torres VE, et al. [Ca2+]i reduction increases cellular proliferation and apoptosis in vascular smooth muscle cells: relevance to the ADPKD phenotype. Circ Res. 2005;96:873–80.
Spinelli AM, Trebak M. Orai channel-mediated Ca2+ signals in vascular and airway smooth muscle. Am J Physiol Cell Physiol. 2016;310:C402–13.
Giachini FR, Chiao CW, Carneiro FS, Lima VV, Carneiro ZN, Dorrance AM, et al. Increased activation of stromal interaction molecule-1/Orai-1 in aorta from hypertensive rats: a novel insight into vascular dysfunction. Hypertension. 2009;53:409–16.
Smith KA, Ayon RJ, Tang H, Makino A, Yuan JX. Calcium-sensing receptor regulates cytosolic [Ca2+] and plays a major role in the development of pulmonary hypertension. Front Physiol. 2016;7:517.
Zhao R, Zhou M, Li J, Wang X, Su K, Hu J, et al. Increased TRPP2 expression in vascular smooth muscle cells from high-salt intake hypertensive rats: the crucial role in vascular dysfunction. Mol Nutr Food Res. 2015;59:365–72.
Streeter E, Hart J, Badoer E. An investigation of the mechanisms of hydrogen sulfide-induced vasorelaxation in rat middle cerebral arteries. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:991–1002.
Chen M, Li J, Jiang F, Fu J, Xia X, Du J, et al. Orai1 forms a signal complex with BKCa channel in mesenteric artery smooth muscle cells. Physiol Rep. 2016;4:e12682.
Lee HJ, Dietrich HH, Han BH. Development of an ex vivo model for the study of cerebrovascular function utilizing isolated mouse olfactory artery. J Korean Neurosurg S. 2015;57:1–5.
Connolly MJ, Prieto-Lloret J, Becker S, Ward JP, Aaronson PI. Hypoxic pulmonary vasoconstriction in the absence of pretone: essential role for intracellular Ca2+ release. J Physiol. 2013;591:4473–98.
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91.
Cui G, Wang Y, Yu S, Yang L, Li B, Wang W, et al. The expression changes of vacuolar protein sorting 4B (VPS4B) following middle cerebral artery occlusion (MCAO) in adult rats brain hippocampus. Cell Mol Neurobiol. 2013;34:83–94.
Cui Y, Liu X, Li X, Yang H. In-depth proteomic analysis of the hippocampus in a rat model after cerebral ischaemic injury and repair by danhong injection (DHI). Int J Mol Sci. 2017;18:1355.
Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–76.
Ruan L, Huang HS, Jin WX, Chen HM, Li XJ, Gong QJ. Tetrandrine attenuated cerebral ischemia/reperfusion injury and induced differential proteomic changes in a MCAO mice model using 2-D dige. Neurochem Res. 2013;38:1871–79.
Spirli C, Locatelli L, Fiorotto R, Morell CM, Fabris L, Pozzan T, et al. Altered store operated calcium entry increases cyclic 3’,5’-adenosine monophosphate production and extracellular signal-regulated kinases 1 and 2 phosphorylation in polycystin-2-defective cholangiocytes. Hepatology. 2012;55:856–68.
Qian Q, Hunter LW, Li M, Marin-Padilla M, Prakash YS, Somlo S, et al. Sieck Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum Mol Genet. 2003;12:pp1875–80.
Narayanan D, Bulley S, Leo MD, Burris SK, Gabrick KS, Boop FA, et al. Smooth muscle cell transient receptor potential polycystin-2 (TRPP2) channels contribute to the myogenic response in cerebral arteries. J Physiol. 2013;591:5031–46.
Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 2008;135:110–22.
Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13:549–65.
Zhou Y, Srinivasan P, Razavi S, Seymour S, Meraner P, Gudlur A, et al. Initial activation of STIM1, the regulator of store-operated calcium entry. Nat Struct Mol Biol. 2013;20:973–81.
Putney JW. Capacitative calcium entry: from concept to molecules. Immunol Rev. 2009;231:10–22.
Sammels E, Devogelaere B, Mekahli D, Bultynck G, Missiaen L, Parys JB, et al. Polycystin-2 activation by inositol 1,4,5-trisphosphate-induced Ca2+ release requires its direct association with the inositol 1,4,5-trisphosphate receptor in a signaling microdomain. J Biol Chem. 2010;285:18794–805.
Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002;4:191.
Li Y, Wright JM, Qian F, Germino GG, Guggino WB. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J Biol Chem. 2005;280:41298–306.
Gao F, Wang DH. Impairment in function and expression of transient receptor potential vanilloid type 4 in Dahl salt-sensitive rats: significance and mechanism. Hypertension. 2010;55:1018–25.
Gao F, Sui D, Garavito RM, Worden RM, Wang DH. Salt intake augments hypotensive effects of transient receptor potential vanilloid 4: functional significance and implication. Hypertension. 2009;53:228–35.
Qian Q, Hunter LW, Li M, Marin-Padilla M, Prakash YS, Somlo S, et al. Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum Mol Genet. 2003;12:1875–80.
Menè P, Teti A, Pugliese F, Cinotti GA. Calcium release-activated calcium influx in cultured human mesangial cells. Kidney Int. 1994;46:122–8.
Chuang TY, Au LC, Wang LC, Ho LT, Yang DM, Juan CC. Potential effect of resistin on the ET-1-increased reactions of blood pressure in rats and Ca2+ signaling in vascular smooth muscle cells. J Cell Physiol. 2012;227:1610–18.
Courjaret R, Machaca K. STIM and Orai in cellular proliferation and division. Front Biosci. 2012;4:331–41.
Simo-Cheyou ER, Tan JJ, Grygorczyk R, Srivastava AK. STIM-1 and ORAI-1 channel mediate angiotensin-II-induced expression of Egr-1 in vascular smooth muscle cells. J Cell Physiol. 2017;232:3496–509.
Tanwar J, Trebak M. Cardiovascular and hemostatic disorders: role of STIM and Orai proteins in vascular disorders. Adv Exp Med Biol. 2017;993:425–52.
Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–7.
Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, Wakamori M, et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. PNAS. 2006;103:16704–9.
Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. PNAS. 2007;104:9301–6.
Feldman CH, Grotegut CA, Rosenberg PB. The role of STIM1 and SOCE in smooth muscle contractility. Cell Calcium. 2017;63:60–5.
Sours-Brothers S, Ding M, Graham S, Ma R. Interaction between TRPC1/TRPC4 assembly and STIM1 contributes to store-operated Ca2+ entry in mesangial cells. Exp Biol Med. 2009;234:673–82.
Guo RW, Yang LX, Li MQ, Pan XH, Liu B, Deng YL. Stim1- and Orai1-mediated store-operated calcium entry is critical for angiotensin II-induced vascular smooth muscle cell proliferation. Cardiovasc Res. 2012;93:360–70.
Nishimoto M, Mizuno R, Fujita T, Isshiki M. Stromal interaction molecule 1 modulates blood pressure via NO production in vascular endothelial cells. Hypertens Res. 2018;41:506–14.
Shinya K, Nakayama T, Nakayama T, Yamamoto T. A case-control study between the STIM1 gene and hypertensive disorders of pregnancy. Hypertens Res. 2018;41:39–44.
Kwon J, An H, Sa M, Won J, Shin JI, Lee CJ. Orai1 and Orai3 in combination with Stim1 mediate the majority of store-operated calcium entry in astrocytes. Exp Neurobiol. 2017;26:42.
Acknowledgements
The present study was supported by a grant (2016YFC1300600, 2016YFC1306400) from the National Key Research and Development Program of China; the Natural Science Foundation of China (Grant No. 81371284, 81570403, U1732157); the Anhui Provincial Natural Science Foundation (1108085J11). The funder had no role in the study design, collection, analysis or interpretation of the data, the writing of the report, or the decision to submit the article for publication.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jiang, W., Ye, L., Yang, Y. et al. TRPP2 associates with STIM1 to regulate cerebral vasoconstriction and enhance high salt intake-induced hypertensive cerebrovascular spasm. Hypertens Res 42, 1894–1904 (2019). https://doi.org/10.1038/s41440-019-0324-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-019-0324-5
Keywords
This article is cited by
-
Role of thrombospondin-1 in high-salt–induced mesenteric artery endothelial impairment in rats
Acta Pharmacologica Sinica (2023)
-
Exploring the association of long noncoding RNA expression profiles with intracranial aneurysms, based on sequencing and related bioinformatics analysis
BMC Medical Genomics (2020)
