
Abstract
Drought stress limits the growth and development of grapevines, thereby reducing productivity, but the mechanisms by which grapevines respond to drought stress remain largely uncharacterized. Here, we characterized a group A bZIP gene from “Kyoho” grapevine, VlbZIP30, which was shown to be induced by abscisic acid (ABA) and dehydration stress. Overexpression of VlbZIP30 in transgenic Arabidopsis thaliana enhanced dehydration tolerance. Transcriptome analysis revealed that a major proportion of ABA-responsive and/or drought-responsive genes are transcriptionally regulated by VlbZIP30 during ABA or mannitol treatment at the cotyledon greening stage. We identified an A. thaliana G-box motif (CACGTG) and a potential grapevine G-box motif (MCACGTGK) in the promoters of the 39 selected A. thaliana genes upregulated in the transgenic plants and in the 35 grapevine homologs, respectively. Subsequently, using two grapevine-related databases, we found that 74% (23/31) and 84% (21/25) of the detected grapevine genes were significantly upregulated by ABA and drought stress, respectively, suggesting that these genes are involved in ABA or dehydration stress and may be regulated by VlbZIP30 in grapevine. We propose that VlbZIP30 functions as a positive regulator of dehydration-responsive signaling in the ABA core signaling pathway.
Similar content being viewed by others

Introduction
Grapevines are among the world’s major fruit crops, and their fruits can be consumed fresh or dried, be processed into wines, spirits, and vinegar, or be transformed into pharmaceutical products that promote human health1. However, abiotic stress, such as drought, perturb the metabolism and growth of grapevines, leading to a loss of yield and reduced fruit quality2. Consequently, increasing the resistance of grapevines to drought stress is an important factor in ensuring yield stability.
Stress signaling in plants can be transduced by various signaling components, including second messengers, signal transduction factors, hormones such as abscisic acid (ABA), and transcription factors (TFs). Such signaling associated with drought has been shown to cause changes in physiological, morphological, and molecular processes, including the activation of many drought-related genes and the accumulation of a range of proteins, reflecting a drought stress response3.
ABA is considered to be a stress hormone and is associated with drought tolerance, as well as involved in various developmental processes, including seed germination and seedling growth3,4. In the context of a drought response it has been shown to mediate stomatal closure and to promote cuticular wax biosynthesis5. ABA-mediated drought tolerance involves complex signaling networks, the core components of which have been identified. Briefly, when ABA is present, it binds to the ABA receptors PYR/PYL/RCAR (PYRABACTIN RESISTANCE1/PYR1-like/REGULATORY COMPONENT OF ABA RECEPTOR1), which interact with the PP2C (PROTEIN PHOSPHATASE 2C) proteins, forming a complex and releasing the inhibitory effect of PP2Cs on SnRK2 (SUCROSE-NONFERMENTING1-RELATED PROTEIN KINASE2) protein kinases. The activated SnRK2s proteins subsequently phosphorylate different downstream TFs, such as AREB1 (ABA-RESPONSE-ELEMENT BINDING1) and ABI5 (ABA INSENSITIVE5), which regulate the expression of ABA-responsive genes6,7.
TFs are generally identified according to conserved sequences, known as the DNA-binding domains. One of the largest TF families in higher plants is the bZIP family, members of which are characterized by a basic region/leucine zipper domain8. Previous studies have shown that bZIP proteins function as regulators of signaling networks by specifically binding cis-elements containing a core ACGT, such as the ABA-responsive element (ABRE; PyACGTGGC), the G-box (CACGTG), and the C-box (GACGTC)9,10, in the promoters of their target genes, to either activate or repress their expression11.
A number of studies have shown that bZIP TFs are important regulators of drought stress signaling. The involvement of bZIPs (ABF1, AREB1/ABF2, ABF3, AREB2/ABF4) in the regulation of drought responses was first reported in the model plant Arabidopsis thaliana12,13,14. Following these studies, drought-related bZIP genes have been identified in a range of other species, including OsABI5 in rice (Oryza sativa)15, LIP19 in wheat (Triticumaestivum)16, ABP9 in maize (Zea mays)17, and so on.
In order to improve the drought resistance of grapevines, researchers have focused their attention on the identification of drought-related TFs. Several, such as WRKY11, ERF1/2/3, and NAC26, have been identified and their overexpression in A. thaliana has been shown to enhance drought resistance18,19,20. However, to date, only a few grapevine bZIP TFs have been functionally characterized during a drought stress response21,22,23,24, and their regulatory mechanisms are not well understood.
Early studies indicated that the bZIP TFs (especially group A) play an important role in the ability of plants to resist abiotic stresses25. Hereafter, we identified 47 bZIP genes in the grape genome, and of these it was found that the expression profile of bZIP30 (a group A bZIP TF) was upregulated in response to drought conditions23, suggesting that it may be associated with tolerance to drought stress. In this current study, we cloned the VlbZIP30, from “Kyoho” grapevine and ectopically expressed it in A. thaliana. The results of physiological and transcriptomic analyses of the transgenic lines are presented and its putative function in dehydration-responsive signaling via the ABA signaling pathway in grapevine is discussed.
Materials and methods
Plant material and growth conditions
Two-year-old “Kyoho” grapevine (Vitis labrusca × V. vinifera) plants used in this study were grown in the grapevine germplasm resource orchard of the Northwest A&F University, Yangling, Shaanxi, China (34°20′N, 108°24′E). The “Kyoho” grapevine was a Chinese variety from number 24635 in the Vitis International Variety Catalog. A. thaliana ecotype Columbia (Col-0) plants used as both wild-type (WT) and for transgenic experiments were grown in a greenhouse at 21 °C under long-day conditions (16 h light/8 h dark).
Dehydration stress and ABA treatment of grapevine leaves
For dehydration treatments, grapevine shoots with three well-developed leaves were detached and immediately placed on dry filter paper in an illumination incubator with 16 h fluorescent light (12,000 lux)/8 h dark photoperiod at 25 °C, with a relative humidity of 60–70%. For ABA treatments, leaves were sprayed with 100 μM ABA (MP Biomedicals, LLc) while the shoots were immersed in water about 8 cm, and the plants were then placed under the same ambient conditions as above. Leaves from the same position were collected from three independent replicates of each treatment at 1, 2, 4, 6, 9, 12, and 24 h after initiating treatment. The 0 h samples were collected before each treatment was initiated and used as control samples according to the method described previously20. All samples were immediately frozen in liquid nitrogen and stored at −80 °C until further analysis.
Bioinformatic analysis
Full-length amino acid sequences of bZIPs from A. thaliana and grapevine were obtained from The Arabidopsis Information Resource (TAIR; http://www.arabidopsis.org/index.jsp) and EnsemblPlants (http://plants.ensembl.org/index.html), respectively. Multiple amino acid sequence alignments were generated using DNAMAN software (Version 5.2.2.0, LynnonBiosoft, USA) with default parameters, and a phylogenetic tree was constructed using the neighbor-joining method and MEGA software (version 5.05), with 1000 bootstrap replicates, as previously described21. The predicted phosphorylation sites (C1, C2, C3, and C4) and highly conserved bZIP domain were analyzed as previously described12.
Transformation and characterization of transgenic plants
The plant transformation vectors 35S:VlbZIP30 and ProVlbZIP30:GUS (β-glucosidase, details of vector construction are supplied in Supplementary Method S1) were transformed into A. thaliana by the floral dip method using Agrobacterium tumefaciens (strain GV3101)26. T3 homozygous lines from three independent transgenic lines were analyzed.
Histochemical GUS assay
An in situ GUS activity assay was performed as previously described21.
Osmotic stress and ABA treatment of transgenic seedlings
WT and transgenic seeds were harvested at the same time. For seed germination and cotyledon greening analyses, approximately 100 seeds from WT and each 35S:VlbZIP30 line (OE1, OE6, and OE23) were grown on Murashige–Skoog (MS) medium27 (Sigma) solidified with 0.7% agar containing 2% sucrose with or without mannitol (300 and 350 mM) or ABA (0.5 and 1 μM), at 21 °C with a 16 h light/8 h dark cycle. Germination and cotyledon greening rates were defined as the obvious emergence of the seedling radicle through the seed coat and green coloration of cotyledons, respectively. The seedlings were sampled after counting to measure the endogenous ABA concentration.
For the osmotic stress and ABA treatments, 7-day-old WT and transgenic seedlings were transferred from MS medium plates into MS agar medium, or MS agar medium supplemented with 300 or 350 mM mannitol, or MS agar medium supplemented with 50 or 100 μM ABA. The root lengths were measured 7 days after the transfer. The relative electrolyte leakage and malondialdehyde (MDA) content were measured as previously described21, as was ABA concentration22.
Transcriptome analysis and identification of differentially expressed genes (DEGs)
Seeds from WT and transgenic lines were cultivated on MS agar medium, with or without stress treatment (0.5 μM ABA or 300 mM mannitol) for 7 days, and collected for RNA extraction. For each RNA purification biological replicate, 300 seedlings of WT or the OE lines from three MS agar plates were pooled to form a single sample. Three independent RNA samples were used for each experiment.
Total RNA was extracted using the E.Z.N.A. Plant RNA Kit (Omega Bio-Tek, USA, R6827-01), according to the manufacturer’s protocol (Invitrogen). RNA concentration and integrity were confirmed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). The construction of RNA-Seq libraries and sequencing were performed by the Biomarker Biotechnology Corporation (Beijing, China). The libraries were generated using the NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA) following the manufacturer’s recommendations. Sequencing of the purified libraries was carried out using an Illumina HiseqXten platform (Illumina, NEB, USA) generating paired-end reads. The raw reads were cleaned by removing reads containing adapter sequences, reads containing poly-N, and low-quality reads. The cleaned reads from each sample were aligned to the A. thaliana reference genome from TAIR using the Tophat2 software28. Gene expression levels were determined by fragments per kilobase of transcript per million fragments mapped (FPKM), and the DEGs were identified using edgeR software29, with a threshold of false discovery rate (FDR) < 0.05 and absolute log2FC (fold change) > 1. All raw sequence data in this study have been submitted to the NCBI Short Read Archive under BioProject accession number PRJNA419694.
Transcriptome data analysis
The Venn diagrams were made using the BMK Cloud platform (www.biocloud.net). Annotations for DEGs were retrieved from TAIR. Gene ontology (GO) enrichment analyses were performed for the functional categorization of DEGs based on the PageMan profiling tool30 and Arabidopsis Functional Modules Supporting Data31. The grapevine orthologs of the A. thaliana genes were identified using TBLASTX software32 with the highest score. Motif predictions were performed using the promoter region 1500 bp upstream of the start codons of the A. thaliana (AT) and grapevine (VIT) genes using DREME software (http://meme-suite.org/tools/dreme). The heat maps were constructed using HemI software33. To identify the predicted grapevine genes, two grapevine-related ABA1 and drought stress34 databases were downloaded from the National Center for Biotechnology Information (NCBI) under BioProject accession number PRJNA369777 and the Gene Expression Omnibus database under the number GSE57669.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from the grapevine leaves after ABA and dehydration treatment using the E.Z.N.A._Plant RNA Kit (Omega Bio-Tek, USA, R6827-01) following the manufacturer’s instructions. The qRT-PCR was analyzed as previously described21. The expression levels of the grape ACTIN1 (VIT_04s0044g00580) or A. thaliana ACTIN2 (AT3G18780) genes were used as references. The specific primers for qRT-PCR are listed in Supplementary Table S1.
Statistical analysis
Data analysis was performed using Microsoft Excel (Microsoft Corporation, USA). The data were plotted using Sigmaplot (v. 10.0, Systat Inc., CA, USA). One-way ANOVA followed by Fisher’s least significant difference (LSD) or Student’s t-test analysis were performed to assess significant differences using the SPSS statistics 17.0 software (IBM China Company Ltd., Beijing, China). All experiments were repeated three times as independent analyses.
Results
Identification of VlbZIP30, a group A bZIP TF from grapevine
The VlbZIP30 (VIT_13s0175g00120) cDNA is 978 bp long and encodes a protein of 325 amino acids. Amino acid sequence analysis showed that, in common with the eight members of the A. thaliana ABF/DPBF bZIP subfamily, VlbZIP30 contains a basic leucine zipper domain25 and conserved domains predicted as phosphorylation sites (C1, C2, C3, and C4) involved in drought stress or ABA signaling12 (Supplementary Fig. S1). A phylogenetic analysis indicated that VlbZIP30 is most closely related to the group A ABF/DPBF TFs, which have previously been shown to be involved in ABA and drought stress signaling in A. thaliana12,13,14,35, and grapevine22,36 (Fig. 1a).

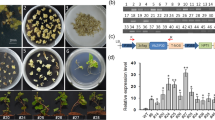
a The phylogenetic tree represents VlbZIP30 (black circle) and other bZIP amino acid sequences from Arabidopsis thaliana (AT) and grapevine (Vitis vinifera, VIT). The clustering of the group A bZIP proteins and the other groups of bZIP proteins (group H, AtHY5; I, AtbZIP29; E, AtbZIP34; L, AtbZIP76; S, AtGBF5; B, bZIP28; D, AtTGA6; J, AtbZIP62; K, AtbZIP60; F, AtbZIP19; and C, AtBZO2H2) have previously been reported25,36. b Expression profiles of VlbZIP30 in grapevine following abscisic acid (ABA) and dehydration treatments. Data represent the mean values ± SE from three independent experiments. Asterisks indicate statistical significance (*0.01 < P < 0.05, **P < 0.01, Student’s t-test) between the treated and untreated control plants. c Patterns of VlbZIP30 promoter-driven GUS (β-glucosidase) expression in A. thaliana at different growth stages. Mature embryos cultivated on Murashige–Skoog (MS) agar medium (a), or MS agar medium supplemented with 300 mM mannitol (b), or 0.5 μM ABA (c) for 2 days. Scale bar = 500 μm. Seven-day-old seedlings cultivated on MS agar medium (d), or MS agar medium supplemented with 300 mM mannitol (e), or 0.5 μM ABA (f) for 7 days. Scale bar = 500 μm. Fourteen-day-old seedlings transferred from MS medium plates into MS agar medium (g) or MS agar medium supplemented with 300 mM mannitol (h), or 100 μM ABA (i) for 7 days. Scale bar = 2 mm. 3-week-old plant (j). 3-week-old plant after dehydration for 2 h (k). Guard cells of 3-week-old plant (l). Scale bar = 50 μm. Guard cells of 3-week-old plant after dehydration for 2 h (m). Scale bar = 50 μm. Inflorescence (n). Scale bar = 2 mm. Leaf (o). Scale bar = 200 μm. Flower (p). Scale bar = 200 μm. Silique (q). Scale bar = 2 mm
Expression of VlbZIP30 is induced by drought and ABA treatment
To test whether VlbZIP30 is involved in ABA and drought stress signaling, we first evaluated the expression levels of VlbZIP30 in grapevine following ABA or dehydration treatments using qRT-PCR. As shown in Fig. 1b, ABA caused an increase in VlbZIP30 expression at 4 and 6 h shortly after initiation of the treatment. The expression peaked at 12 h, before decreasing for the next 24 h. Dehydration caused an increase in VlbZIP30 expression at 24 h.
Next, to investigate the temporal and spatial expression patterns of VlbZIP30 in more detail, histochemical GUS reporter experiments were performed with transgenic plants (ProVlbZIP30:GUS) grown under ABA and dehydration stress, as well as under normal conditions. Low levels of GUS staining were observed in 2-day seeds at the germination stage, as well as in 7 and 14-day-old seedlings (Fig. 1c, a, d, and g), and GUS activity was significantly enhanced after mannitol (Fig. 1c, b, e, and h) and ABA (Fig. 1c, c, f, and i) treatments at the same stages. In mature plants, GUS staining was obviously detected in stems, trichomes, flowers, and siliques (Fig. 1c, n–q), while only slight staining was detected in leaf petioles (Fig. 1c, j), and no staining was detected in guard cells (Fig. 1c, l). However, after dehydration for 2 h, the leaf petioles and guard cells showed an increase in GUS staining (Fig. 1c, k and m). The dehydration treatments had no effect on the size of the stomatal aperture.
Overexpressing VlbZIP30 in A. thaliana reduces mannitol and ABA sensitivity during seed germination and post-germination growth
Three homozygous transformed lines (OE1, OE6, and OE23) with the highest levels of VlbZIP30 expression were selected based on qRT-PCR analysis (Supplementary Fig. S2). Sterilized seeds of the transgenic lines and WT plants were cultivated on MS agar medium with or without mannitol (300 and 350 mM), or ABA (0.5 and 1 μM). The seed germination rates and the cotyledon greening rates of the transgenic lines were significantly higher than those of WT after 3 days (Supplementary Fig. S3A, B) and 7 days (Fig. 2a–d) under mannitol and ABA treatments, respectively. Given that ABA controls seed germination, and that its biosynthesis can be affected by abiotic stress37,38, we measured endogenous ABA concentrations. However, there was no significant difference in ABA concentrations between WT and transgenic lines under 350 mM mannitol and 1 μM ABA treatments at the seed germination stage (Supplementary Fig. S3C), as well as at the cotyledon greening stage (Fig. 2e).

a Greening cotyledons from WT and transgenic lines 7 days after seeds were cultivated on Murashige–Skoog (MS) agar medium, with or without 300 or 350 mM mannitol, or 0.5 or 1 μM ABA. b Cotyledon greening rates of WT and transgenic lines 7 days after cultivation on MS agar medium with or without 300 or 350 mM mannitol, or 0.5 or 1 μM ABA. c, d Cotyledon greening rates of WT and transgenic lines grown on MS basal medium containing 1 μM ABA (c) or 350 mM mannitol (d). e Endogenous ABA levels of WT and transgenic lines 7 days after cultivationon MS agar medium, or MS agar medium containing 350 mM mannitol or 1 μM ABA. Three independent experiments were performed with about 100 seeds per experiment. Data represent mean values ± SE from three independent experiments. Statistically significant differences are indicated by different lowercase letters according to Fisher’s LSD test (P < 0.05)
To further characterize morphological changes of WT and transgenic seedlings in response to the mannitol and ABA treatments, the various seed genotypes described above were grown on MS agar medium, with or without 300 mM mannitol, or 0.5 μM ABA for 14 days. As shown in Fig. 3a, the size of cotyledons and roots of the transgenic seedlings were significantly bigger and longer than those of WT following both treatments, while there was no differences observed under control conditions.

a Photographs of morphology in WT and transgenic lines 14 days after seeds were cultivated on Murashige–Skoog (MS) agar medium with or without 300 mM mannitol or 0.5 μM ABA. b Root length of WT and transgenic lines after 7 days of growth with or without mannitol (300 or 350 mM) or ABA (50 or 100 μM). c Photographs of 14-day-old seedlings transferred from MS agar medium to MS agar medium or MS agar medium supplemented with mannitol (300 or 350 mM) or ABA (50 or 100 μM) for 7 days. Electrolyte leakage (d) and malondialdehyde (MDA) content (e) of 14-day-old seedlings transferred from MS agar medium to MS agar medium or MS agar medium supplemented with mannitol (300 or 350 mM) or ABA (50 or 100 μM) for 7 days. In all cases, data represent mean values ± SE from three independent experiments. Statistically significant differences are indicated by different lowercase letters according to Fisher’s LSD test (P < 0.05)
To determine whether the transgenic lines had longer primary roots as a consequence of precocious germination or development of post-germination growth, 7-day-old transgenic lines and WT seedlings grown on MS agar medium were transferred to MS agar medium with or without mannitol or ABA, and grown for another 7 days. When grown on MS agar medium, the primary root lengths were similar; however, after treatments, the transgenic lines had relatively longer primary roots than WT seedlings under mannitol (300 and 350 mM) treatment, while there was no significant difference in response to the ABA (50 and 100 μM) treatment (Fig. 3b, c). This indicated that VlbZIP30 plays a role in suppressing the retardation of germination mediated by ABA, but not in root growth inhibition.
Then, we measured electrolyte leakage and MDA levels in the transgenic and WT seedlings after 300 or 350 mM mannitol treatment and saw that electrolyte leakage levels between the transgenic lines and WT were no significant difference, but the MDA levels in the transgenic lines were significantly lower than in WT, indicating that the degree of membrane and tissue damage was less as a result of VlbZIP30 overexpression (Fig. 3d, e).
The expression of many ABA-responsive or drought-responsive genes is induced in VlbZIP30-overexpressing lines
To examine the possible roles of VlbZIP30 in transcriptional regulation in response to ABA and osmotic stress, we performed a global transcriptome analysis to identify DEGs between the WT and VlbZIP30-overexpressing lines using RNA-Seq. Seeds from WT and OE1 transgenic lines were cultivated on MS agar medium with or without 0.5 μM ABA or 300 mM mannitol for 7 days, and the seedlings were then collected for transcriptome analysis (Fig. 2a, scheme summarized in Fig. 4a). DEGs were defined based on a threshold of 2-fold change (FDR < 0.05). We identified 10 genes that were upregulated and 10 that were downregulated in the OE lines compared with WT plants under control conditions (OEC/WTC, Fig. 4b, c). Details of these 20 genes, including their annotation and their expression levels are listed in Supplementary Table S2. After treatments, a total of 1735 and 2203 upregulated genes and 1734 and 1764 downregulated genes were identified in WT plants subjected to ABA (WTA/WTC) and mannitol (WTM/WTC) stress, respectively (Fig. 4b, c). A total of 1510 and 1494 upregulated genes, and 1058 and 729 downregulated genes were found in the OE lines subjected to ABA (OEA/OEC) and mannitol (OEM/OEC) stress, respectively (Fig. 4b, c). We also identified 359 and 139 genes that were upregulated and downregulated, respectively, in the OE lines compared with WT plants when treated with ABA (OEA/WTA, Fig. 4b, c), while 783 and 344 genes were upregulated and downregulated, respectively, when treated with mannitol (OEM/WTM, Fig. 4b, c).

a Experimental set up: WT and OE transgenic seeds were cultivated on Murashige–Skoog (MS) agar medium (WTC, OEC) or MS agar medium supplemented with 0.5 μM ABA (WTA, OEA) or 300 mM mannitol (WTM, OEM) for 7 days. b, c show the numbers of overlapping downregulated and upregulated genes, respectively. The numbers in brackets represent the total numbers of DEGs in different comparisons. The DEGs in red circles were selected for further analysis. d The selected upregulated genes in the comparisons (OE/WT) under control conditions, ABA or mannitol stress in Arabidopsis thaliana. e The grapevine (Vitis vinifera) homologs of the selected A. thaliana DEGs from (d)
To deduce the possible functions of the VlbZIP30-induced genes, we performed a GO analysis of the genes, whose expression levels were significantly altered in the OE lines compared with the WT plants in response to ABA (OEA/WTA) or mannitol stress (OEM/WTM), using the PageMan profiling tool30. This revealed that some genes encode TFs, and some genes are putatively involved in photosynthesis, stress, signaling, transport, development, and several other metabolic pathways involving hormones, amino acids, and lipids (Supplementary Fig. S4).
To better understand the role of VlbZIP30 in ABA and osmotic stress signaling, the OEA/OEC and OEA/WTA intersecting genes (upregulated, 29 + 53; downregulated, 8 + 11) and the OEM/OEC and OEM/WTM intersecting genes (upregulated, 47 + 115; downregulated, 5 + 16) were selected for further analysis (Fig. 4b, c). We identified all of the 248 (210 upregulated, 38 downregulated) genes mentioned above, and found that 38% (95/248) genes had been identified in A. thaliana. Among them, 54 genes were involved in abiotic stress, including stress-responsive genes (RD20, RD22, RD26, SIS, and ERD10), ABA signaling genes (AFP1, AFP3, HB7, HB12, MAPKKK18, PYL6), PP2C genes (ABI1, ABI2, HAI1, HAI2, HAB1, and PP2CA), TFs (ABF2, ABF3, ABF4, DREB1A, NFYA5, NFYB2, NAP, MYB74, WRKY28, ERF053, and bHLH129) (Table 1). The other 41 genes were involved in other processes such as wax biosynthesis, photosynthesis, transport, hormone signaling (brassinosteroids, ethylene, and cytokinins), and mineral homeostasis (Fe, Ca, phosphate, and sulfur) (Supplementary Table S3).
To confirm the RNA-Seq results, we examined the expression of 20 drought-responsive genes by qRT-PCR and saw that the expression changes of all these genes were similar in the RNA-Seq (Table 1) and qRT-PCR data (Supplementary Fig. S5).
Identification of a potential G-box motif in VlbZIP30-induced genes
To identify candidate VlbZIP30 target genes, promoter analyses of the DEGs between OE lines and WT plants under control conditions (10 upregulated and 10 downregulated genes) and during ABA or mannitol treatment (210 upregulated and 38 downregulated genes) were performed using the DREME motif tool. We identified a potential G-box cis-element (ACGTGKV; E-value, 2.5e-017) including a 4-bp core sequence (ACGT) that is known to be a bZIP binding motif, as being significantly enriched in the promoters of the upregulated genes (Fig. 5a), of which 187 of the 220 genes carried this motif in their upstream 1500 bp promoter region. The G-box motif was not enriched in the downregulated genes. The number and location of the G-box motifs in the 187 gene promoters were analyzed, and many genes carried 1–3 G-box motifs, and that the highest G-box frequency was within the first 300 bp from the start codon site (Fig. 5c, e).

a, b DREME motif analysis, showing the predicted G-box (ACGTGKV) motif (a) in the promoter regions of selected Arabidopsis thaliana upregulated genes from the overexpressing (OE) lines, and the potential G/C-box (MCACGTS) motif (b) in the promoter regions of the corresponding grapevine (Vitis vinifera) homologs. c, d Number of predicted G-box motifs in the promoters of the selected A. thaliana genes (c), and the number of predicted G/C-box motifs in the promoter of the corresponding grapevine homologs (d). Predicted VlbZIP30-induced A. thaliana and grapevine genes with at least three G-box or G/C-box motifs were selected for further analyses (highlighted in blue). e, f Frequency of the predicted G-box motif in the promoters of the selected A. thaliana genes with respect to their distance from the start codon site (e), and the frequency of the potential G/C-box motif in the promoters of the corresponding grapevine homologs with respect to their distance from the start codon site (f)
Then, we searched the grapevine genome for homologs of the identified A. thaliana upregulated (220) and downregulated (48) genes were identified (Fig. 4d, e; Supplementary Data S1) and a promoter analysis performed using the DREME motif discovery tool. Homologs were found of 195 of the upregulated and 44 of the downregulated genes (Supplementary Data S1). Using the same analytical method, surprising, a potential G/C-box cis-element motif (MCACGTS; E-value, 8.3e-018) including the core sequence (ACGT) was found to be significantly enriched in the homologs to the upregulated genes (Fig. 5b). We determined that 108 of the 195 genes had at least one G/C-box motif in their upstream 1500 bp promoter region, while this motif was not enriched in the downregulated genes. Many of the 195 genes carried 1 or 2 G/C-box motifs, and the highest frequency was within the 300 bp from start codon site (Fig. 5d, f), similar to the A. thaliana genes (Fig. 5c, e).
Genes with three or more G-box (A. thaliana) or G/C-box (grapevine) motifs were selected for further investigation39. We found that 52% (97/187) of the A. thaliana genes and 50% (54/108) of the grapevine genes had at least three motifs and that all of the latter had G-box, but not C-box motifs. We found that 61 of the A. thaliana genes were homologous to 54 grapevines genes. Of the 97 and 61 A. thaliana genes, we identified 39 with three or more G-box motifs in their promoters shown as Venn diagram (Fig. 6a). Another DREME promoter analysis of these 39 genes revealed 4 enriched motifs, including a perfect G-box (CACGTG, E-value: 9.0e-012), a GAGA-box (DAGAGAGA, E-value: 1.1e-005), an AAGAAAAR motif (E-value: 7.9e-004), and a TATA-box (ABATATAT, E-value: 9.9e-004) (Fig. 6b). The frequencies of the G-box in the VlbZIP30-induced 39 and 220 A. thaliana genes were 89.7 and 70.0%, respectively, while the frequency in the whole genome was 51.5%, suggesting an enrichment in the VlbZIP30-induced genes. Such an enrichment was not found for the other three motifs (Fig. 6b).

a Venn diagram showing the selected A. thaliana genes with three or more predicted G-box elements and the A. thaliana homologs of selected grapevine genes with three or more predicted G-box motifs. c Venn diagrams of the selected grapevine genes with three or more predicted G-box motifs and the grapevine homologs of the selected A. thaliana genes with three or more predicted G-box motifs. b, d The 1500 bp promoter regions of the overlapping 39 A. thaliana genes from (a) and 35 grapevine genes from (c) were analyzed using the DREME motif enrichment tool. The potential A. thaliana G-box and grapevine G-box motif enrichments were identified using the whole-genome sequences from A. thaliana and grapevine as a reference
In grapevine, 88 homologs of the 97 A. thaliana genes were found. Among the 88 and 54 grapevine genes, we identified 35 with at least three G-box motifs in their promoters shown as Venn diagram (Fig. 6c). Three enriched motifs, including a potential grapevine G-box (MCACGTGK, E-value: 1.9e-009), a GAGA-box (RAGAGARA, E-value: 2.4e-007), and a CACSTC motif (E-value: 3.5e-004) were identified in these 35 genes (Fig. 6d). Among them, the frequencies of the G-box in the predicted VlbZIP30-induced 35 and 196 grapevine genes were 100.0 and 77.4%, respectively, while the frequency of the G-box in the whole genome was 59.0%, again indicating an enrichment in the predicted VlbZIP30-induced genes. These results suggest that the 35 grapevine genes may be regulated by VlbZIP30 via the potential G-box. The names and gene IDs of the 35 grapevine genes and 39 A. thaliana genes are listed in Supplementary Data S2.
The expression of the predicted VlbZIP30-induced genes with at least three potential G-box motifs was upregulated in grapevine following both ABA and drought treatments
To investigate the potential roles of the 35 identified grapevine genes in ABA and drought stress, two different grapevine-related databases for ABA1 and drought stress34 treatments were analyzed. RNA-Seq analysis was performed of grapevine berry skins with or without ABA treatment for 20 and 44 h. A GrapeGene GeneChips® data analysis was performed on leaves from two Vitis vinifera L. varieties (Trincadeira, TR, and Touriga Nacional, TN) grown under control and drought greenhouse conditions, as well as fully irrigated and non-irrigated field conditions. We compared the genes in those datasets with the 35 predicted grapevine genes, and found that 31 and 25 appeared in the RNA-Seq and GeneChips® data (Fig. 7a). Of these, 74% (23/31) and 84% (21/25) were significantly upregulated following the ABA and drought treatments, respectively (Fig. 7a). Six genes uniquely responded to ABA and 4 specifically responded to drought, while 17 genes responded to both treatments (Fig. 7a). The detailed expression data for these 27 genes (Fig. 7a) are shown in heat map diagrams in Fig. 7b.

a Venn diagram of the selected upregulated genes in two different grapevine-related databases with the 35 predicted grapevine genes. Thirty-one and 25 predicted grapevine genes were present in the RNA-Seq and GeneChips® data. The numbers in brackets represent the numbers of upregulated genes in the two different grapevine-related databases. b Heat maps showing the expression of the 27 genes found in (a). RNA-Seq analysis was performed of transcripts expressed in the skin of berries from grapevines treated, or not, with ABA for 20 and 44 h. The GrapeGene GeneChips® data was derived from an analysis of the leaves of two V. vinifera L. varieties (Trincadeira, TR, and Touriga Nacional, TN) grown under control and drought conditions in a greenhouse, as well as under fully irrigated and non-irrigated conditions in the field. Heat map color gradation in red indicates the increase in expression (log2 fold change). c Gene expression profiles of randomly selected VlbZIP30-induced grapevine candidate genes analyzed using qRT-PCR. For each gene, the expression level in the 0 h sample from the ABA and dehydration treatments was defined as 1.0. The VvActin1 gene was used as an internal control. Data represent mean values ± SE from three independent experiments. Asterisks indicate statistical significance (*0.01 < P < 0.05, **P < 0.01, Student’s t-test) between the treated and untreated control plants
We randomly selected 16 genes of the 27 genes induced by ABA or drought stress, for confirmatory qRT-PCR expression analysis, using the previously mentioned grapevine leaf samples subjected to ABA or dehydration treatment. The results were consistent with the results previously described (Fig. 7c; Supplementary Fig. S6).
Discussion
Early studies identified three bZIP-type ABRE-binding proteins, AREB1/2/3 from A. thaliana, using an ABRE motif40, and AREB1, AREB2, and ABF3 were shown to play important roles in response to ABA and drought stress signaling41. In addition, Furihata et al. showed that exogenous ABA activates the SnRK2 protein kinases, and that the activated SnRK2 proteins phosphorylate a Ser/Thr residue in the conserved domains (C1, C2, C3, and C4) of the downstream AREB1 gene, allowing it to bind to the cis-acting ABRE element of downstream drought-related genes42. The mechanism involving AREB1 in the ABA core signaling pathway in A. thaliana is also present in economically important crops, such as rice43,44,45. These studies suggest that similar regulatory responses to stress are evolutionarily conserved in plants. In this study, we identified a group A bZIP TF, VlbZIP30, from grapevine. The results suggest that is positively regulates plant dehydration tolerance through regulation of the downstream drought-related genes via G-box cis-element (MCACGTGK) in ABA signaling pathway.
Previous studies have demonstrated that overexpressing drought-induced genes in A. thaliana can cause hypersensitivity to ABA and increase tolerance to drought stress12. However, in this study, overexpressing VlbZIP30 in A. thaliana did not make the plants more sensitive to ABA, but increased their osmotic stress tolerance during germination and post-germination growth. Given that VlbZIP30 is a TF, its role in osmotic stress response is likely to involve regulating downstream gene expression. Indeed, a majority of the ABA-induced and drought-induced genes tested were induced in VlbZIP30-overexpressing A. thaliana following ABA and mannitol treatment. Ninety-five (38%) of the 248 genes identified (Fig. 4) have previously been identified and of these 54 were involved in abiotic stress, including stress marker genes (RD20, RD22, RD26), ABA core signaling components (6 PP2Cs: ABI1, ABI2, HAI1, HAI2, HAB1, PP2CA, and PYL6), TFs (ABF2, ABF3, ABF4) (Table 1). Nine A. thaliana PP2Cs belonging to cluster A have been identified46, and studies have shown that six of them (ABI1/2, HAI1/2, HAB1, and PP2CA) function as negative regulators of ABA signaling, with their mutants showing hypersensitivity to ABA during seed germination and seedling growth6,47,48,49. In addition, the ABI1/2 and HAI1/2 genes are known to act in a negative feedback regulatory loop of the ABA signaling pathway47,49. A sextuple mutant impaired in six PYR/PYL receptors was shown to be very insensitive to ABA during seed germination and seedling growth50. Here, we found that the transcript levels of the six PP2Cs were all significantly higher in the VlbZIP30-overexpressing lines in response to osmotic stress, while the expression of PYL6 was lower (Table 1). Consistent with this, the OE lines were found to be insensitive to ABA. These results suggested that VlbZIP30 may play a role in a negative feedback regulatory loop of the ABA core signaling pathway under osmotic stress conditions in A. thaliana.
The ABRE (PyACGTGGC) and G-box (CACGTG) elements were identified as bZIP TF cis-binding elements regulating gene expression in response to ABA or drought stress in many plants, including A. thaliana40, rice51, wheat52, apple53, and so on. In this study, we identified 39 A. thaliana genes and 35 predicted grapevine genes (Supplementary Data S2) that may be directly or indirectly regulated by VlbZIP30. Seventeen (43.6%) of the 39 A. thaliana genes have been found to be involved in drought stress, including RD26, AFP1/3, PP2CA, HAI1/2, ABF3, NAP, MYB74, WRKY28, and PUB19 (Table 1), implying that our analytical methods and results are credible. Other genes found here that have not been previously characterized may therefore also be involved in drought stress signaling. In contrast, there has been little characterization of the 35 grapevine genes, and only 3 were identified as being involved in ABA or drought stress signaling. ABF2/bZIP39 (VIT_18s0001g10450), which was characterized as being involved in ABA signaling in grapevine cell culture, has been reported to transiently trans-activate the expression of NAC17 (VIT_19s0014g03290) and PUB19 (VIT_17s0000g08080) following ABA treatment1,36. In addition, overexpression of this gene in A. thaliana enhances tolerance to drought stress through the ABA signaling pathway22, suggesting that NAC17 and PUB19 may enhance drought stress in grapevine.
Subsequently, a perfect A. thaliana G-box (CACGTG) and a putative grapevine G-box (MCACGTGK) element were significantly enriched in the promoter of the 39 A. thaliana genes and 35 predicted grapevine genes (Fig. 6b, d). The highly conserved G-box motif (CACGTG) is regulated by bZIP TFs in organisms ranging from yeast to humans54. Ezer et al. constructed an available gene expression network (www.araboxcis.org) for prediction of genes regulating the G-box, or a set of genes regulated by the G-box. They identified approximately 2000 seedling-expressed genes expressed in 229 RNA-Seq samples of 7-day-old A. thaliana seedlings that are highly likely to be regulated by a perfect G-box motif (CACGTG) in their promoter, and predicted how bZIP proteins might regulate these genes54. These results suggest that VlbZIP30 is likely to enhance A. thaliana dehydration tolerance by regulating downstream genes containing a perfect G-box (CACGTG). Large-scale transcriptome analyses also show that the G-box (CACGTG) was highly enriched in stress-responsive genes in grapevines55. These results suggest a general conservation in promoter framework, gene expression dynamics, and gene regulatory networks across species. We also used two grapevine-related databases to gain support for the potential roles of the 35 grapevine genes in ABA and drought stress.
We noted that 74 and 84% (a total of 27) candidate genes were significantly upregulated under ABA or drought treatment, respectively (Fig. 7a, b), and that the expression of which was up to 64-fold induced (Fig. 7b). In addition, the expression levels of 16 randomly selected genes from the 27 genes (including VvPP2C9, VvPP2C37, and VvABF2) were significantly upregulated by ABA or dehydration treatment by qRT-PCR analysis (Fig. 7c; Supplementary Fig. S6), supporting the accuracy of the RNA-Seq data. These results suggest that VlbZIP30 may be involved in drought stress signaling in grapevine via regulation of the 27 grapevine genes containing the grapevine G-box (MCACGTGK). This conclusion is supported by the observation that 17 of the 39 A. thaliana homologous genes have previously been found to be involved in drought stress. Further studies be required to elucidate the functions of these regulated ABA and drought stress regulated grapevine genes.
References
Pilati, S. et al. Abscisic acid is a major regulator of grape berry ripening onset: new insights into ABA signaling network. Front. Plant Sci. 8, 1–16 (2017).
Ferreira, R. B., Monteiro, S. S., Picarra-Pereira, M. A. & Teixeira, A. R. Engineering grapevine for increased resistance to fungal pathogens without compromising wine stability. Trends Biotechnol. 22, 168–173 (2004).
Tang, N., Zhang, H., Li, X. H., Xiao, J. H. & Xiong, L. Z. Constitutive activation of transcription factor OsbZIP46 improves drought tolerance in rice. Plant Physiol. 158, 1755–1768 (2012).
Finkelstein, R. R., Gampala, S. S. L. & Rock, C. D. Abscisic acid signaling in seeds and seedlings. Plant Cell 14, S15–S45 (2002).
Nambara, E. & Kuchitsu, K. Opening a new era of ABA research. J. Plant Res. 124, 431–435 (2011).
Saez, A. et al. Enhancement of abscisic acid sensitivity and reduction of water consumption in Arabidopsis by combined inactivation of the protein phosphatases type 2C ABI1 and HAB1. Plant Physiol. 141, 1389–1399 (2006).
Danquah, A., de Zelicourt, A., Colcombet, J. & Hirt, H. The role of ABA and MAPK signaling pathways in plant abiotic stress responses. Biotechnol. Adv. 32, 40–52 (2014).
Van Leene, J. et al. Functional characterization of the Arabidopsis transcription factor bZIP29 reveals its role in leaf and root development. J. Exp. Bot. 67, 5825–5840 (2016).
Foster, R., Izawa, T. & Chua, N. H. Plant bZIP proteins gather at ACGT elements. FASEB J. 8, 192–200 (1994).
Yamaguchi-Shinozaki, K., Mino, M., Mundy, J. & Chua, N. H. Analysis of an ABA-responsive rice gene promoter in transgenic tobacco. Plant Mol. Biol. 15, 905–912 (1990).
Mitsuda, N. & Ohme-Takagi, M. Functional analysis of transcription factors in Arabidopsis. Plant Cell Physiol. 50, 1232–1248 (2009).
Fujita, Y. et al. AREB1 is a transcription activator of novel ABRE-dependent ABA signaling that enhances drought stress tolerance in Arabidopsis. Plant Cell 17, 3470–3488 (2005).
Kang, J. Y., Choi, H. I., Im, M. Y. & Kim, S. Y. Arabidopsis basic leucine zipper proteins that mediate stress-responsive abscisic acid signaling. Plant Cell 14, 343–357 (2002).
Yoshida, T. et al. Four Arabidopsis AREB/ABF transcription factors function predominantly in gene expression downstream of SnRK2 kinases in abscisic acid signalling in response to osmotic stress. Plant Cell Environ. 38, 35–49 (2015).
Zou, M. J., Guan, Y. C., Ren, H. B., Zhang, F. & Chen, F. A bZIP transcription factor, OsABI5, is involved in rice fertility and stress tolerance. Plant Mol. Biol. 66, 675–683 (2008).
Kobayashi, F. et al. Development of abiotic stress tolerance via bZIP-type transcription factor LIP19 in common wheat. J. Exp. Bot. 59, 891–905 (2008).
Zhang, X. et al. Maize ABP9 enhances tolerance to multiple stresses in transgenic Arabidopsis by modulating ABA signaling and cellular levels of reactive oxygen species. Plant Mol. Biol. 75, 365–378 (2011).
Liu, H. Y. et al. Ectopic expression of a grapevine transcription factor VvWRKY11 contributes to osmotic stress tolerance in Arabidopsis. Mol. Biol. Rep. 38, 417–427 (2011).
Zhu, Z. G. et al. Three ERF transcription factors from Chinese wild grapevine Vitis pseudoreticulata participate in different biotic and abiotic stress-responsive pathways. J. Plant Physiol. 170, 923–933 (2013).
Fang, L. C. et al. Expression of Vitis amurensis NAC26 in Arabidopsis enhances drought tolerance by modulating jasmonic acid synthesis. J. Exp. Bot. 67, 2829–2845 (2016).
Tu, M. X. et al. Expression of a grape (Vitis vinifera) bZIP transcription factor, VIbZIP36, in Arabidopsis thaliana confers tolerance of drought stress during seed germination and seedling establishment. Plant Sci. 252, 311–323 (2016).
Tu, M. X. et al. Expression of a grape bZIP transcription factor, VqbZIP39, in transgenic Arabidopsis thaliana confers tolerance of multiple abiotic stresses. Plant Cell Tiss. Org. 125, 537–551 (2016).
Gao, M. et al. Evolutionary and expression analyses of basic zipper transcription factors in the highly homozygous model grape PN40024 (Vitis vinifera L.). Plant Mol. Biol. Rep. 32, 1085–1102 (2014).
Liu, J. et al. Genome-wide analysis and expression profile of the bZIP transcription factor gene family in grapevine (Vitis vinifera). BMC Genom. 15, 281 (2014).
Jakoby, M. et al. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 7, 106–111 (2002).
Clough, S. J. & Bent, A. F. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Murashige, T. & Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant 15, 473–497 (1962).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Usadel, B. et al. PageMan: an interactive ontology tool to generate, display, and annotate overview graphs for profiling experiments. BMC Bioinformatics 7, 535 (2006).
Heyndrickx, K. S. & Vandepoele, K. Systematic identification of functional plant modules through the integration of complementary data sources. Plant Physiol. 159, 884–901 (2012).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Deng, W., Wang, Y., Liu, Z., Cheng, H. & Xue, Y. HemI: a toolkit for illustrating heatmaps. PLoS ONE 9, e111988 (2014).
Rocheta, M. et al. Transcriptomic comparison between two Vitis vinifera L. varieties (Trincadeira and Touriga Nacional) in abiotic stress conditions. BMC Plant Biol. 16, 224 (2016).
Kim, S. Y., Ma, J., Perret, P., Li, Z. & Thomas, T. L. Arabidopsis ABI5 subfamily members have distinct DNA-binding and transcriptional activities. Plant Physiol. 130, 688–697 (2002).
Nicolas, P. et al. The basic leucine zipper transcription factor ABSCISIC ACID RESPONSE ELEMENT-BINDING FACTOR2 is an important transcriptional regulator of abscisic acid-dependent grape berry ripening processes. Plant Physiol. 164, 365–383 (2014).
Iuchi, S. et al. Regulation of drought tolerance by gene manipulation of 9-cis-epoxycarotenoid dioxygenase, a key enzyme in abscisic acid biosynthesis in Arabidopsis. Plant J. 27, 325–333 (2001).
Fujii, H. & Zhu, J. K. Arabidopsis mutant deficient in 3 abscisic acid-activated protein kinases reveals critical roles in growth, reproduction, and stress. Proc. Natl Acad. Sci. USA 106, 8380–8385 (2009).
Piya, S., Kihm, C., Rice, J. H., Baum, T. J. & Hewezi, T. Cooperative regulatory functions of miR858 and MYB83 during cyst nematode parasitism. Plant Physiol. 174, 1897–1912 (2017).
Uno, Y. et al. Arabidopsis basic leucine zipper transcription factors involved in an abscisic acid-dependent signal transduction pathway under drought and high-salinity conditions. Proc. Natl Acad. Sci. USA 97, 11632–11637 (2000).
Kim, S., Kang, J. Y., Cho, D. I., Park, J. H. & Kim, S. Y. ABF2, an ABRE-binding bZIP factor, is an essential component of glucose signaling and its overexpression affects multiple stress tolerance. Plant J. 40, 75–87 (2004).
Furihata, T. et al. Abscisic acid-dependent multisite phosphorylation regulates the activity of a transcription activator AREB1. Proc. Natl Acad. Sci. USA 103, 1988–1993 (2006).
Kagaya, Y., Hobo, T., Murata, M., Ban, A. & Hattori, T. Abscisic acid-induced transcription is mediated by phosphorylation of an abscisic acid response element binding factor, TRAB1. Plant Cell 14, 3177–3189 (2002).
Kobayashi, Y. et al. Abscisic acid-activated SNRK2 protein kinases function in the gene-regulation pathway of ABA signal transduction by phosphorylating ABA response element-binding factors. Plant J. 44, 939–949 (2005).
Chae, M. J. et al. A rice dehydration-inducible SNF1-related protein kinase 2 phosphorylates an abscisic acid responsive element-binding factor and associates with ABA signaling. Plant Mol. Biol. 63, 151–169 (2007).
Schweighofer, A., Hirt, H. & Meskiene, I. Plant PP2C phosphatases: emerging functions in stress signaling. Trends Plant Sci. 9, 236–243 (2004).
Bhaskara, G. B., Nguyen, T. T. & Verslues, P. E. Unique drought resistance functions of the highly ABA-induced clade A protein phosphatase 2Cs. Plant Physiol. 160, 379–395 (2012).
Yoshida, T. et al. ABA-hypersensitive germination3 encodes a protein phosphatase 2C (AtPP2CA) that strongly regulates abscisic acid signaling during germination among Arabidopsis protein phosphatase 2Cs. Plant Physiol. 140, 115–126 (2006).
Merlot, S., Gosti, F., Guerrier, D., Vavasseur, A. & Giraudat, J. The ABI1 and ABI2 protein phosphatases 2C act in a negative feedback regulatory loop of the abscisic acid signalling pathway. Plant J. 25, 295–303 (2001).
Gonzalez-Guzman, M. et al. Arabidopsis PYR/PYL/RCAR receptors play a major role in quantitative regulation of stomatal aperture and transcriptional response to abscisic acid. Plant Cell 24, 2483–2496 (2012).
Liu, C. et al. OsbZIP71, a bZIP transcription factor, confers salinity and drought tolerance in rice. Plant Mol. Biol. 84, 19–36 (2014).
Wang, J., Li, Q., Mao, X., Li, A. & Jing, R. Wheat transcription factor TaAREB3 participates in drought and freezing tolerances in Arabidopsis. Int. J. Biol. Sci. 12, 257–269 (2016).
An, J.-P. et al. The bZIP transcription factor MdHY5 regulates anthocyanin accumulation and nitrate assimilation in apple. Hortic. Res. 4, 17023 (2017).
Ezer, D. et al. The G-Box transcriptional regulatory code in Arabidopsis. Plant Physiol. 175, 628–640 (2017).
Wong, D. C. J., Lopez Gutierrez, R., Gambetta, G. A. & Castellarin, S. D. Genome-wide analysis of cis-regulatory element structure and discovery of motif-driven gene co-expression networks in grapevine. DNA Res. 24, 311–326 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31572110), as well as the Program for Innovative Research Team of Grape Germplasm Resources and Breeding (2013KCT-25). We thank PlantScribe (www.plantscribe.com) for careful editing of this manuscript.
Author information
Authors and Affiliations
Contributions
X.W. and M.T. designed the study. M.T. and X.H.W. contributed to the experiments, X.H.W. and D.W. constructed the vectors, Y.Z. performed the qRT-PCR analysis, M.T. and D.W. performed data analysis. M.T., X.H.W., X.Z., and Y.C. performed transcriptome data analysis, Z.L., Y.W., Y.L., and M.G. assisted with the data analysis. M.T. and X.W. wrote the manuscript. All of the authors approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tu, M., Wang, X., Zhu, Y. et al. VlbZIP30 of grapevine functions in dehydration tolerance via the abscisic acid core signaling pathway. Hortic Res 5, 49 (2018). https://doi.org/10.1038/s41438-018-0054-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-018-0054-x
This article is cited by
-
Transcriptional regulation of KCS gene by bZIP29 and MYB70 transcription factors during ABA-stimulated wound suberization of kiwifruit (Actinidia deliciosa)
BMC Plant Biology (2022)
-
The apple DNA-binding one zinc-finger protein MdDof54 promotes drought resistance
Horticulture Research (2020)
-
Grapevine VlbZIP30 improves drought resistance by directly activating VvNAC17 and promoting lignin biosynthesis through the regulation of three peroxidase genes
Horticulture Research (2020)
-
Overexpression of an ABA-dependent grapevine bZIP transcription factor, VvABF2, enhances osmotic stress in Arabidopsis
Plant Cell Reports (2019)
