
Abstract
The Lori-Bakhtiari fat-tailed sheep is one of the most important heavyweight native breeds of Iran. The breed is robust and well-adapted to semi-arid regions and an important resource for smallholder farms. An established nucleus-based breeding scheme is used to improve their production traits but there is an indication of inbreeding depression and loss of genetic diversity due to selection. Here, we estimated the inbreeding levels and the distribution of runs of homozygosity (ROH) islands in 122 multi-generational female Lori-Bakhtiari from different half-sib families selected from a breeding station that were genotyped on the 50k array. A total of 2404 ROH islands were identified. On average, there were 19.70 ± 1.4 ROH per individual ranging between 6 and 41. The mean length of the ROH was 4.1 ± 0.14 Mb. There were 1999 short ROH of length 1–6 Mb and another 300 in the 6–12 Mb range. Additionally long ROH indicative of inbreeding were found in the ranges of 12–24 Mb (95) and 24–48 Mb (10). The average inbreeding coefficient (FROH) was 0.031 ± 0.003 with estimates varying from 0.006 to 0.083. Across generations, FROH increased from 0.019 ± 0.012 to 0.036 ± 0.007. Signatures of selection were identified on chromosomes 2, 6, and 10, encompassing 55 genes and 23 QTL associated with production traits. Inbreeding coefficients are currently within acceptable levels but across generations, inbreeding is increasing due to selection. The breeding program needs to actively monitor future inbreeding rates and ensure that the breed maintains or improves on its current levels of environmental adaptation.
Similar content being viewed by others

Introduction
Sheep were first domesticated around 10,500 years ago in the northern part of the Fertile Crescent between south-eastern Anatolia and the Iranian Zagros Mountains, where their wild relatives are still extant (Alberto et al., 2018). The wild Asiatic mouflon (Ovis orientalis) is believed to be the direct ancestor of the domestic sheep breeds in this area and from there spread out across south-west Asia, covering the geographical regions that are now northern Syria, northern and eastern Iraq, western Iran, and central and eastern Anatolia (Yeomans et al., 2017). There are now hundreds of breeds across the world adapted to the most diverse environments and production systems being raised primarily for wool, meat, and milk. Sheep are one of the world’s main animal genetic resources, an important source of income and the most numerous livestock species in the Middle East (not including chickens).
Since the 1970’s Iran has been among the top sheep producers in the world. The country currently has the 6th largest flock with 46.6 million sheep, and it is the largest sheep producer among the Middle Eastern countries (FAOstat 2020 data; https://www.fao.org/faostat/en/#data/QCL). In terms of numbers, sheep are the largest livestock species in Iran, followed by goats with around 16.7 million animals, and cattle at 5.3 million (FAOstat 2020 data; https://www.fao.org/faostat/en/#data/QCL). Both, the sheep and goat sectors are getting smaller, with animal numbers estimated to have decreased between 10–20% since their heights at the turn of the century, with adequate sources of feed and water being a major concern, especially in the drier regions in the south and the central region’s desert zone. Most of the sheep stock is extensively grazed and kept by small farmers (the average flock size is 38) as a complementary source of income for plant-based farming. Although large areas of Iran have arid or semi–arid climate, there is still a large diversity of agroecological conditions across the country that have led to the formation of 27 locally adapted indigenous sheep breeds and ecotypes (Sheikhlou & Abbasi, 2016). These native sheep breeds that now inhabit the original center of domestication have largely been defined using conformation traits and there is still limited genomic information available for them. Although the evolutionary history of most of these breeds is still largely unknown, a recent study by Eydivandi et al. (2020) has shed some light on the evolutionary history of five of these Iranian native breeds (Afshari, Lori-Bakhtiari, Moghani, Qezel, and Zel) and their relationship with the wild Asiatic mouflon and other regional and commercial breeds. In line with what has already been reported in other countries such as Italy (Ciani et al., 2014), Portugal (Pereira et al., 2006), Spain (Pedrosa et al., 2007), Algeria (Ghernouti et al., 2017) and other European countries (Peter et al., 2007) these breeds seem to have a complex history of past gene flow and admixture; they are however clearly distinct from other non-Iranian sheep and more related to the wild Asiatic mouflon than breeds from other regions (Eydivandi et al., 2020).
Except for the Zel breed, all Iranian sheep breeds are fat-tailed with the Lori-Bakhtiari being the most fat-tailed sheep breed in Iran. It is one of the heaviest meat sheep breeds in the Middle East and it is mainly reared in semi-extensive systems in the western and south-western parts of Iran. It is the least admixed (34.8%) of the few Iranian native breeds that have been genetically categorized (Eydivandi et al., 2020). There are around 1.7 million Lori-Bakhtiari in Iran, comprising 3.6% of the national flock. The breed is well adapted to the hilly and mountain ranges of the Zagros mountains, and ecologically interesting as it is raised in the same environment where the ancestral wild Asiatic mouflon is still found. In 1989 a selection program for Lori-Bakhtiari was established at the Shooli Breeding Station in Shahrekord (Iran) in collaboration with smallholders to improve their reproductive and growth performance and to reduce its fat-tail size. The breeding program for Lori-Bakhtiari adopts a nucleus-based breeding scheme centered at the Shooli station, the flock is managed under a semi-migratory or village system (Vatankhah et al., 2016) and superior animals selected at the station are then disseminated into local flocks (Vatankhah et al., 2019). Recent work demonstrated however that the breed, probably due to increased inbreeding and a reduction of its effective population size in recent generations, may have started to suffer from inbreeding depression and a loss of genetic diversity (Rashedi Dehsahraei et al., 2013; Keshavarzpour et al., 2018; Vatankhah et al., 2019). In domestic species, loss of genetic diversity has important economic and ecological implications (Lawson Handly et al., 2007). A meta-analysis conducted across a broad range of selected traits in seven livestock species, including sheep (Leroy, 2014), reported that, on average, a 1% increase in inbreeding had a negative effect of 0.137% on the mean of a trait; this was even worse for production traits which on average decreased 0.351%. Knowledge of the genetic diversity and population structure of domestic sheep breeds is an important tool to develop and monitor sustainable breed management programs that properly balance genetic improvement with the conservation of genetic resources and maintenance of their environmental adaptation.
The inbreeding coefficient (F) calculated from pedigree information (FPED) is a classic metric used to estimate the inbreeding levels of a population (Wright, 1922). Nowadays with the wide adoption of genomic technologies, inbreeding coefficients can be estimated directly through runs of homozygosity (ROH) calculated from genotypic data. ROH are long homozygous genomic regions (Gibson et al., 2006) that are assumed to be identical DNA segments inherited by descent from a common ancestor, and that serve as an indicator of genomic autozygosity, consanguinity, selection and population size reduction (Szpiech et al., 2013). Simulation studies have shown that in populations undergoing selection the pedigree-based inbreeding coefficient estimates tend to underestimate autozygosity levels, whilst ROH-based estimates of inbreeding can more accurately measure autozygosity across the genome (Forutan et al., 2018). The length and frequency of these ROH are important parameters to determine the forces driving genomic changes in populations (Dixit et al., 2020) and, through their exploration and characterization, it is possible to understand the genetic structure of populations, elucidate the demographic history of how they evolved over time and identify signatures of natural and/or artificial selection events (Purfield et al., 2012; Dixit et al., 2020).
The aims of this study are to characterize the autozygosity levels and distribution of genome-wide ROH in Iranian Lori-Bakhtiari fat-tailed sheep from the nucleus Sholi Breeding Station, estimate their inbreeding levels, identify genomic regions potentially targeted by selection in the breeding flock and gleam some insights into how the breeding program is changing the genetic composition of this native Iranian breed.
Materials and Methods
Samples and Genotypic Information
A total of 124 female Lori-Bakhtiari sheep were genotyped with the Ovine SNP50 genotyping array (Illumina Inc.), which includes 54,241 SNP. Most genomic regions are well represented and there is, on average, one marker at every 46 kb. Samples for genotyping were collected from animals from the nucleus Sholi Sheep Breeding Station in Iran and based on the available pedigree information, came from 63 sires and 110 dams. Details of the breeding program and flock management system employed by the Sholi Station is given in Vatankhah et al. (2016) and Vatankhah et al. (2019), but briefly there were 10,645 animals with pedigree information spanning 13 generations. There were 732 animals in the base population, and the effective number of founders (fe) was 109. The average generation interval was 4.11 years; sires had an average of 26.45 offspring (the most used sire had 151) and the average for females was 3.6. Genotyped animals spanned across 7 generations (generations 5 through 12) and were more skewed towards the younger generations (Fig. 1). Farm management practices, data collection and blood sampling methods were approved by the Ethics Committee of the University of Guilan.

Genotyped animal number per generation were: 2, 10, 8, 13, 28, 41, 19, and 1, spanning generations 5 through 12. Dots correspond to the individual inbreeding coefficients and the line is the mean trend across generations.
Samples with call rates lower than 95% were excluded (2 animals). SNPs with no known genomic coordinates were also excluded from the genotype data. Remaining samples and SNPs were then re-clustered and standard quality control filters using R (R Core Team, 2020) and Plink (Chang et al., 2015) were applied to remove SNPs with low GenCall (GC) scores (<0.6), SNPs with minor allele frequency lower than 0.05, call rates <95% and large Hardy-Weinberg equilibrium deviations (P-value<1E − 6).
The admixture model implemented in the ADMIXTURE software, with an ancestral population (K) size ranging from 2 to 6 and 10,000 iterations for each run (Alexander et al., 2009) was used to evaluate population stratification. The admixture analysis was carried out based on 21,903 SNPs after LD pruning. SNPs were pruned with Plink by removing any SNP that had an r2 > 0.2 with another SNP within a 50-SNP window and this window was then shifted across the chromosome 10 SNPs at a time. The genomic correlations of the population were further evaluated in R with a singular value decomposition (SVD) of the genomic relationship matrix (GRM) defined as \(G = M^\prime M/\mathop {\sum}\nolimits_{j = 1}^m {2p_jq_j}\), \(p_j^\prime s\) were the minor allele frequencies of the SNP genotypes \((q_j = 1 - p_j)\), and M the SNPs genotype matrix (VanRaden, 2008). The SVD is \(G = UDV\prime\) where \(D = diag(d_1,d_2, \ldots ,d_{n - 1},d_n)\) is a diagonal matrix of the n singular-values, such that \(d_1 \ge \ldots \ge d_n \ge 0\) with \(d_i = 0\) for every \(i > rank(G)\); \(U_{n \times n} = [U_1 \ldots U_n]\) and \(V_{m \times n} = [V_1 \ldots V_n]\) are matrices of unitary eigen-vectors, such that \(U^\prime U = UU^\prime = I_n\) and \(V^\prime V = I_n\). Each of the components \(d_1^2, \ldots ,d_n^2\) explains a proportion of the variation in G and the principal components scores are \(UD\).
The sheep genome map (OvineSNP50 v3.1) released by the International Sheep Genomics Consortium (https://www.sheephapmap.org/) was used for mapping the SNP coordinates. After quality control, a total of 122 animals and 45,402 SNP on 27 chromosomes were used for the downstream analyses.
Runs of homozygosity
ROH and their distributions for each sample were estimated with the R package detectRUNS using the consecutive SNP-based run detection method (Biscarini et al., 2018). ROH detection methods such as the one implemented in Plink (Purcell et al., 2007) generally use a sliding-window approach; the consecutive runs method, first proposed by Marras et al. (2015), is however a window-free method that directly scans the genome SNP by SNP. The ROH analysis parameters used were set as per Dzomba et al. (2021): minSNP equal to 30 (the minimum number of SNP that constitute a ROH), maxGap equal to 250 Kb (the maximum gap between consecutive homozygous SNP), minLenghtBps equal to 1 Mb (the minimum length that constitutes the ROH), maxOppRun equal to 1 (maximum number of SNP with opposite genotype) and maxMissRun equal to 1 (maximum number of missing genotypes).
The identified ROH were then grouped into bins based on cut-offs of 1–6, 6–12, 12–24 and 24–48 Mb in length. Summary statistics were calculated: ROH count per individual, ROH count per chromosome, ROH percentage per individual, ROH percentage per chromosome, ROH mean per chromosome and ROH mean per class-size.
Inbreeding coefficients
From the runs of homozygosity, individual inbreeding coefficients (FROH) were calculated as:
where ∑LROH is the sum of the length of all ROH detected in an individual, and Lgenome is the total length of the genome covered by the array markers (total genome length was 2,583,493,571 bp). For comparison purposes, two alternative measurements of the inbreeding coefficient were calculated. The first one was obtained by subtracting 1 from the diagonal elements of the genomic relationship matrix (GRM) estimated using a base allele frequency of 0.5 (FGRM; VanRaden, 2008). The second one was estimated from the pedigree (FPED) using the R package pedigreemm (Vazquez et al., 2010), which contained records of 10,645 animals.
An additional statistic was calculated to estimate changes in heterozygosity across generations. Heterozygosity for each animal was calculated as \({\sum} {het/n}\), where \({\sum} {het}\) is the sum of the number of heterozygous sites in the individual and n is the number of markers. The observed heterozygosity (Ho) per generation was the average of the heterozygosity estimates of individuals in a generation. Similarly, expected heterozygosity was calculated as \(He = {\sum} {2pq/n}\), where p and q are the allele frequencies per generation and inbreeding was calculated as \(F_H = (H_e - H_0)/H_e\).
The rates of change for FROH, FGRM, FPED and Ho across generations was estimated using a linear regression model \(F_{stat} = b_0 + b_1gen\), where \(F_{stat}\) were each of the inbreeding coefficients and Ho. The slope (\(b_1\)) of the regression denoted the trend of the statistic over generations.
Functional annotation of ROH islands
The frequency with which SNP appeared in a ROH were used to detect genomic regions potentially under selection by the breeding program. ROH that appeared in more than 15% of the individuals were considered as being significant ROH islands. The BioMart-Ensembl (www.ensembl.org/biomart) database was used to identify the sheep genes annotated to the genome under the ROH islands and the functional annotation of the gene ontology terms and pathways associated with these genes was obtained using DAVID (Huang et al., 2009a; Huang et al., 2009b). We also explored the sheep quantitative trait locus database (Sheep QTLdb) to identify any overlap between the candidate regions and previously reported QTL data (http://www.animalgenome.org/QTLdb/sheep).
Results
Quality control and population structure
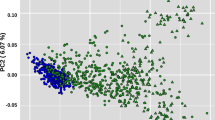
After quality control, 122 samples and 45,402 variants remained for downstream analysis. The overall genotyping call rate for these samples was 0.99. The first two principal components (PCA) from the genomic relationship matrix were used to visualize the population structure of the 122 sheep (Fig. 2), with little stratification discernable across generations. PCs 1 and 2 accounted for 7.03% and 5.57% of the total genotypic variance, respectively. With ADMIXTURE the lowest CV error (0.57102) was for K = 2, with also no clear pattern of cross-generational stratification (Fig. 3) beyond the familial genetic relationships shown in Fig. 4.

First two principal components of the GRM. Individuals are color coded by the generation they belong to.

Each sheep sorted by generation for K = 2.

Relationships between individuals color-coded by generation.
ROH distribution and inbreeding
A total of 2404 ROH were found across all samples, and each individual had on average 19.70 ± 1.4 ROH, varying between a minimum of 6 and a maximum of 41 and a standard deviation of 7.6. The mean length of the ROH was 4.1 ± 0.14 Mb. The number of ROH in each of the bins was 1999, 300, 95, and 10 for length intervals of 1–6, 6–12, 12–24, and 24–48 Mb, respectively. Most of the ROH identified (83.15%) were in the 1–6 Mb length category. Per chromosome, the lowest number of ROH were observed on OAR27/X (n = 18 or 0.74%) and the highest on OAR2 (n = 272 or 11.31%), respectively. The lowest ROH mean per chromosome was also OAR27/X (2.53) and the highest was OAR16 (5.35). The ROH means per bin size were as follows: 1–6 Mb (2.80 ± 0.05), 6–12 Mb (8.41 ± 0.19), 12–24 Mb (15.18 ± 0.50), and 24–48 Mb (29.05 ± 3.87).
The average genomic inbreeding coefficient estimated using ROH (FROH) was 0.031 ± 0.003 with estimates varying from 0.006 to 0.083. Average, minimum, and maximum FGRM values were 0.051 ± 0.005, 0.0, and 0.115, and for FPED 0.019 ± 0.004, 0.0, and 0.137. FROH values per generation varied between 0.019 ± 0.012 and 0.036 ± 0.007, for FGRM between 0.046 ± 0.007 and 0.085 ± 0.020, and for FPED between 0.001 ± 0.001 and 0.028 ± 0.008. The FH values were all negative ranging between −0.358 and −0.0285. The boxplots of the inbreeding levels for FROH, FGRM, and FPED is shown in Fig. 5. The regression coefficients (slopes) of the linear model used to estimate inbreeding per generation were FROH = 0.0024, FGRM = 0.0057 and FPED = 0.006 (p-values < 0.01) and for Ho it was −0.0013 (p-value = 0.003) with Ho estimates varying between 0.371 in generation 5 and 0.362 in generation 11. At the chromosome level, the lowest mean inbreeding coefficient was observed on OAR27/X (0.023) and the highest on OAR26 (0.134). Boxplot of inbreeding levels per chromosome are shown in Fig. 6.

Boxplots of estimated inbreeding levels for FROH, FGRM and FPED.

FROH inbreeding levels for each chromosome.
Genomic regions within ROH islands
Significant ROH islands (ROH repeated in more than 15% of the individuals) were found in 3 genomic regions located on chromosomes OAR2, OAR6 and OAR10 (Fig. 7 and Table 1). The lengths of these regions were 1.77 Mb for OAR2, 0.22 Mb for OAR6 and 1.83 Mb for OAR10 (Table 1). Figure 7 resembles the traditional GWAS Manhattan plot and shows the proportion of times each SNP fell inside a ROH. Figures 8 and 9 detail the ROH in the three chromosomes with significant regions; Fig. 8 shows the cumulative ROH per SNP and Fig. 9 shows all runs detected for each individual along the three chromosomes.

Plot of the incidence (%) of common runs per SNP and per chromosome across animals.

Stacked runs of the significant peaks on chromosomes 2, 6, and 10.

Runs detected for each animal along the genomic coordinates of chromosomes 2, 6, and 10.
Within these genomic regions with a high level of autozygosity, 55 genes, and 23 ovine QTL were identified (Table 1). Out of the 55 genes in Ensembl, 38 are annotated and have an Entrez gene identifier (Table S1) and 33 are associated to a gene ontology term and/or a KEGG pathway (Table S2). The same regions surrounding the identified genes in the ovine genome also contain 23 previously reported QTL that, among others, are related to body weight, meat and muscle characteristics, milk yield, milk contents, bone characteristics, and fecal egg count. (Table 1).
Discussion
The population used for this study was a female flock of Lori-Bakhtiari sheep from the Shooli sheep breeding station in Shahrekord, Iran. Lori-Bakhtiari sheep are fat-tailed and are an important local meat source for Iran (Vatankhah et al., 2008). The population structure of Lori-Bakhtiari sheep in the breeding flock was previously detailed by Vatankhah et al. (2019) using pedigree records of 10,644 animals from 1989 through 2016 (27 years). At the time, the mean inbreeding coefficient measured from the pedigree records (FPED) was 0.025 in the inbred animals and 0.012 across the whole population with an annual increase of 0.09% per year and 0.87% per generation. The effective population size (Ne) over the period was around 143 but quite reduced in later generations (Vatankhah et al. 2019). Eydivandi et al. (2020) estimated a Ne5 of 84 using genomic data from 46 Lori-Bakhtiari (the estimate with our data was 148). These results suggested that the inbreeding levels per generation and effective population sizes were not yet within critical levels for genetic erosion, i.e., an inbreeding rate of more than 1% per generation as per FAO guidelines (FAO, 1998) and the recommendation of Bijma 2000. However, the continuous use of a small number of breeding animals is contributing to an increased inbreeding rate and a decreased effective population size (Vatankhah et al., 2019).
The traditional inbreeding coefficient estimate based on the pedigree approach (FPED) is still the most common method used to describe inbreeding levels in animal populations, despite common practical problems such as availability of complete and correct pedigree data. These limitations can be overcome with SNP arrays which can be used to estimate inbreeding levels directly from the genotypic data and they also provide the additional benefit of having better granularity at the genomic level which, through runs of homozygosity, allows detection of genomic regions where a reduction in heterozygosity has occurred (Mastrangelo et al., 2017). A simulation study using Holstein dairy cattle data demonstrated that inbreeding coefficients estimated from ROH (FROH) were more accurate than the FPED and FGRM estimates (Forutan et al., 2018). With real data, FROH has been reported as being reliable in cattle (Zhang et al., 2015), goat (Burren et al., 2016) and buffalo (Nascimento et al., 2021). In our study the average inbreeding coefficient estimated from ROH (FROH) was 0.031 ± 0.003 with individual values ranging between 0.006 and 0.083. The average value for FGRM was 0.051 ± 0.005 and for FPED 0.019 ± 0.004. The FPED value of the genotyped animals was slightly higher than the previous estimate of 0.012 reported by Vatankhah et al. (2019) for the whole population. This is probably due to increasing inbreeding across generations since the genotyped animals were sampled from later generations and the inbreeding coefficients are reflecting the population’s inbreeding trend. At this point all inbreeding estimates are still relatively low but as already noted by Vatankhah et al. (2019) there is evidence of mounting inbreeding due to selection across generations which is increasing at estimated rates of 0.24% (FROH), 0.57% (FGRM) and 0.6% (FPED) per generation. This is also supported by the small reduction observed in the heterozygosity estimates across generations (−0.13% per generation), which varied from 0.371 in generation 5 to 0.362 in generation 11. Currently however, there does not seem to be any particular concern beyond appropriate management of inbreeding through the breeding program. It does however highlight the challenging dynamics of balancing productivity increases to improve the livelihood of smallholder farms with the need to conserve genetic resources; and, of growing importance, the need to ensure that the breeding program does not have a negative impact on the natural adaptation of the breed to harsh environments.
ROH studies of sheep populations have been widely used to determine the forces driving genetic changes, to identify signatures of natural and/or artificial selection and to understand their demographic history and genetic structure. A recent comprehensive study of ROH in 68 global sheep populations from Africa, Asia, America and Europe identified 60,301 ROH across all breeds (Nosrati et al., 2021). Genomic inbreeding estimates were on the lower end with an average FROH of 0.09. The 465 ROH islands detected in the autosomes harbored 257 genes with known functions in pathways associated with growth, body weight, meat production and quality, wool production and pigmentation. Similarly, Purfield et al. (2017) used six commercial meat sheep breeds (Belclare, Beltex, Charollais, Suffolk, Texel and Vendeen) to show that ROH islands were a common occurrence across all breeds, but the length and frequency of the ROH mostly differed between breeds. The most frequent ROH island was found on OAR2 and the likely candidate genes within this region included MSTN, ITGAV, BIN1 and NUP35, all of which are involved in muscle differentiation (Purfield et al., 2017). For each breed, most of the ROH were under 10 Mb in length and, putatively, there were approximately 11 genomic regions under directional selection across all breeds on OAR 2, 4, 5, 17 and 22.
In another study with eight local Swiss sheep breeds (Bundner Oberlander, Engadine Red, Saaser Mutten, Swiss Black Brown Mountain, Swiss Mirror, Swiss White Alpine, Valais Blacknose and Valais Red), a total of 17,719 ROH with lengths ranging between 1.88 and 103.25 Mb were identified. Similarly, ROH with lengths between 1 and 10 Mb were the most frequent in each breed (Signer-Hasler et al., 2019) and the estimated average inbreeding levels (0.061, 0.021, 0.045, 0.064, 0.076, 0.045, 0.10 and 0.097, respectively) are in line with the results obtained in our study of Iranian fat-tailed sheep. ROH islands were in the vicinity of genes associated with body size, litter size, milk production, coat color and horn status (Signer-Hasler et al., 2019).
A study with 13 South African sheep breeds from very different production systems (Dzomba et al., 2021) identified a very large number of ROH (121,399) which is reflective of the extensive variation in selective forces applied to populations in different pastoral environments. The mean number of ROH per animal per breed ranged from 800 in African White Dorper to 15,097 in Australian Poll Dorset and, in line with other studies, the size of most of the ROH were in the 1–6 Mb interval (88.2%) for all breeds. The Nguni and Blackhead Persian breeds had the highest FROH at 0.031. In common across breeds, the most frequent ROH island was on OAR10 in the vicinity of genes related to metabolic and immune response traits and the horned phenotype.
A single breed ROH study with Valle del Belice sheep (Mastrangelo et al., 2017) identified 11,629 ROH and the mean value of F estimated from ROH longer than 1 Mb was 0.084, OAR24 had the highest number of ROH and OAR1 the lowest; chromosomes 1, 2 and 3 had the longest ROH. None of the genes within the ROH islands overlapped with any previously reported ovine QTL, but a few genomic regions harbored candidate genes known to affect specific production traits. Most of the genes were however associated with multiple cellular and biochemical pathways involved in modulating response to environmental stress (Mastrangelo et al., 2017). In another study, Sobieraj-Kmiecik et al. (2020) showed that the Southern and Central populations of Polish Olkuska sheep had different average FROH – 0.124 and 0.082, respectively. In total 1974 ROH were detected, and the average length was 7.77 Mb. The highest number of ROH were observed on OAR1, OAR2 and OAR3, and most of ROH were also reported to be short in length (<5 Mb). Genomic regions within the ROH harbored genes related to growth, meat, and milk traits.
As would be expected, ROH patterns differ considerably between sheep breeds as a reflection of their geographical regions, demographic history, evolutionary trajectory and of course, in response to breeding selection objectives and management decisions. There are some commonalities however, as many ROH islands and the genes within these genomic regions are associated with biological processes of relevance in livestock production such as growth, body weight, meat and muscle characteristics, milk yield and milk contents, and immune system traits. Natural selection has made Lori-Bakhtari sheep well adapted to hilly landscapes and the semi-arid temperate climate of the Zagros mountains (Vatankhah et al., 2019) while artificial selection in the breeding program has focused on reproductive efficiency, growth traits and decreased fat-tail size. The candidate genes identified in the ROH islands did not immediately suggest a direct link with the selection traits, possibly due to lack of information about their, if any, relevance to production traits. On the other hand, QTL reported in these regions are associated with production, reproduction and immune response traits that are in line with the breeding program’s objective for the breed.
FROH values reported for sheep populations are generally low, especially when compared to a highly selected population such as Holstein dairy cattle (FROH = 0.15, Forutan et al., 2018); this broadly suggests that most sheep breeds still have relatively low levels of autozygosity/inbreeding and maintain adequate levels of genetic diversity. Our results with Lori-Bakhtari align well with these other reports and further suggest that there is currently limited cause for concern about loss of diversity in the breed due to recent artificial selection in the breeding station. Nonetheless, the increase in FROH across generations is noticeable and control of inbreeding rates need to be incorporated into the future breeding decisions made by the Shooli Station.
Climate change resulting from anthropogenic activities is increasing global temperatures (Pasqui and Di Giuseppe, 2019) and is also increasing the frequency of extreme weather events such as the number of hot days and heat waves. This is putting pressure on the thermal comfort of livestock animals and jeopardizing their welfare and productivity, which in turn can lead to greater food insecurity, particularly in tropical environments. The Lori-Bakhtari breeding program of the Shooli Station has followed the same path of other modern breeding efforts by being largely centered on increasing productivity with little concern for improving environmental adaptation and thermal stress resistance (Collier et al., 2019). It is unknown if the breeding program has had a negative effect on the adaption of the breed but in other highly selected livestock systems these selection priorities have led to large genetic gains in productivity at the expense of reduced thermal tolerance due to an increase in their internal heat loads (Collier et al., 2019). As adaptation to harsher environments became a pressing concern, there has been a renewed interest in the characterization of genetic diversity of indigenous livestock breeds, such as the Lori-Bakhtari, that are already well adapted to a range of more extreme environments. This can provide us with a better understanding of the genetic basis of adaptation and help redirect future selection efforts for improved resilience in livestock species to better withstand the impacts of climate change. This is also of practical importance for decision making in breeding programs of these adapted breeds to ensure that long term selection efforts for increased productivity is properly balanced with at least maintaining or preferably increasing their adaptation.
Conclusion
The frequency and distribution of ROH in the genome of Iranian fat-tailed Lori-Bakhtari sheep from a nucleus breeding station were investigated. ROH patterns revealed genomic regions under selection that harbor QTL associated with production, reproduction, and immune response traits in line with the selection priorities of the breeding program. It is however important to point out that the small number of animals available for this work rather limits extrapolation across the breed and results should be treated with some caution. Nonetheless, this study does improve our understanding of an interesting and not well characterized sheep breed that is well adapted to semi-arid environments – it will be interesting to see how these results generalize across the breed and in other sheep breeding stations as well. The average inbreeding coefficients were within acceptable levels and similar to what has been reported in other sheep populations. Although inbreeding is increasing across generations due to selection, it still within acceptable levels of traditional breeding programs. It is however important to monitor future inbreeding rates and, even more importantly, ensure that the breed maintains or improves on its current levels of environmental adaptation to be able to withstand the increasingly hotter temperatures expected in the region in the future.
Data access
The datasets analyzed during the current study are available on Dryad https://datadryad.org/stash/share/vYeYNPi52ZA0uRcnzEC2KQL-EE9YihVphBPHx1WDntM.
References
Alberto FJ, Boyer F, Orozco-terWengel P, Streeter I, Servin B, De Villemereuil P et al. (2018) Convergent genomic signatures of domestication in sheep and goats. Nat Commun 9:813
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Bijma P (2000) Long-term Genetic Contributions: Predictions of Rates of Inbreeding and Genetic Gain in Selected Populations. Wageningen University, Veenendaal, the Netherlands
Biscarini F, Cozzi P, Gaspa G, Marras G (2018) detectRUNS: Detect runs of homozygosity and runs of heterozygosity in diploid genomes. CRAN. The Comprehensive R Archive Network
Burren A, Neuditschko M, Signer-Hasler H, Frischknecht M, Reber I, Menzi F et al. (2016) Genetic diversity analyses reveal first insights into breed-specific selection signatures within Swiss goat breeds. Anim Genet 47:727–39.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 25; 4:7.
Ciani E, Crepaldi P, Nicoloso L, Lasagna E, Sarti FM, Moioli B et al. (2014) Genome-wide analysis of Italian sheep diversity reveals a strong geographic pattern and cryptic relationships between breeds. Anim Genet 45:256–266
Collier RJ, Baumgard LH, Zimbelman RB, Xiao Y (2019) Heat stress: physiology of acclimation and adaptation. Anim Front 9(1):12–19
Dixit SP, Singh S, Ganguly I, Bhatia AK, Sharma A, Kumar NA et al. (2020) Genome-wide runs of homozygosity revealed selection signatures in Bos indicus. Front Genet 11:92
Dzomba EF, Chimonyo M, Pierneef R, Muchadeyi FC (2021) Runs of homozygosity analysis of South African sheep breeds from various production systems investigated using OvineSNP50k data. BMC Genomics 22:7
Eydivandi S, Sahana G, Momen M, Moradi M, Schönherz A (2020) Genetic diversity in Iranian indigenous sheep vis-à-vis selected exogenous sheep breeds and wild mouflon. Anim Genet 51:772–787
FAO (1998) Secondary Guidelines for Development of National Farm Animal Genetic Resources Management Plans. United Nations, Rome, Italy.
Forutan M, Ansari Mahyari S, Baes C, Melzer N, Schenkel FS, Sargolzaei M (2018) Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genomics 19:98
Ghernouti N, Bodinier M, Ranebi D, Maftah A, Petit D, Gaouar SBS (2017) Control Region of mtDNA identifies three migration events of sheep breeds in Algeria. Small Rumin Res 155:66–77.
Gibson J, Morton NE, Collins A (2006) Extended tracts of homozygosity in outbred human populations. Hum Mol Genet 15:789–795
Huang DW, Sherman BT, Lempicki RA (2009a) Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc 4(1):44–57
Huang DW, Sherman BT, Lempicki RA (2009b) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37(1):1–13
Keshavarzpour M, Bahreini Behzadi MR, Muhaghegh, Dolatabadi M (2018) Pedigree analysis and inbreeding investigation in Lori-Bakhtiari Sheep. Iran J Anim Sci Res 9:376–386
Lawson Handly LJ, Byrne K, Santucci F, Townsend S, Taylor M, Bruford MW et al. (2007) Genetic structure of European sheep breeds. Heredity 99:620–631
Leroy G (2014) Inbreeding depression in livestock species: review and meta-analysis. Anim Genet 45:618–628
Marras G, Gaspa G, Sorbolini S, Dimauro C, Ajmone‐Marsan P, Valentini A et al. (2015) Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim Genet 46:110–121
Mastrangelo S, Tolone M, Sardina MT, Sottile G, Sutera AM, Di Gerlando R et al. (2017) Genome‑wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet Sel Evol 49:84
Nascimento AV, Cardoso DF, Santos DJA, Romero ARS, Scalez DCB, Borquis RRA et al. (2021) Inbreeding coefficients and runs of homozygosity islands in Brazilian water buffalo. J Dairy Sci 104(2):1917–1927
Nosrati M, Asadollahpour Nanaei H, Javanmard A, Esmailizadeh A (2021) The pattern of runs of homozygosity and genomic inbreeding in world-wide sheep populations. Genomics 113:1407–1415
Pasqui M, Di Giuseppe E (2019) Climate change, future warming, and adaptation in Europe. Anim Front 9(1):6–11
Pedrosa S, Arranz JJ, Brito N, Molina A, San Primitivo F, Bayón Y (2007) Mitochondrial diversity and the origin of Iberian sheep. Genet Sel Evol 39:91
Pereira F, Davis SJM, Pereira L, McEvoy B, Bradley DG, Amorim A (2006) Genetic signatures of a Mediterranean Influence in Iberian Peninsula Sheep Husbandry. Mol Biol Evol 23(7):1420–1426
Peter C, Bruford M, Perez T, Dalamitra S, Hewitt G, Erhardt G, the ECONOGENE Consortium (2007) Genetic diversity and subdivision of 57 European and Middle-Eastern sheep breeds. Anim Genet 38:37–44
Purcell SN, Todd-Brown K, Thomas L, Ferreira M, Bender D, Maller J et al. (2007) PLINK: a toolset for whole-genome association and population-based linkage analysis [Online]. Am J Hum Gen 81:559–575
Purfield DC, Berry DP, McParland S, Bradley DG (2012) Runs of homozygosity and population history in cattle. BMC Genet 13:70
Purfield DC, McParland S, Wall E, Berry DP (2017) The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 12:e0176780
R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
Rashedi Dehsahraei A, Fayazi J, Vatankhah M (2013) Investigating inbreeding trend and its impact on growth traits of Lori-Nakhtiari sheep. J Rumin Res 1:65–78
Sheikhlou M, Abbasi MA (2016) Genetic diversity of Iranian Lori-Bakhtiari sheep assessed by pedigree analysis. Small Rumin Res 141:99–105
Signer-Hasler H, Burren A, Ammann P, Drögemüller C, Flury C (2019) Runs of homozygosity and signatures of selection: a comparison among eight local Swiss sheep breeds. Anim Genet 50(5):512–525
Sobieraj-Kmiecik A, Drobik-Czwarno W, Nowak-Życzyńska Z, Wojciechowska M, Demars J, Bodin L et al. (2020) Genome analysis in local breeds: A case study on Olkuska sheep. Livest Sci 231:103880
Szpiech ZA, Xu J, Pemberton T, Peng W, Zöllner S, Rosenberg NA et al. (2013) Long runs of homozygosity are enriched for deleterious variation. Am J Hum Genet 93(1):90–102
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
Vatankhah M, Sigdel A, Abdollahi-Arpanahi R (2019) Population structure of Lori-Bakhtiari sheep in Iran by pedigree analysis. Small Rumin Res 174:148–155
Vatankhah M, Talebi MA, Blair H (2016) Genetic analysis of Lori-Bakhtiari lamb survival rate up to yearling age for autosomal and sex-linked. Small Rumin Res 136:121–126
Vatankhah M, Talebi MA, Edriss MA (2008) Estimation of genetic parameters for reproductive traits in Lori–Bakhtiari sheep. Small Rumin Res 74:216–220
Vazquez AI, Bates DM, Rosa GJ, Gianola D, Weigel KA (2010) Technical note: an R package for fitting generalized linear mixed models in animal breeding. J Anim Sci 88(2):497–504
Wright S (1922) Coefficients of inbreeding and relationship. Am Nat 56(645):330–338
Yeomans L, Martin L, Richter T (2017) Expansion of the known distribution of Asiatic mouflon (Ovis orientalis) in the Late Pleistocene of the Southern Levant. R Soc Open Sci 4:170409
Zhang Q, Calus MPL, Guldbrandtsen B, Lund MS, Sahana G (2015) Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet 16:88
Acknowledgements
The authors are grateful to Guilan and Bu-Ali Sina Universities for providing financial support for this study.
Author information
Authors and Affiliations
Contributions
Conceptualization: RA; analyses: RA, SZM, CG, MS & MHF; draft preparation: RA, SZM, NGHZ, PZ & MHM; review and editing: CG, MS, SZM & NGHZ. All authors have read and agreed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor Christine Baes.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Abdoli, R., Mirhoseini, S.Z., Ghavi Hossein-Zadeh, N. et al. Runs of homozygosity and cross-generational inbreeding of Iranian fat-tailed sheep. Heredity 130, 358–367 (2023). https://doi.org/10.1038/s41437-023-00611-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-023-00611-y
This article is cited by
-
High-throughput DNA sequence analysis elucidates novel insight into the genetic basis of adaptation in local sheep
Tropical Animal Health and Production (2024)
