
Abstract
Saline–alkali stress is a severely adverse abiotic stress limiting plant growth. Malus halliana Koehne is an apple rootstock that is tolerant to saline–alkali stress. To understand the molecular mechanisms underlying the tolerance of M. halliana to saline–alkali stress, an integrated metabolomic and proteomic approach was used to analyze the plant pathways involved in the stress response of the plant and its regulatory mechanisms. A total of 179 differentially expressed proteins (DEPs) and 140 differentially expressed metabolites (DEMs) were identified. We found that two metabolite-related enzymes (PPD and PAO) were associated with senescence and involved in porphyrin and chlorophyll metabolism; six photosynthesis proteins (PSAH2, PSAK, PSBO2, PSBP1, and PSBQ2) were significantly upregulated, especially PSBO2, and could act as regulators of photosystem II (PSII) repair. Sucrose, acting as a signaling molecule, directly mediated the accumulation of D-phenylalanine, tryptophan, and alkaloid (vindoline and ecgonine) and the expression of proteins related to aspartate and glutamate (ASP3, ASN1, NIT4, and GLN1−1). These responses play a central role in maintaining osmotic balance and removing reactive oxygen species (ROS). In addition, sucrose signaling induced flavonoid biosynthesis by activating the expression of CYP75B1 to regulate the homeostasis of ROS and promoted auxin signaling by activating the expression of T31B5_170 to enhance the resistance of M. halliana to saline–alkali stress. The decrease in peroxidase superfamily protein (PER) and ALDH2C4 during lignin synthesis further triggered a plant saline–alkali response. Overall, this study provides an important starting point for improving saline–alkali tolerance in M. halliana via genetic engineering.
Similar content being viewed by others


Introduction
Soil salinization–alkalization is a major abiotic stress that affects plant growth1. More than 831 million hectares of agricultural land is threatened by salinization and alkalinization worldwide2, and this problem continues to worsen. In recent years, extensive studies have focused on the mechanisms underlying the tolerance of plants to salt stress or alkali stress. However, studies on saline–alkali stress have rarely been conducted3,4. In northwestern China, saline–alkali stress has become a major limiting factor for apple growth and productivity5. The selection of saline–alkali-tolerant rootstocks is an effective strategy to minimize soil salinization and alkalinization problems6. Malus halliana is an indigenous apple species that, throughout its long evolutionary history, has already developed suitable mechanisms to adapt to saline–alkali environments. Our recent study also confirmed that M. halliana was more resistant to saline–alkali stress than other apple rootstocks in terms of physiological responses7. Consequently, exploration of the possible molecular mechanisms of how M. halliana responds to saline–alkali stress is urgently needed.
In plants, salt stress induces ionic stress, osmotic stress, and oxidative stress7. Many studies demonstrate that these stresses reduce the plant uptake of water and interfere with photosynthesis8, thereby affecting osmotic balance and leading to the production of reactive oxygen species (ROS)4,5. For instance, high-salt stress damages the photosynthetic apparatus and disturbs the expression levels of proteins related to photosynthesis, thus reducing photosynthesis and causing premature senescence9. Similarly, salt stress also causes dysregulation of various metabolic pathways, including signal transduction, energy metabolism, and hormone synthesis10,11. However, saline–alkali stress is more complex than salt stress, due to the addition of high pH stress12. Guo et al.3 have revealed that high pH disrupts ion homeostasis and accelerates the accumulation of ROS, which contributes to severe damage to cellular structures13. Therefore, under saline–alkali stress, plants should have more sophisticated mechanisms to re-establish osmotic and ROS homeostasis.
Numerous enzymatic scavengers play critical roles in mitigating damage induced by ROS in plants under salt stress, such as superoxide dismutase, dehydroascorbate reductase, and ascorbate peroxidase14,15. Zhang et al.16 reported that 143 proteins, which are ROS scavenging-related proteins, were induced by salinity in 24 plant species. These proteins are mainly involved in the glutathione–ascorbate cycle, catalase (CAT) pathway, PrxR/Trx pathway, and GPX pathway to alleviate ROS damage and enhance salt tolerance17. Moreover, in Arabidopsis, Hsp17.6CII (a chaperone protein) activates CAT2 activity and increases abiotic stress resistance by scavenging ROS18. MPK3 and MPK6 phosphorylate heat- shock factor A4A (HSFA4A) to regulate the homeostasis of ROS19. Intriguingly, ROS can also function as signaling molecules; plants perceive stress signals and can then transmit ROS to the cellular structures to activate defense responses that are completed by regulating related genes, protein expression, and metabolite accumulation20. For example, ABA pathways are important in terms of stress response signaling17. In apple, MdBT2, an ABA-responsive protein, interacts directly with MdbHLH93 and induces the ubiquitination and degradation of the MdbHLH93 protein, which play a significant role in delaying leaf senescence21. Several proteins, such as mitogen-activated protein kinases (MAPKs), PM-located protein (OSCA1), and MscS-like 8, have been to be involved in various signal transduction pathways22,23,24. In rice, MAPKs (MKK1–MPK4 cascade) play a vital role in the regulation of the expression of transcription factor genes under salt stress25.
Stress signals also induce the biosynthesis and accumulation of compatible osmolytes, including sugars, amino acids, and secondary metabolites. These metabolites can remove excessive ROS and lower the osmotic potential in cells26, which is a primary adaptive strategy in response to stress. Studies on lotus have shown that amino acids such as proline, phenylalanine, and glycine, significantly increase under salt stress27. A similar result was obtained by Asha et al.11, who reported that the accumulation of sucrose in halophytes resulted in the activation of antioxidant enzymes and decrease in ROS production, thus improving plant tolerance to salt stress. Importantly, recent studies have demonstrated that these metabolites also directly stimulate the related enzymes to regulate salt responses14. For instance, in Arabidopsis, free unsaturated fatty acids enhance the activity of the plasma membrane (PM) H+-ATPase under salt stress, which contributes to modulating protein functions and signal transduction28. In apple, glucose improves salt tolerance by regulating the expression of the glucose sensor MdHXK129. Some of these metabolites, such as sucrose and fructose, are widely recognized as signaling molecules that trigger downstream salt-stress responses30. In addition, some small molecules, including proline, c-aminobutyric acid (GABA), and melatonin, mediate salt-stress signaling by stimulating the expression of related proteins31.
Molecular responses to stress depend on various interactions, such as those between metabolites and proteins and those between enzymes and metabolites32. Studies of omics, including those involving proteomics and metabolomics, offer an effective approach to obtain comprehensive insight and to connect the nodes of the molecular networks underlying the abiotic tolerance mechanisms of plants33. Many salt-responsive proteins and metabolites that contribute to various functions, such as photosynthesis, signal transduction, and scavenging systems of ROS, have been identified via proteomic technologies. In addition, most related differentially expressed proteins (DEPs) and metabolites are involved in metabolic processes in response to salt stress, including glycolysis (the energy metabolism pathway (EMP)), carbohydrate metabolism, the TCA cycle, and secondary metabolic pathways11. In recent years, significant research advances have been made in understanding plant salt-tolerance mechanisms13,31. However, few studies have focused on saline–alkali stress in plants. Therefore, in this study, we integrated metabolomic and proteomic analyses to identify the different proteins, metabolites, and pathways involved in the response to saline–alkali stress in plants. The objective was to gain a comprehensive understanding of how M. halliana regulates and responds to molecular mechanisms under saline–alkali stress, to provide an effective engineering strategy to improve plant saline–alkali tolerance.
Results
Selection of time points for proteomic and metabolomic analyses
To determine suitable time points for our proteomic and metabolomic analyses, we observed the phenotypic changes in M. halliana under saline–alkali stress. The applied saline–alkali stress did not significantly alter the phenotypes of the plants for 0–2 days, as the plants showed little sign of damage. Chlorosis and dehydration in the older leaves were observed, but new leaves grew normally after 4 days of exposure to saline–alkali conditions. New leaves were chlorotic after 6 days of stress, and desiccation and crinkling of the older leaves occurred, which reflected severe stress-induced damage and even led to M. halliana death (Fig. 1).

Phenotypic changes in M. halliana under saline–alkali stress
Proteomic analyses of M. halliana leaves in response to saline–alkali stress
The M. halliana leaves were assessed by iTRAQ quantitative proteomics. Quantitative analysis showed that 19,092 unique peptides corresponding to 2454 proteins were identified via LC-MS/MS determination, and the data were searched against the UniProt database. Saline–alkali stress was examined with an adjusted fold change >2 or fold change <0.5 and p-value <0.05 as the threshold to screen significant changes in the abundance of DEPs. We identified 179 DEPs in the leaves; among these DEPs, 74 were downregulated, and 105 were upregulated (Supplementary Table S1).
To investigate the functional features of these DEPs, Gene Ontology (GO) term enrichment analysis was performed using the GO and UniProt databases. The 179 DEPs were classified into three categories, including biological process (BP), cellular component (CC), and molecular function (MF) categories (Fig. 2a). In the BP category, the most abundant groups included response to a stimulus, single-organism metabolic process, and response to stress. The CC category of DEPs was significantly enriched for the cytoplasmic part, cytoplasm, and chloroplast. For the MF category, oxidoreductase activity, metal ion binding, and cation binding were the most abundant groups.

a GO classification of the identified proteins under saline–alkali stress. The results for the three main GO categories are summarized: BP, CC, and MF. b KEGG pathways of the differentially expressed proteins involved in the response to saline–alkali stress in the leaves of M. halliana
Furthermore, the proteomic results via KEGG analysis revealed that 70 pathways were enriched (Supplementary Table S2). We observed that these DEPs were involved mainly in the following metabolic pathways: amino sugar and nucleotide sugar metabolism; glyoxylate and dicarboxylate metabolism; photosynthesis; ascorbate and aldarate metabolism; phenylpropanoid biosynthesis; carbon (C) metabolism; C fixation in photosynthetic organisms; tryptophan metabolism; and alanine, aspartate, and glutamate metabolism (Fig. 2b). These results showed that proteins involved in the synthesis of metabolites also regulate various metabolic pathways in response to saline–alkali stress.
Metabolic analyses of M. halliana leaves in response to saline–alkali stress
In this study, 11,135 kinds of metabolites were identified via LC–MS. Notably, 140 differentially expressed metabolites (DEMs), which included mainly sugars, glucosides, amino acids, organic acids and secondary metabolites, were selected that passed the criteria of an OPLS-DA model VIP >2.5 and had a p-value <0.05 in M. halliana leaves (Supplementary Table S3). To select marker metabolites accurately and investigate changes in the related metabolic processes, the selected DEMs were analyzed by hierarchical clustering based on the expression of significantly different metabolites in each group of samples (Fig. 3a). There was a clear separation of metabolites between the upregulated and downregulated M. halliana leaves. In addition, a total of 59 metabolites were upregulated, and 81 were downregulated.

a Hierarchical cluster heat map of different metabolites under saline–alkali stress of M. halliana leaves; b KEGG pathways of the different metabolites involved in the response to saline–alkali stress in the leaves. “CK” represents 0 day. “T” represents 4 days. “1–8” represents repetitions
All of the DEMs affected by saline–alkali stress were mapped to the KEGG database to analyze the relevant pathways. The results showed that 35 pathways were enriched with DEMs (Supplementary Table S4). The top ten most enriched items were as follows: indole alkaloid biosynthesis; phenylalanine biosynthesis; tropane, piperidine and pyridine alkaloid biosynthesis; phenylalanine metabolism; ABC transporters; shikimate pathway-derived alkaloid biosynthesis; carbohydrate digestion and absorption; biotin metabolism; mineral absorption; and phenylalanine, tyrosine and tryptophan biosynthesis (Fig. 3b). The changes in these metabolites and metabolic pathways provide important information on how M. halliana responds to saline–alkali stress.
Integrated proteomic and metabolomic analyses of M. halliana leaves in response to saline–alkali stress
By conducting a KEGG mapping analysis based on the differences in proteins and metabolites, we found that saline–alkali stress-induced changes in the relative amounts of proteins and metabolites associated with 11 metabolic pathways, including the following: 2-oxocarboxylic acid metabolism; amino acid biosynthesis; alpha-linolenic acid metabolism; glycine, serine, and threonine metabolism; phenylalanine, tyrosine, and tryptophan biosynthesis; phenylalanine metabolism; secondary metabolite biosynthesis; tropane, piperidine, and pyridine alkaloid biosynthesis; tryptophan metabolism; phenylpropanoid biosynthesis; and metabolic pathways (Table 1).
In addition, we also performed a KEGG mapping analysis based on the differences in metabolite-related enzymes and DEPs. In total, 70 metabolic pathways exhibited changes (Supplementary Table S5), and 16 enzymes were assigned to these pathways (Supplementary Table S6). Further analysis of these metabolic pathways suggested that these proteins and enzymes were involved mainly in energy metabolism as well as in dicarboxylate metabolism; secondary metabolite biosynthesis; alanine, aspartate, and glutamate metabolism; phenylpropanoid biosynthesis; and thiamine metabolism (Fig. 4a).

a KEGG pathways of the metabolite-related enzymes and DEPs involved in the response to saline–alkali stress in the leaves. b Network analysis of the differential proteins, metabolites, and enzymes performed using Ingenuity Pathway Analysis (IPA) software. The solid lines indicate direct interactions or regulations, while the dashed lines indicate effects mediated by additional molecules
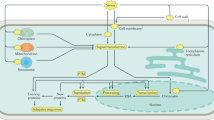
Using an integrated analysis of DEPs, DEMs, and enzymes, we constructed an interactive diagram of the response of M. halliana to saline–alkali stress. In the network, the BGLU13 protein not only had the greatest expression fold change but also directly interacted with XYL2, CWINV6, and At1g73050, and BGLU13 was involved in both starch and sucrose metabolism and the phenylpropanoid biosynthesis pathway. In addition, metabolic pathways, glyoxylate and dicarboxylate metabolism and amino sugar and nucleotide sugar metabolism had the largest −log(p-value). PSBP1 can directly interact with PSBQ, PSBO2, PSAH2, and PSAK, each of which regulates the photosynthesis of M. halliana leaves. Only one metabolite, sucrose, which interacts with XYL2, RFSA, and AGAL3, clustered into the integrated network (Fig. 4b).
Analysis of the comprehensive systemic metabolic pathways diagram
To explore the molecular mechanisms of M. halliana in regulating and adapting to saline–alkali stress further, we constructed a comprehensive systemic metabolic pathway diagram by combining the KEGG pathways of Table 1 and Fig. 5a. As shown in Fig. 5, 31 proteins and 13 metabolites were mapped to the diagram. The expression of UGE3, XYL2, At2g31390, AMY1, BGLU13, BGLU44, and sucrose, which are involved in starch and sucrose metabolism, was upregulated. Notably, the upregulation of UGE3 catalyzes the interconversion of UDP-glucose and UDP-galactose, which is the first step in the conversion of sucrose. The expression of BGLU13 increased by as much as 13.965-fold; sucrose increased by as much as 7.105-fold. In contrast, the expression of PMM, At5g28840, and VTC4, which are involved in mannose and galactose metabolism, was downregulated under saline–alkali stress (Table 2). In addition, we found high expression of PDH2 (2.246-fold), ALDH2B4 (3.503-fold), and EMB2024 (6.350-fold) in relation to the EMP and the pentose phosphate pathway (PPP). EMB2024 is involved in step 2 and synthesizes 6-phospho-d-gluconate from glucose-6-phosphate in the PPP, the former of which is a precursor to pyruvate for entry into the TCA cycle. Moreover, ALDH2B4 and PDH2 catalyze the overall conversion of pyruvate into acetyl-CoA and CO2 in EMP.

Proteins (↑) and metabolites (□) were mapped to a comprehensive systemic metabolic pathways diagram by combining the KEGG pathways of the DEMs, enzymes, and proteins. The DEPs and metabolites are marked in bold; red indicates upregulation; green indicates downregulation
Significant upregulation of d-phenylalanine (5.873-fold) and downregulation of ASP3, phenylacetylglycine, and l-hyoscyamine, which are involved in phenylalanine metabolism, were detected (Table 3). In addition, the expression of BGLU13 and BGLU44, which are involved in phenylpropanoid biosynthesis, was upregulated, while the expression of coumarin and peroxidase superfamily protein (PER)12, PER53, PER52, PER17, and PER3, was downregulated in this pathway. BGLU13 and BGLU44 catalyzed the conversion of β-d-glucosyl-2-coumarinate into coumarinate and the subsequent conversion into coumarine. PER is one of the key proteins involved in lignin synthesis, and can catalyze the use of lignin monomer precursors to produce guaiacyl (G), syringyl (S), and p-hydroxyphenyl (H) lignin (Fig. 5, Table 4).
We further observed that the expression of kaempferol and CYP75B1, which are related to flavonoid biosynthesis, was upregulated under saline–alkali conditions (Table 4). Similarly, tryptophan (7.364-fold) and vindoline (6.220-fold), which are involved in indole alkaloid biosynthesis, significantly accumulated, but TAR1, CAT2, and ajmaline levels decreased. T31B5_170 was found to be involved in plant hormone signal transduction and was highly expressed up to 5.822-fold (Table 5). The expression of PSBO2, PSBP1, PSBQ2, PSAH2, and PSAK was significantly upregulated with respect to photosynthesis, but the expression of PSBR was downregulated; moreover, the expression of PPD, PAO, and pheophorbide a was upregulated with respect to porphyrin and Chl metabolism (Table 6).
Verification of DEG analysis results by qRT-PCR
To further evaluate the validity of our results, 16 proteins involved in sugar metabolism, amino acid metabolism, phenylpropanoid metabolism, indole and tropane, piperidine, and pyridine alkaloid metabolism, and photosynthesis and chlorophyll metabolism were selected for quantitative real-time PCR (qRT-PCR). The results indicated that 15 genes (93.75%) showed similar trends between the qRT-PCR results and the iTRAQ analysis, but ASP3 analyzed by qRT-PCR was not consistent with iTRAQ data (Fig. 6). Interestingly, we found that ASP3 was downregulated in phenylalanine metabolism and upregulated in aspartate and glutamate synthesis under saline–alkali stress (Fig. 5). These results revealed that the iTRAQ analysis was highly reliable.

qRT-PCR confirmation of 16 proteins at two timepoints: 0 and 4 days
Discussion
Physiological indices, photosynthesis-related proteins, and Chl metabolism
Soil salinization–alkalization is a major abiotic stress that affects plant growth, production, and crop yield34. Saline–alkali stress leads to damage of the plant photosynthetic apparatus and even death35. Our physiological data showed that Pn, Gs, Tr, Chl a, Chl b, Fm, and Fv/Fm in M. halliana decreased under saline–alkali stress, but that F0 and Y(NO) increased. The decreased Fv/Fm implied that the photosystem II (PSII) reaction center induced photoinhibition, leading to a reduction in light energy use efficiency36. The increases in F0 and Y(NO) indicated that the photosynthetic system gradually suffered from damage37. This damage eventually caused an imbalance in the electron transport chain in the chloroplasts, thereby accelerating the formation of ROS and causing oxidative damage38,39. However, plants have evolved a number of photoprotective mechanisms. Increases in NPQ, qN, and Car are mainly thought to reflect the energy dissipation mechanism that protects the photosynthetic system by which excess energy is dissipated as heat and prevents oxidative damage7. In this study, the NPQ and qN in M. halliana did not definitively change throughout the entire stress duration, suggesting that the energy dissipation mechanism was not triggered. Therefore, we hypothesize that M. halliana may have triggered another stress sensing-response mechanism to avoid the production of excessive ROS in the chloroplasts.
To verify this hypothesis, we constructed a comprehensive systemic metabolic pathways diagram by combining proteomic and metabolomic approaches to explore the molecular regulatory mechanisms in response to saline–alkali stress in M. halliana (Fig. 5). The diagram shows that accumulated pheophorbide a was converted into both pyropheophorbide a and red Chl by PPD and PAO, respectively, which is a key step in Chl breakdown. In Arabidopsis, repression of ACD1 expression leads to an unusual pheophorbide a accumulation related to leaf senescence, and its accumulation ultimately results in cell death40. These results provide evidence that the phenotypic changes in M. halliana leaves may be caused by the accumulation of pheophorbide a. Chl is the main photosynthetic pigment because it is capable of harvesting, transmitting, and transforming light energy41. Thus, the decomposition of Chl reduced its content in the M. halliana leaves, thereby affecting photosynthetic efficiency and aggravating photoinhibition.
The photosystems include both photosystem I (PSI) and photosystem II (PSII)42. Many photosynthetic proteins have been identified in plants; for example, the LHC chlorophyll a/b-binding protein (LHC-CAB), oxygen-evolving enhancer protein (OEE), and PS II D1 protein are involved in salinity tolerance16. In this study, six proteins were found to be related to photosynthesis in M. halliana in response to saline–alkali stress, including four PSII proteins (PSBR, PSBO2, PSBP1, and PSBQ2) and two photosystem I (PSI) proteins (PSAH2 and PSAK) (Table 6). Xue et al.43 reported that the decrease in PSBR expression decreased the magnitude of light-induced nonphotochemical quenching (NPQ). As expected, the small change in NPQ in M. halliana may be a result of the downregulation of PSBR, reducing the extent of NPQ. In addition, PSBO2, PSBP1, and PsbQ2 are the oxygen-evolving enhancer (OEE) proteins of eukaryotic PSII and are required for photosynthetic water oxidation44. Previous studies demonstrated that PSBO2 also participates in the D1 repair cycle by regulating the turnover of the PSII D1 protein45, and PSBP1 and PSBQ2 are essential for the regulation of PSII assembly and/or activity46. These results suggested that OEE proteins play an important role in maintaining the stability and repairing the injury of the PSII reaction center to alleviate saline–alkali stress. A similar result was obtained by Zhu et al.47, who reported that upregulation of OEE2 repairs the injury of the PSII complex. Studies on the effects of oxidative stress on the photodamage of plants have demonstrated that ROS inhibits the repair of photodamaged PSII but does not accelerate damage to PSII48. Nevertheless, our study confirmed that saline–alkali stress enhanced the photoinhibition of PSII, indicating that ROS directly damages PSII. The significant upregulation of OEE proteins revealed that M. halliana tried to repair PSII by regulating the turnover of the D1 protein. In addition, our data showed that PSBP1 interacts with PSBQ, PSBO2, PSAH2 and PSAK (Fig. 4b); thus, we believe that PSAH2 and PSAK might be PSI-protective proteins, but their function is still unknown, which encourages additional studies to strengthen the knowledge about the changes induced by saline–alkali stress. In short, the upregulation of photosynthesis-related proteins contributed to maintaining the photosynthesis level to cope with saline–alkali stress.
Sugar metabolism plays significant roles in many metabolic processes under saline–alkali stress
Sucrose is the primary product of photosynthesis and is widely considered an energy source for metabolism activity in plants31. In this study, saline–alkali stress induced a significant accumulation of sucrose and expression of UGE3 protein (Fig. 5 and Table 2). Evidence has revealed that sucrose functions as compatible osmolytes for osmotic adjustment and detoxification31,49, suggesting that sucrose accumulation under saline–alkali stress may be a primary strategy, by which plants protect themselves from damage by ROS and adapt to osmotic stress. In Arabidopsis, UGE3 plays a very critical role in the regulation of sucrose synthesis50. However, the photosynthesis of M. halliana was reduced under saline–alkali stress (Fig. 7a). These results suggested that the upregulation of UGE3 promotes the degradation of polysaccharides, thereby leading to the generation of additional sucrose to counteract saline–alkali stress. This result is similar to the result of Xu et al.50. In addition, sucrose also acts as a signaling molecule that regulates biotic and abiotic stress responses30. Sucrose-based regulation has also been observed in Arabidopsis, as sucrose regulates iron deficiency by promoting auxin signaling51. Rosa et al.52 confirmed that sucrose is a primary messenger that controls signaling by regulating the expression of different proteins and genes. In fact, experiments on Camellia sinensis have shown that sucrose induces polyphenol biosynthesis by altering the expression of transporters GST, ABC, and MATE53. These findings provide evidence that sucrose, which acts as a signaling molecule, might regulate many vital metabolic processes and activate other resistance pathways.

a net photosynthetic rate (Pn); b stomatal conductance (Gs); c transpiration rate (Tr); d Chl a content (Chl a); e Chl b content (Chl b); f carotenoid content (Car); g initial fluorescence (F0); h maximum fluorescence (Fm); i maximal photochemical efficiency (Fv/Fm); j nonregulatory energy dissipation (Y(NO)); k light-induced nonphotochemical quenching (NPQ); l photochemical quenching coefficient (qN)
Recent studies have shown that glucose also acts as a direct and central signaling molecule in plants; this molecule was shown to improve salt tolerance by mediating the glucose sensor MdHXK1 and the vacuolar Na+/H+ transporter MdNHX1 in “Royal Gala” apple29. In our study, 4 significantly upregulated proteins, including XYL2, AMY1, BGLU13 and BGLU44, were involved in glucose synthesis in three different pathways (Fig. 5, Table 2). This result suggested that the pathways of glucose synthesis were enhanced under saline–alkali stress. Interestingly, in this study, glucose was not detected in M. halliana. Barpeled and O’Neill54 reported that glucose, as the energy material, was involved in various primary and secondary metabolic pathways. Studies on rice have demonstrated that EMP and PPP are important mechanisms of glucose degradation55. As expected, the expression of three proteins, ALDH2B4, PDH2, and EMB2024, involved in the EMP and PPP, was significantly upregulated under saline–alkali stress (Fig. 5, Table 2), which indicated that these proteins promote glucose degradation. Similar regulation was also observed by Sobhanian et al.56, who reported that salt stress induced the upregulation of EMP-related proteins in Aeluropus lagopoides. Moreover, EMP and PPP play pivotal roles in the production of both energy and the C skeletons of primary and secondary metabolites57. Höper et al.58 found that the upregulation of fructokinase-1 (FRK) contributed to glucose breakdown for energy generation to resist salt stress. These results revealed that glucose, as the main energy source, provided substrates and energy for the synthesis of other metabolites in M. halliana, which also explained why glucose did not accumulate under saline–alkali stress.
Amino acid metabolism plays key roles in improving saline–alkali tolerance in M. halliana
Many studies have revealed that sugar metabolism provides sufficient energy to amino acid metabolism, and that intermediates from the EMP can also be utilized as precursors for the synthesis of amino acids59. In this study, d-phenylalanine and tryptophan, which are synthesized via the shikimic acid pathway, significantly accumulated under saline–alkali stress (Table 3). Notably, we found that the levels of all related proteins and metabolites, such as PMM, At5g28840, VTC4 and AOC2 and stearidonic acid, were downregulated in galactose metabolism and alpha-linolenic acid metabolism (Fig. 5, Table 2), which indicated that the two pathways were inhibited by saline–alkali stress in M. halliana. This phenomenon might be caused by a decrease in the consumption of galactose and stearidonic acid synthesis and an increase in energy supplies for amino acid metabolism. In higher plants, amino acids accumulate in response to various stresses and have multiple functions in plant growth60. However, our results showed no altered protein levels (Fig. 5). Similar results were obtained in alfalfa, in which l-tyrosine and phenylalanine were upregulated but in which upregulated levels of related proteins were not detected. Fan et al.32 inferred that this upregulation may have resulted from protein hydrolysis. In potato tubers, it has also been postulated that amino acid accumulation is a mechanism to compensate for cellular osmolarity caused by reductions in sucrose concentration61. Sugar metabolism analyses have revealed that sucrose, as a signaling molecule, can transmit stress signals to other pathways and regulate the expression of related proteins and metabolites to respond to adversity52. We therefore speculate that the accumulation of both d-phenylalanine and tryptophan may be induced by sucrose signals in response to saline–alkali stress.
In this study, d-phenylalanine is reversibly converted into phenylalanine, leading to the production of phenylacetylglycine after a series of reactions in phenylalanine metabolism. The downregulation of ASP3 results in a decrease in phenylacetylglycine, suggesting that M. halliana limits phenylalanine metabolism to resist saline–alkali conditions (Fig. 5, Table 3). In contrast, the upregulation of ASP3, which is involved in aspartate and glutamate synthesis, indicates that ASP3 might be a signaling-related protein. The upregulated expression of ASP3 can initiate amino acid defense reactions to resist saline–alkali stress62. In addition, the accumulation of amino acids (proline, serine, phenylalanine, and threonine as osmolytes) can improve plant tolerance to salt stress by mediating the removal of ROS and damage control and repair13,27. In addition, these amino acids serve as precursors for a large array of secondary metabolites, including pigments, alkaloids, hormones, and cell wall components63.
Sucrose signaling regulates ROS homeostasis by inducing phenylpropanoid biosynthesis pathway and flavonoid synthesis
Considerable evidence exists that phenylalanine, which is required for the biosynthesis of flavonoids and lignin, is an important precursor of phenylpropanoids57. In this study, kaempferol was significantly upregulated and was subsequently converted into quercetin by upregulated CYP75B1, thus enhancing flavonoid biosynthesis (Fig. 5, Table 4). Similarly, GmMYB173 has been reported to improve soybean salt tolerance by regulating flavonoid biosynthesis64. Quercetin and kaempferol are well-known flavonol glycosides that play a positive role in scavenging excess ROS65. Notably, Dubos et al.66 found that sucrose signaling induced flavonoid biosynthesis by activating the expression of MYBL2 in Arabidopsis. This finding might provide strong evidence that sucrose signaling regulates flavonoid synthesis to control the homeostasis of ROS under saline–alkali stress.
Furthermore, our results showed that most of the proteins and metabolites were downregulated in the phenylpropanoid biosynthesis pathway, especially the proteins involved in lignin synthesis, such as PER3, PER17, PER12, PER52, PER53 (PER, peroxidase superfamily protein), and ALDH2C4 (Fig. 5, Table 4). This finding suggested that lignin synthesis was inhibited by saline–alkali stress in M. halliana. Lignin provides mechanical strength to plant secondary cell walls67, which not only protect cells from abiotic stress but also are serve as the structures that first perceive and respond to environmental stress68. In alfalfa, antisense downregulation of the enzyme hydroxycinnamoyl-CoA: shikimate hydroxycinnamoyl transferase (HCT) led to decreased lignin content. Gallego-Giraldo et al.69 reported that the downregulation of lignin biosynthesis might be a defense mechanism against abiotic stress. These results demonstrated that cell wall damage and altered components in response to salt stress are possible salt-sensing mechanisms that would further trigger plant salt responses31. In addition, studies in trees have described that phenylpropanoids represent a significant pathway for C and energy flow during lignin biosynthesis70. Hence, a decrease in phenylpropanoid biosynthesis might conserve C skeletons and energy for flavonoid biosynthesis.
In this study, BGLU13 and BGLU44 were upregulated in the phenylpropanoid biosynthesis pathway, which is involved in the synthesis of coumarine. However, coumarine was downregulated by saline–alkali stress (Fig. 5, Table 4). This result indicated that BGLU13 and BGLU44 might be defense-related proteins and that their upregulation might protect plants against saline–alkali stress. Previous studies have demonstrated that defense proteins play a pivotal role in repairing oxidative damage71. These results revealed that phenylpropanoid metabolism is a key defense signaling pathway.
Sucrose signaling regulates ROS homeostasis by inducing alkaloid metabolism and auxin synthesis
Alkaloids, which are secondary metabolites that play a role in defense against stresses, such as salinity72 and water73 stress, are synthesized from tryptophan and glutamate. Studies on Catharanthus roseus have demonstrated that the upregulated expression of sgd, ORCA3, and t16h may contribute to the observed increases in ajmalicine, vindoline, and catharanthine following binary stress74. In the present study, vindoline was highly upregulated, but no related proteins were identified in the indole alkaloid pathways (Fig. 5, Table 5), indicating that the accumulation of vindoline may be a defense response to saline–alkali stress. Similar regulation was also observed by Li et al.75 who reported that vindoline accumulation increased the scavenging capacity of ROS, thereby providing protection against oxidative injury and reducing metabolic disturbance in plants. However, in our study, ajmaline levels were downregulated, and no related proteins were found in the pathway (Fig. 5). One possible explanation is that tryptophan is the first step of indole alkaloid synthesis and that its accumulation provides skeletons for vindoline and ajmaline76. The reduced ajmaline synthesis may provide energy for vindoline synthesis in M. halliana. Our results showed that the response mechanism of ecgonine and hyoscyamine was typically consistent with that of vindoline and ajmaline. As a result, we believe that the changes in alkaloid levels may be regulated by sucrose signaling, and vindoline and ecgonine, as nonenzymatic scavengers, can mitigate damage induced by ROS to increase saline–alkali tolerance in M. halliana.
In this study, sucrose signaling also mediated auxin biosynthesis in M. halliana. Auxins, of which indole-3-acetic acid (IAA) is the main active form in higher plants, regulate many aspects of plant growth and development, including seed germination, root architecture, and leaf formation77,78. However, several recent studies have highlighted that auxin also regulates plant responses to stresses, such as drought and salinity tolerance79. We observed that the expression of T31B5_170, an auxin-responsive GH3 family protein, was significantly upregulated under saline–alkali stress and was involved in IAA synthesis (Fig. 5, Table 5). GH3 family proteins play critical roles in the regulation of auxin homeostasis80. In Arabidopsis, several members of the GH3 protein family activate IAA by adenylation81. Recent studies have demonstrated that auxin homeostasis under alkaline stress contributes to the adaptation of citrus to alkaline stress. These results indicated that T31B5_170 may induce the auxin signaling pathway that enhanced the resistance of M. halliana to saline–alkali stress.
Conclusions
In summary, saline–alkali stress induced the accumulation of ROS. To adapt to stress, M. halliana relied on signals and pathways to re-establish the homeostasis of ROS. The upregulated expression of photosynthesis-related proteins enhanced the capacity of photosynthesis in M. halliana, which contributed to maintaining the photosynthesis level to cope with saline–alkali stress. Sucrose acted as a signaling molecule and directly mediated amino acid metabolism; phenylpropanoid metabolism; and indole, tropane, piperidine, and pyridine alkaloid metabolism. These pathways play a central role in maintaining osmotic balance and removing ROS. In addition, sucrose signaling induced flavonoid biosynthesis by activating the expression of CYP75B1 to regulate the homeostasis of ROS and promoted auxin signaling by activating the expression of T31B5_170 to enhance the resistance of M. halliana to saline–alkali stress. The decrease in PER and ALDH2C4 during lignin synthesis further triggered the plant saline–alkali response (Fig. 8).

Signal transduction of ROS during the plant response to saline–alkali stress
This is the first study that has used an integrated approach to determine the possible molecular mechanisms of how M. halliana adapts to saline–alkali environments. Moreover, this study provides the basis for an improved understanding of saline–alkali tolerance responses in apples and provides an important starting point for future analyses. Thus, identifying specific saline–alkali stress sensors/receptors and exploring their underlying mechanisms via genetic engineering remain exciting challenges.
Materials and methods
Plant material and stress treatments
The experiment was carried out at Gansu Agricultural University (Gansu Province, China) in May 2018. M. halliana seeds were sterilized with 0.2% KMnO4 for 30 min and then washed with running water for 12 h. Subsequently, seeds in sand were germinated at 4 °C for 35 days and shown in 16.8-cm-diameter plastic pots that contained 2.5 kg of nutrient soil. Healthy seedlings with eight true leaves were selected and transferred to foam boxes containing Hoagland’s solution (pH = 6.8 ± 0.2)7. Five seedlings were grown in each box. The nutrient solution was changed every 3 days, and oxygen was continuously ventilated by an electric ventilation pump during the cultivation period. After 12 days, ten boxes with uniform seedlings were selected and subjected to saline–alkali stress, which involved treatment with Hoagland’s solution supplemented with 100 mM NaCl + NaHCO3 (1:1 M ratio of NaCl:NaHCO3) (pH = 8.2). To avoid any salt-shock reaction, an increase of 25 mM concentration per day was applied at the beginning of the stress. All physiological parameters were measured after reaching the 100 mM concentration.
Determination of photosynthetic parameters
The photosynthetic characteristics of functional leaves at 0, 1, 2, and 4 days were measured from 9:00 to 11:00 a.m. using an LI-6400XT portable open-flow gas-exchange system (Li-COR Biosciences, Lincoln, USA). After 30 min of the darkness treatment, we used an Imaging-Pam Chl fluorimeter and Imaging WinGegE software (Walz, Effeltrich, Germany) to determine the chlorophyll (Chl) fluorescence parameters of leaves. The Chl content was extracted with 10 mL of acetone for 48 h in darkness and measured at 440, 645, and 663 nm spectrophotometrically. The calculations used the methods of Lichtenthaler82.
iTRAQ analysis methods
Protein extraction, protein digestion, and iTRAQ labeling
Proteins were extracted from three biological replicates per treatment, and 0.1 g of frozen leaf tissue was placed into a cold mortar. A mortar and pestle were used to grind tissue into a fine powder, and 1 mL of phenol extraction buffer was added to incubate at room temperature for 10 min. Then, 1 mL of phenol saturated was added, and the mixture was shaken for 40 min at 4 °C. Tubes were centrifuged at 5000 g for 15 min at 4 °C, and the upper phenolic phase was collected. Subsequently, cold 0.1 M ammonium acetate–methanol solution was added at −20 °C using five volumes of the collected phenolic phase. The sediment was collected after centrifugation at 12,000 g for 10 min at 4 °C. Then, the sediment was collected, repeating this step one more time, and dried at room temperature for 2 min. Subsequently, the sediment was resuspended in 300 μL of lysate solution for 3 h. Finally, the supernatant was the extracted protein solution after centrifugation of mixtures at 12,000 g for 10 min at room temperature. Measurements of protein were performed by the Bradford method83. iTRAQ labeling was performed according to the FASP method84.
Mass spectrometry analysis
All samples were analyzed by a Triple TOF 5600 mass spectrometer (SCIEX, USA). The flow rate of the Eksigent nanoLC-1D plus system (SCIEX, USA) was 300 nL/min, and the linear gradient was 90 min (from 5 to 85% B over 67 min; mobile phase A = 2% ACN/0.1% FA and B = 95% ACN/0.1% FA). A rolling collision energy voltage was used for CID fragmentation for MS/MS spectra acquisitions. Mass was dynamically excluded for 22 s.
Bioinformatics analysis
To analyze the functional characteristics of the selected DEPs, GO functional annotations and enrichment analysis of the proteins were performed by using the cloud platform of OmicsBean. For this analysis, the online Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp/) was used to classify the identified proteins. In addition, noncommercial databases, including metabolite pathways, were searched on KEGG (http://www.genome.jp/KEGG/pathway.html).
LC/MS untargeted metabolomics analysis methods
Sample preparation
A total of 80 mg of accurately weighed sample was transferred to a 1.5-mL Eppendorf tube. Then, 1 mL of a mixture of methanol and water (7/3, vol/vol) was added to each tube, and 20 μL of 2-chloro-l-phenylalanine (0.3 mg/mL) was dissolved in methanol to serve as the internal standard. Samples were ground at 60 Hz for 2 min, ultrasonicated at room temperature for 30 min after vortexing, and then placed at 4 °C for 10 min. After centrifugation at 13,000 rpm at 4 °C for 15 min, the supernatants (200 μL) from each tube were collected.
LC/MS analysis
LC/MS analysis was performed using an Acquity UHPLC system (Waters Corporation, Milford, USA) coupled with an AB Sciex Triple TOF 5600 System (AB Sciex, Framingham, MA). Data acquisition was conducted in full-scan mode (the m/z ranged from 70 to 1000) in combination with information-dependent acquisition mode. The parameters were set as follows: ion spray voltage, 5500 V (+) and 4500 V (−); ion source temperature, 550 °C (+) and 550 °C (−); collision energy, 10 eV (+) and −10 eV (−); curtain gas of 35 PSI; interface heater temperature, 550 °C (+) and 600 °C (−).
Data preprocessing and statistical data analysis
The raw data were converted to common data format (mzML) files using the software program MSconverter, and metabolomic data were obtained using the software XCMS 1.50.1 version. The positive and negative data were combined to obtain a combined data set that was imported into the SIMCA software package (version 14.0, Umetrics, Umeå, Sweden). All samples were tested to visualize the metabolic alterations by principal component analysis and (orthogonal) partial least-squares-discriminant analysis (O)PLS-DA. Variable importance in the projection (VIP) ranks the overall contribution of each variable to the OPLS-DA model, and those variables with VIP >1 were considered relevant for group discrimination. Reference material databases built by Dalian Institute of Chemical Physics, Chinese Academy of Sciences, and Dalian ChemData Solution Information Technology Co., Ltd., HMDB and METLIN, were used. In addition, metabolite pathways were searched on noncommercial databases (KEGG, http://www.genome.jp/KEGG/pathway.html).
Quantitative real-time PCR
The expression of 16 proteins was measured by performing real-time quantitative PCR (qRT-PCR). cDNA was synthesized from total RNA using the PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) (TaKaRa, Dalian, China) according to the manufacturer’s instructions. GAPDH was used as an internal control gene and qRT-PCR method based on Pan et al.85. The qRT-PCR analysis of each sample was performed in triplicate. Primers for the quantitative real-time PCR are listed in Table 7.
Statistical analysis
Statistical differences were tested by analyses of variance, and the mean differences were compared using Duncan’s test (P < 0.05). Statistical analyses were analyzed using SPSS (version 22.0, IBM, Armonk, NY, USA), and figures were made using Origin 9.1 software (OriginLab, Hampton, MA, USA).
References
Martínez-Cuenca, M. R., Iglesias, D. J., Forner Giner, M. A., Primo-Millo, E. & Legaz, F. The effect of sodium bicarbonate on plant performance and iron acquisition system of FA-5 (Forner-Alcaide 5) citrus seedlings. Acta Physiol. Plant. 35, 2833–2845 (2013).
Jin, H. et al. Comparative EST profiles of leaf and root of Leymus chinensis, a xerophilous grass adapted to high pH sodic soil. Plant Sci. 170, 1081–1086 (2006).
Guo, R. et al. Comparative metabolic responses and adaptive strategies of wheat (Triticum aestivum) to salt and alkali stres. BMC Plant Biol. 15, 170–182 (2015).
Guo, R. et al. Ionomic and metabolic responses to neutral salt or alkaline salt stresses in maize (Zea mays L.) seedlings. BMC Plant Biol. 17, 41–53 (2017).
Kawanabe, S. & Zhu, T. C. Degeneration and conservation of aneurolepidium chinense grassland in Northern China. J. Jpn. Grassl. Sci. 37, 91–99 (1991).
Martínez-Ballesta, M. C., Alcaraz-López, C., Muries, B., Mota-Cadenas, C. & Carvajal, M. Physiological aspects of rootstock–scion interactions. Sci. Hortic. 127, 112–118 (2010).
Jia, X. M. et al. Comparative physiological responses and adaptive strategies of apple Malus halliana to salt, alkali and saline–alkali stress. Sci. Hortic. 245, 154–162 (2018).
Hasegawa, P. M., Bressan, R. A., Zhu, J. K. & Bohnert, H. J. Plant cellular and molecular responses to high salt. Annu. Rev. Plant Physiol. Plant Mol. Biol. 51, 463–499 (2000).
Yu, J. et al. Physiological and proteomic analysis of salinity tolerance in Puccinellia tenuiflora. J. Proteome Res. 10, 3852–3870 (2011).
Fricke, W., Akhiyarova, G., Veselov, D. & Kudoyarova, G. Rapid and tissue-specific changes in ABA and in growth rate response to salinity in barley leaves. J. Exp. Bot. 55, 1115–1123 (2004).
Asha, K. et al. Proteomics, metabolomics, and ionomics perspectives of salinity tolerance in halophytes. Front. Plant Sci. 6, 537–556 (2015).
Li, Q., Yang, A. & Zhang, W. H. Efficient acquisition of iron confers greater tolerance to saline–alkaline stress in rice (Oryza sativa L.). J. Exp. Bot. 22, 6431–6444 (2016).
Yang, Y. & Guo, Y. Unraveling salt stress signaling in plants. J. Integr. Plant Biol. 9, 796–804 (2018).
Bose, J., Rodrigo-Moreno, A. & Shabala, S. ROS homeostasis in halophytes in the context of salinity stress tolerance. J. Exp. Bot. 65, 1241–1257 (2013).
Gupta, B. & Huang, B. Mechanism of salinity tolerance in plants: physiological, biochemical, and molecular characterization. Int. J. Genom. 2014, 1–18 (2014).
Zhang, H. et al. Mechanisms of plant salt response: insights from proteomics. J. Proteome Res. 11, 49–67 (2012).
Zhang, H. X., Hodson, J. N., Williams, J. P. & Blumwald, E. Engineering salt-tolerant Brassica plants: characterization of yield and seed oil quality in transgenic plants with increased vacuolar sodium accumulation. Proc. Natl Acad. Sci. USA. 98, 12832–12836 (2001).
Li, G., Li, J., Hao, R. & Guo, Y. Activation of catalase activity by a peroxisome-localized small heat shock protein Hsp17.6CII. J. Genet. Genom. 44, 395–404 (2017).
Perez-Salamo, I. et al. The heat shock factor A4A confers salt tolerance and is regulated by oxidative stress and the mitogen-activated protein kinases MPK3 and MPK6. Plant Physiol. 165, 319–334 (2014).
Yan, S. P., Tang, Z. C., Su, W. A. & Sun, W. N. Proteomic analysis of salt stress-responsive proteins in rice root. Proteomics 5, 235–244 (2005).
An, J. P. et al. MdbHLH93, an apple activator regulating leaf senescence, is regulated by ABA and MdBT2 in antagonistic ways. New Phytol. 222, 735–751 (2019).
Mishra, N. S., Tuteja, R. & Tuteja, N. Signaling through MAP kinase networks in plants. Arch. Biochem. Biophys. 452, 55–68 (2006).
Yuan, F. et al. OSCA1 mediates osmotic-stress-evoked Ca2+ increases vital for osmosens-ing in Arabidopsis. Nature 514, 367–371 (2014).
Hamilton, E. S. Mechanosensitive channel MSL8 regulates osmotic forces during pollen hydration and germination. Science 350, 438–441 (2015).
Wang, F., Jing, W. & Zhang, W. The mitogen-activated protein kinase cascade MKK1-MPK4 mediates salt signaling in rice. Plant Sci. 227, 181–189 (2014).
Blumwald, E. Engineering salt tolerance in plants. Curr. Opin. Biotechnol. 13, 261–275 (2003).
Sanchez, D. H. Comparative ionomics and metabolomics in extremophile and glycophytic Lotus species under salt stress challenge the metabolic pre-adaptation hypothesis. Plant Cell Environ. 34, 605–617 (2011).
Han, X. et al. Abioassay-guided fractionation system to identify endogenous small molecules that activate plasma membrane H+-ATPase activity in Arabidopsis. J. Exp. Bot. 68, 2951–2962 (2017).
Sun, M. H. et al. The glucose sensor MdHXK1 phosphorylates a tonoplast Na+/H+ exchanger to improve salt tolerance. Plant Physiol. 176, 01472.2017 (2018). pp.
Dasgupta, K. et al. Expression of sucrose transporter cDNAs specifically in companion cells enhances phloem loading and long-distance transport of sucrose but leads to an inhibition of growth and the perception of a phosphate limitation. Plant Physiol. 165, 715–731 (2014).
Yang, Y. & Guo, Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 217, 523–539 (2018b).
Fan, W. Q. et al. Proteomics integrated with metabolomics: analysis of the internal causes of nutrient changes in alfalfa at different growth stages. BMC Plant Biol. 18, 78–92 (2018).
Bassel, G. W. Systems analysis of plant functional, transcriptional, physical interaction, and metabolic networks. Plant Cell. 24, 3859–3875 (2012).
Iqbal, M. & Ashraf, M. Changes in hormonal balance: a possible mechanism of pre-sowing chilling-induced salt tolerance in spring wheat. J. Agron. Crop Sci. 96, 440–454 (2010).
Abbasi, G. H. et al. Potassium application mitigates salt stress differentially at different growth stages in tolerant and sensitive maizehybrids. Plant Growth Regul. 76, 111–125 (2015).
Sultana, N., Ikeda, T. & Itoh, R. Effect of NaCl salinity on photosynthesis and dry matter accumulation in developing rice grains. Environ. Exp. Bot. 42, 211–220 (1999).
Demmig-Adams, B. & Adams, W. W. III: Photoprotection and other responses of plants to high light stress. Annu. Rev. Plant Physiol. Plant Mol. Biol. 43, 599–626 (1992).
Fahnenstich., H., Scarpeci, T. E., Valle, E. M., Flugge, U. I. & Maurino, V. G. Generation of hydrogen peroxide in chloroplasts of Arabidopsis overexpressing glycolate oxidase as an inducible system to study oxidative stress. Plant Physiol. 148, 719–729 (2008).
Affenzeller, M. J., Darehshouri, A., Andosch, A., Lutz, C. & Lutz-Meindl, U. Salt stress-induced cell death in the unicellular green alga Micrasterias denticulata. J. Exp. Bot. 60, 939–954 (2009).
Tanaka, R., Hirashima, M., Satoh, S. & Tanaka, A. The A rabidopsis-accelerated cell death gene ACD1 is involved in oxygenation of pheophorbide a: inhibition of the pheophorbide a oxygenase activity does not lead to the “stay-green” phenotype in Arabidopsis. Plant Cell Physiol. 44, 1266–1274 (2003).
Zuo, Z., Chen, Z. & Zhu, Y. Effects of NaCl and Na2CO3 stresses on photosynthetic ability of Chlamydomonas reinhardtii. Biologia 69, 1314–1322 (2014).
Wang, Y. et al. Transcriptional and physiological analyses of short-term iron deficiency response in apple seedlings provide insight into the regulation involved in photosynthesis. BMC Genom. 19, 461–472 (2018).
Xue, H. et al. PSBR is required for efficient binding of LHCSR3 to photosystem II-light-harvesting supercomplexes in Chlamydomonas reinhardtii. Plant Physiol. 167, 1566–1578 (2015).
Yi, X. P., Mcchargue, M., Laborde, S., Frankel, L. K. & Bricker, T. M. The manganese-stabilizing protein is required for photosystem II assembly/stability and photoautotrophy in higher plants. J. Biol. Chem. 280, 16170–16174 (2005).
Allahverdiyeva, Y. et al. Comparison of the electron transport properties of the psbo1 and psbo2 mutants of Arabidopsis thaliana. Biochim. Biophys. Acta (BBA) - Bioenerg. 1787, 1230–1237 (2009).
Suorsa, M. et al. PsbR, a missing link in the assembly of the oxygen-evolving complex of plant photosystem II. J. Biol. Chem. 281, 145–150 (2006).
Tada, Y. & Kashimura, T. Protection analysis of salt-responsive proteins in the mangrove plant, Bruguiera gymnorhiza. Plant Cell Physiol. 50, 439–446 (2009).
Nishiyama., Y., Allakhverdiev, S. I. & Murata, N. A new paradigm for the action of reactive oxygen species in the photoinhibition of photosystem II. Biochim. Biophys. Acta (BBA) - Bioenerg. 1757, 742–749 (2006).
Singh, M., Kumar, J., Singh, S., Singh, V. P. & Prasad, S. M. Roles of osmoprotectants in improving salinity and drought tolerance in plants: a review. Rev. Environ. Sci. Bio-technol. 14, 407–426 (2015).
Barber, C. et al. Distinct properties of the five UDP-D-glucose/UDP-D-galactose 4-epimerase isoforms of Arabidopsis thaliana. J. Biol. Chem. 281, 17276–17285 (2006).
Lin, X. Y., Ye, Y. Q., Fan, S. K., Jin, C. W. & Zheng, S. J. Increased sucrose accumulation regulates iron-deficiency responses by promoting auxin signaling in Arabidopsis plants. Plant Physiol. 170, 907–920 (2016).
Park, H. J., Kim, W. Y. & Yun, D. J. A new insight of salt stress signaling in plant. Mol. Cell. 39, 447–459 (2016).
Qian, Y. et al. Effects of vitro sucrose on quality components of tea plants (Camellia sinensis) based on transcriptomic and metabolic analysis. BMC Plant Biol. 18, 121–140 (2018).
Barpeled, M. & O’Neill, M. A. Plant nucleotide sugar formation, interconversion, and salvage by sugar recycling. Annu Rev. Plant Biol. 62, 127–155 (2011).
Shu, L. et al. Genetic, proteomic and metabolic analysis of the regulation of energy storage in rice seedlings in response to drought. Proteomics 11, 4122–4138 (2011).
Sobhanian, H., Motamed, N., Jazii, F. R., Nakamura, T. & Komatsu, S. Salt stress induced differential proteome and metabolome response in the shoots of Aeluropus lagopoides (Poaceae), a halophyte C4 plant. J. Proteome Res. 9, 2882–2897 (2010).
Cramer, G. R. et al. Proteomic analysis indicates massive changes in metabolism prior to the inhibition of growth and photosynthesis of grapevine (Vitis vinifera L.) in response to water deficit. BMC Plant Biol. 13, 49–70 (2013).
Höper, D., Bernhardt, J. & Hecker, M. Salt stress adaptation of Bacillus subtilis: a physiological proteomics approach. Proteomics 6, 1550–1562 (2006).
Wang, L. et al. Metabolomic and proteomic profiles reveal the dynamics of primary metabolism during seed development of Lotus (Nelumbo nucifera). Front. Plant Sci. 7, 750–761 (2016).
Less, H. & Galili, G. Principal transcriptional programs regulating plant amino acid metabolism in response to abiotic stresses. Plant Physiol. 147, 316–330 (2008).
Fernie, A. R., Tiessen, A., Stitt, M., Willmitzer, L. & Geigenberger, P. Altered metabolic fluxes result from shifts in metabolite levels in sucrose phosphorylase expressing potato tubers. Plant Cell Environ. 25, 1219–1232 (2002).
Cheng, L. X. et al. Comparative proteomics illustrates the complexity of drought resistance mechanisms in two wheat (Triticum aestivum L.) cultivars under dehydration and rehydration. BMC Plant Biol. 16, 188–210 (2016).
Watanabe, M. et al. Comprehensive dissection of spatiotemporal metabolic shifts in primary, secondary, and lipid metabolism during developmental senescence in Arabidopsis. Plant Physiol. 162, 1290–1310 (2013).
Pi, E. et al. Quantitative phosphoproteomic and metabonomic analyses reveal GmMYB173 optimizes flavonoid metabolism in soybean under salt stress. Mol. Cell. Proteom. 17, mcp.RA117.000417 (2018).
Petkovšek, M. M., Štampar, F. & Veberič, R. Accumulation of phenolic compounds in apple in response to infection by the scab pathogen, Venturia inaequalis. Physiol. Mol. Plant Pathol. 74, 60–67 (2010).
Dubos, C. G. J. et al. MYBL2 is a new regulator of flavonoid biosynthesis in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 55, 940–953 (2008).
Li, X., Bonawitz, N. D., Weng, J. K. & Chapple, C. The growth reduction associated with repressed lignin biosynthesis in Arabidopsis thaliana is independent of flavonoids. Plant Cell. 22, 1620–1632 (2010).
Parrotta., L., Guerriero, G., Sergeant, K., Cai, G. & Hausman, J. F. Target or barrier? The cell wall of early- and later-diverging plants vs cadmium toxicity: differences in the response mechanisms. Front. Plant Sci. 6, 133–148 (2015).
Gallego-Giraldo, L., Jikumaru, Y., Kamiya, Y., Tang, Y. & Dixon, R. A. Selective lignin downregulation leads to constitutive defense response expression in alfalfa (Medicago sativa L.). New Phytol. 190, 627–639 (2011).
Novaes, E., Kirst, M., Chiang, V., Winter-Sederoff, H. & Sederoff, R. Lignin and biomass: a negative correlation for wood formation and lignin content in trees. Plant Physiol. 154, 555–561 (2010).
Xu, Y. et al. Proteomic analysis of heat stress resistance of cucumber leaves when grafted onto Momordica rootstock. Hortic. Res. 5, 53 (2018).
Hussein, M. M., Faham, S. Y. & Alva, A. K. Role of foliar application of nicotinic acid and tryptophan on onion plants response to salinity stress. J. Agric. Sci. 6, 41–51 (2014).
Rai, V. K. Role of amino acids in plant responses to stresses. Biol. Plant. 45, 481–487 (2002).
Zhu, W. et al. Binary stress induces an increase in indole alkaloid biosynthesis in Catharanthus roseus. Front. Plant Sci. 6, 582–593 (2015).
Li, C. Y. et al. The ORCA2 transcription factor plays a key role in regulation of the terpenoid indole alkaloid pathway. BMC Plant Biol. 13, 155–171 (2013).
Gao, C., Yang, B., Zhang, D., Chen, M. & Tian, J. Enhanced metabolic process to indole alkaloids in Clematis terniflora, DC. after exposure to high level of UV-B irradiation followed by the dark. BMC Plant Biol. 16, 231–246 (2016).
Zhao, Y. Auxin biosynthesis and its role in plant development. Annu. Rev. Plant Biol. 61, 49–64 (2010).
Liu, X. Auxin controls seed dormancy through stimulation of abscisic acid signaling by inducing ARF-mediated ABI3 activation in Arabidopsis. Proc. Natl Acad. Sci. USA. 110, 15485–15490 (2013).
Verma, V., Ravindran, P. & Kumar, P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 16, 86–95 (2016).
Staswick, P. E., Tiryaki, I. & Rowe, M. L. Jasmonate response locus JAR1 and several related Arabidopsis genes encode enzymes of the firefly luciferase superfamily that show activity on jasmonic, salicylic, and indole-3-acetic acids in an assay for adenylation. Plant Cell 14, 1405–1415 (2002).
Park, J. E. et al. GH3-mediated auxin homeostasis links growth regulation with stress adaptation response in Arabidopsis. J. Biol. Chem. 282, 10036–10046 (2007).
Lichtenthaler, H. K. Chlorophylls and carotenoids: pigments of photosynthetic biomembranes. Methods Enzymol. 148C, 350–382 (1987).
Smith, P. K. et al. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 (1985).
Wisniewski, J. R. et al. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
Pan, L., Gao, H., Xia, W., Zhang, T. & Dong, L. Establishing a herbicide-metabolizing enzyme library in Beckmannia syzigachne to identify genes associated with metabolic resistance. J. Exp. Bot. 67, 1745–1757 (2016).
Acknowledgements
This work was supported by the Special Fund for Discipline Construction of Gansu Agricultural University (GSAU-XKJS-2018-222) and Gansu Provincial Natural Fund (18JR3RA171). We thank Shanghai Luming Biotechnology Co., Ltd. for its valuable effort in data processing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jia, Xm., Zhu, Yf., Hu, Y. et al. Integrated physiologic, proteomic, and metabolomic analyses of Malus halliana adaptation to saline–alkali stress. Hortic Res 6, 91 (2019). https://doi.org/10.1038/s41438-019-0172-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-019-0172-0
This article is cited by
-
Exogenous melatonin strengthens saline-alkali stress tolerance in apple rootstock M9-T337 seedlings by initiating a variety of physiological and biochemical pathways
Chemical and Biological Technologies in Agriculture (2024)
-
Functional identification of CCR1 gene in apple (Malus halliana) demonstrates that it enhances saline-alkali stress tolerance
Chemical and Biological Technologies in Agriculture (2024)
-
Regulatory effects of silicon nanoparticles on the growth and photosynthesis of cotton seedlings under salt and low-temperature dual stress
BMC Plant Biology (2023)
-
Investigation of the response of Platycodongrandiflorus (Jacq.) A. DC to salt stress using combined transcriptomics and metabolomics
BMC Plant Biology (2023)
-
Transcriptome and Metabolome Integrated Analysis Reveals the Mechanism of Cinnamomum bodinieri Root Response to Alkali Stress
Plant Molecular Biology Reporter (2023)
