
Abstract
Hypercalciuria is the most common metabolic risk factor in people with kidney stone disease. Its etiology is mostly multifactorial, although monogenetic causes of hypercalciuria have also been described. Despite the increased availability of genetic diagnostic tests, the vast majority of individuals with familial hypercalciuria remain unsolved. In this study, we investigated a consanguineous pedigree with idiopathic hypercalciuria. The proband additionally exhibited severe skeletal deformities and hyperparathyroidism. Whole-exome sequencing of the proband revealed a homozygous ultra-rare variant in TRPV5 (NM_019841.7:c.1792G>A; p.(Val598Met)), which encodes for a renal Ca2+-selective ion channel. The variant segregates with the three individuals with hypercalciuria. The skeletal phenotype unique to the proband was due to an additional pathogenic somatic mutation in GNAS (NM_000516.7:c.601C>T; p.(Arg201Cys)), which leads to polyostotic fibrous dysplasia. The variant in TRPV5 is located in the TRP helix, a characteristic amphipathic helix that is indispensable for the gating movements of TRP channels. Biochemical characterization of the TRPV5 p.(Val598Met) channel revealed a complete loss of Ca2+ transport capability. This defect is caused by reduced expression of the mutant channel, due to misfolding and preferential targeting to the proteasome for degradation. Based on these findings, we conclude that biallelic loss of TRPV5 function causes a novel form of monogenic autosomal recessive hypercalciuria, which we name renal Ca2+-wasting hypercalciuria (RCWH). The recessive inheritance pattern explains the rarity of RCWH and underscores the potential prevalence of RCWH in highly consanguineous populations, emphasizing the importance of exploration of this disorder within such communities.
Similar content being viewed by others

Introduction
Hypercalciuria is a condition of increased urinary calcium (Ca2+) excretion, present in ~4-10% of the otherwise healthy population [1, 2]. It is defined as >250 mg/24 h urine and >200 mg/24 h urine in men and women respectively, and an increased urinary calcium/creatinine ratio compared to age-matched children [3, 4]. People who suffer from hypercalciuria are mostly asymptomatic but may have nephrocalcinosis or increased bone resorption [5, 6]. Hypercalciuria is multifactorial, with an estimated heritability of ~50% [7]. To date, at least 25 genes have been linked to monogenic hypercalciuria, some accompanied by other systemic findings [8]. Nevertheless, the basis of hypercalciuria is still unidentifiable in the majority [8]. Many of these people have idiopathic hypercalciuria (IH), which is used as an umbrella term for hypercalciuric individuals with normocalcemia and no other systemic diseases, such as hyperparathyroidism [9].
Hypercalciuria can arise due to defects in three organs/systems and is classified according to the primary defect as absorptive (intestine), resorptive (bone), or renal leak type hypercalciuria [10]. These three organs collaborate to maintain a steady blood Ca2+ concentration and the interplay between them is controlled by a hormonal feedback loop mainly involving parathyroid hormone (PTH) and 1,25-dihydroxy vitamin D3 (calcitriol) [8]. The main role of kidneys is to regulate Ca2+ excretion. The fine-tuning of renal Ca2+ reabsorption takes place in the distal convoluted (DCT) and connecting tubules (CNT) [8]. TRPV5 (formerly known as ECaC1, Epithelial Calcium Channel 1), encoded by the TRPV5 gene on 7q34 [MIM: 606679], is the main Ca2+ channel in the apical membranes. TRPV5-dependent Ca2+ transport constitutes the rate-limiting step in DCT/CNT-mediated Ca2+ reabsorption [11]. Knockout Trpv5 mouse models, which suffer from a phenotype of renal Ca2+ wasting, illustrate the importance of this channel in Ca2+ homeostasis [12]. In addition, several human TRPV5 variants have been associated with hypercalciuria and nephrolithiasis in case-control studies [13, 14]. Despite its role in Ca2+ homeostasis, TRPV5 is yet to be linked to a monogenic disorder [11].
In this study, we explored the genetic cause of hypercalciuria driven by renal Ca2+ wasting in a family of consanguineous parents and three affected individuals. We employed a combination of homozygosity mapping and massively parallel sequencing to identify a homozygous missense mutation in TRPV5 in all affected individuals. The proband also demonstrated hyperparathyroidism and severe bone deformities explained by a blended phenotype due to an additional somatic GNAS mutation. Biochemical and functional analyses on the mutated TRPV5 channel demonstrated failure to increase plasma membrane Ca2+ permeability in HEK293 cells and increased proteasomal degradation. Taken together, our data demonstrate that biallelic TRPV5 mutations are responsible for a novel form of monogenic hypercalciuria in humans, which we term renal calcium-wasting hypercalciuria (RCWH).
Subjects and methods
Study participants
A single-family of 8 individuals was evaluated for hypercalciuria by urinary Ca2+/creatinine ratio (uCa/Cr) from either spot or 24-hour urine samples. uCa/Cr levels were compared to age-matched reference values [4]. Additionally, all individuals underwent physical examination and systematic evaluation. Genomic DNA was extracted using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) from the peripheral blood of all family members. Additionally, peripheral whole blood RNA was obtained from the proband (II-2) for splice-site sequencing (Supplementary Information).
The study protocol was approved by the local ethics committees of Hacettepe University and Dr. Sami Ulus Obstetrics and Gynecology, Children Health and Disease Training and Research Hospital (GO21/446, 30.03.2021, and 2012-KAEK-15/2030, 22.01.2020). The study was conducted in accordance with the Declaration of Helsinki and written informed consent was obtained from all participants and/or their parents.
DNA sequencing
Whole-exome sequencing (WES) using peripheral blood DNA from the proband was utilized to uncover the genetic etiology. WES library was generated using Twist Human Core Exome Kit v2 (Twist Bioscience, San Francisco, CA, USA) and was sequenced on the Illumina NextSeq 500 (Illumina, San Diego, CA, USA) platform. Sequence reads were aligned to hg19/GRCh37 and germline/somatic variants were called and analyzed following the pipelines provided in Supplementary Information. The strong implication of identity-by-descent prompted us to perform homozygosity mapping for all family members and focus variant analysis on shared long contiguous stretches of homozygosity (LCSHs) in either all individuals with hypercalciuria (II:1, II:2, II:6) or those unique to the proband (II:2). Furthermore, gene panels were adopted into the pipeline as detailed in Supplementary Information.
Any candidate variants were inspected via Integrative Genomics Viewer (IGV) and sequenced in family members by Sanger sequencing using BigDye Terminator v3.1 (ThermoFisher Scientific, Waltham, MA, USA) on ABI 3500 Genetic Analyzer (ThermoFisher Scientific, Waltham, MA, USA) for evaluating segregation and validation.
Homozygosity mapping
For all eight individuals, genome-wide single nucleotide polymorphism (SNP) genotyping was performed using Infinium HumanCytoSNP-12 v2.1 microarrays, according to the manufacturer’s protocol. GenomeStudio software v2.0 (Illumina, San Diego, CA, USA) was utilized for calling SNP genotypes according to hg19/GRCh37, and obtained genotypes were viewed by MS-Excel to look for ≥2 Mbp LCSHs, indicating homozygosity-by-descent.
Generation of TRPV5 p.(Val598Met) construct
Site-directed mutagenesis was used to introduce a point mutation in the wildtype TRPV5 sequence, using the Q5 hot-start kit (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s protocol. Details of the protocol are presented in Supplementary Information. The resulting construct contained the TRPV5 p.(Val598Met) ORF, CMV promoter, and GFP tag and was validated using Sanger sequencing.
Functional evaluation of TRPV5 in HEK293 cells
HEK293 cells were transfected with the aforementioned constructs using Lipofectamine2000 (Thermo Fisher Scientific, Waltham, MA, USA). TRPV5 protein levels were evaluated in a semi-quantitative manner using Western Blotting, either on total cellular proteins or on cell-surface proteins marked by biotinylation. The effect of the mutation on protein stability was assessed using inhibitors of proteasomal (MG-132 (MG), 5 µM, obtained from Merck, Darmstadt, Germany) and lysosomal (Bafilomycin A1 (Baflo), 100 nM, obtained from Cell Signaling Technology, Beverly, MA, USA) degradation. Finally, the permeability of the TRPV5 channel was evaluated using a previously described radioactive 45Ca2+ uptake protocol [15]. Each assay and statistical method are detailed in Supplementary Information.
Results
Clinical features of the affected individuals
The 10-year old proband has been evaluated at the Pediatric Endocrinology and Medical Genetics Departments of Dr. Sami Ulus Obstetrics and Gynecology, Children Health and Disease Training and Research Hospital for short stature, bone deformities with several fractures, and hypercalciuria. He was the second of six siblings born to consanguineous parents of Syrian origin (Fig. 1A). He appeared unaffected at birth and started to walk independently when he was 1 year old. At 1.5 years, parents noticed unusual bending of the limbs, gradually progressing and becoming painful. During his first visit to the medical genetics clinic at 7 years old, he was not able to walk. He had caput quadratum, thickening of joints, and bent limbs, also documented by radiological imaging (Fig. 1B). Neurodevelopmental milestones throughout his life and cognitive development assessed at 7.5 years were clinically appropriate for his age. Ophthalmologic, audiologic, and dermatologic evaluations were normal, and echocardiography revealed a patent foramen ovale.

A The hypercalciuria and skeletal defects running in the family are indicated on the pedigree. B Photographs and radiological images of the proband demonstrate severe skeletal deformities. Radiographs revealed generalized osteopenia. The severe bowing deformities in the right leg (B1, red arrow) and left arm (B2, red arrow) are seen upon inspection, along with the widening of the left wrist, metacarpophalangeal and interphalangeal joints (B3, red arrows). The bone age is delayed and appropriate for ~4 years (B4). Radiographs (B4-6) reveal extensive POFD-related findings; including the characteristic “ground-glass” appearance; composed of mixed radiolucent (cystic) and sclerotic lesions indicated by orange asterisks, metaphyseal flaring & cupping (green arrows), and bowing of long bones (red arrows). Occasional cortical thinning and medullary enlargement of the long bones are also evident. Vertebrae appear spared (B7). The computed tomography scans also show radiolucent bony lesions with sclerotic rims (B8, orange asterisks) and 3D-reconstructions of CT scans demonstrate bowing in femora (Shepherd’s crook sign) and tibiae (red arrows). Bowing of the bones within radiological images indicates poorly healed new and old appendicular fractures. C Age-dependent change in Ca2+ metabolism-related biochemical parameters of the proband is shown. Any missing measurements are indicated as dashed lines. The three rectangles at the bottom indicate the various treatments the proband received over time. D Urinalysis reveals multiple typical octahedral calcium oxalate crystals in the proband.
The combination of the proband’s skeletal findings and hypercalciuria resembled severe rickets or osteogenesis imperfecta. At 5 years, laboratory tests to evaluate Ca2+ metabolism revealed hypercalciuria, normocalcemia, hypophosphatemia, hyperphosphaturia, elevated serum alkaline phosphatase (ALP) and parathyroid hormone (PTH) but with normal 25-hydroxy vitamin D3 and calcitriol levels (Fig. 1C and Supplementary Table S1). Additionally, the Tc-99m sestamibi scintigraphy in the proband did not reveal any abnormal uptake by parathyroids. This metabolic profile partially coincided with those seen in four disorders; (1) primary hyperparathyroidism, which contradicts proband’s normocalcemia and normal parathyroid scintigraphy; (2) hereditary hypophosphatemic rickets except for proband’s hypercalciuria; (3) hereditary hypophosphatemic rickets with hypercalciuria (HHRH) except for hyperparathyroidism and normal calcitriol levels seen in the proband; (4) Polyostotic fibrous dysplasia (POFD) albeit hyperparathyroidism and hypercalciuria. Although each of these conditions differed from the proband’s metabolic profile to some extent, initially sodium phosphate was administered for HHRH treatment. Additionally, due to low 1,25-(OH)2 Vitamin D levels, calcitriol was supplemented, which did not benefit the calcium metabolism and was eventually discontinued. After the identification of a potentially pathogenic variant in TRPV5, the treatment was tailored towards a renal Ca2+-wasting condition. Prior treatments were replaced by oral Ca2+-lactate, which succeeded in decreasing PTH levels but failed to improve bone phenotype in the short term. The bone turnover parameters under this treatment indicated increased bone formation, represented by continuously high ALP and osteocalcin levels with normal bone resorption characterized by normal urinary deoxypyridinoline excretion. The evolution of Ca2+ metabolism indicators is summarized in Fig. 1C and details can be found in Supplementary Table S1.
Repeated renal ultrasounds during the proband’s follow-up revealed intermittent kidney stones and microscopic urinalysis identified multiple calcium oxalate crystals (Fig. 1D). He consistently had normal urinary pH, and no glucosuria, aminoaciduria, or proteinuria. His siblings were also screened for hypercalciuria (Supplementary Table S1). These measurements demonstrated persistent hypercalciuria in individuals II-1, II-2, and II-6. None of the skeletal abnormalities found in the proband were observed in these individuals. The mother (individual I-2) had nephrolithiasis during her fifth pregnancy at 25 years of age, presumptively suggesting that heterozygosity for the TRPV5 variant may be a predisposing factor for nephrolithiasis.
Genome-wide search for the cause of hypercalciuria in the family identifies biallelic p.(Met598Val) in TRPV5
Considering the parental consanguinity and three hypercalciuric children, we carried out genome-wide homozygosity mapping of the whole family to explore genomic regions identical-by-descent. This revealed 2 shared LCSH regions in affected individuals (II-1, II-2, and II-6), which were located on 1q24.2–1q32.1 (168,891,735-201,400,200) and 7q33–7q35 (141,672,604–145,136,645) (Fig. 2A, B), excluding 98.85% of the genome. When high-quality (GQ ≥ 30 and read depth ≥20), homozygous, and rare (MAF ≤ 0.01 and no homozygous individual in gnomAD v4.0) variants in shared LCSHs from WES data were filtered in only 3 candidate variants remained (Table S2). The variant containing genes, MROH9 and METTL11B, have no renal expression [16]. Additionally, these are unlikely to be disease-causing genes as healthy individuals harboring homozygous loss-of-function variants in MROH9 have already been reported in gnomAD v4.0 and METTL11B-knockout mice have no significant phenotypic abnormality [17]. On the other hand, the only remaining candidate, TRPV5, is exclusively expressed in the kidney and under selection pressure in the healthy human population (LOEUF = 0.97) with no reported homozygous predicted loss-of-function variants in gnomAD v4.0 [16, 18]. The NM_019841.7:c.1792G>A (p.(Val598Met)) variant in the TRPV5 protein disturbed the highly conserved ‘VATVV’ motif (Fig. 2C). Sanger sequencing confirmed this variant in the proband and demonstrated cosegregation with hypercalciuria within the family (Fig. 2D).

A LCSHs (≥2 Mbp) on all autosomes and the X chromosome are shown for each member of the family, specified by the color legend on the right. Gray dashed rectangles on chromosomes 1 (left) and 7 (right) indicated LCSHs shared by all affected individuals, while blue dashed rectangles on 7 genomic loci on 6 chromosomes correspond to LCSHs unique to the proband. B A detailed view of the two genomic loci common in all affected siblings is shown. Each row represents an individual genome and the vertical lines indicate the genotype for the aligned genomic loci. Homozygous and heterozygous loci in the proband (II-2) are indicated in blue and orange, respectively. The same color code is also valid for other individuals with the addition of white and red lines which symbolize contrasting homozygous genotypes and erroneous genotypes, respectively. Note that 4 genes with candidate variants on both chromosomes are marked with red arrowheads. C Evolutionary conservation of the 598th residue of TRPV5 across 10 commonly used model organisms. Note that this position is occupied by either valine or alanine residues in all species, while the p.(Val598Met) variant replaces these with the bulkier methionine residue containing Sulfur. D Sanger electropherograms of the TRPV5:c.1792G>A and GNAS:c.601C>T variants reveal respective genotypes for all family members. Note that the somatic GNAS variant is only present in the proband with distinctively lower peak amplitude.
Furthermore, the proband’s exceptionally severe skeletal phenotype hinted towards genetic variants besides TRPV5 in this individual. For this purpose, we initially explored 7 LCSH regions unique to the proband (Fig. 2A). These regions contain 2 candidate high-quality, homozygous, and rare variants in POP1 and TSPAN6 which are excluded as shown in Table S2. Additionally, the synonymous variant 3 bp to the nearest intron-exon boundary in POP1 did not alter the mRNA splicing (Supplementary Fig. S1). We also investigated the WES data with 2 virtual panels. The virtual Skeletal Disorders panel uncovered 8 candidate variants, which are further excluded (Table S2). On the other hand, the virtual Hypercalciuria/Hyper-PTH panel uncovered only the TRPV5 variant, but this variant being not unique to the proband, cannot explain his severe skeletal phenotype. Lastly, we looked for somatic variants in the virtual Skeletal Disorders panel and identified a well-characterized pathogenic GNAS variant (NM_000516.7:c.601C>T; p.(Arg201Cys)), exclusively present in the proband (Fig. 2D) with a variant allele fraction of 0.12 (Supplementary Fig. S2). In light of this finding, a reevaluation of the clinical and radiological findings established the diagnosis of POFD (OMIM 174800).
TRPV5 p.(Val598Met) mutant channel is not functional
The p.(Val598Met) residue is located in the TRP helix region of TRPV5, which plays an instrumental role in controlling the channel pore gating, folding, and assembly. Initially, we visualized the wildtype and TRPV5 p.(Val598Met) variant structure models in UCSF Chimera, using an existing full-length wildtype TRPV5 structure and Dynamut2 prediction software [19,20,21]. We found that the TRPV5 p.(Val598Met) variant is more tightly contacted by surrounding structures (Fig. 3A, B). DynaMut2 prediction also suggests that the variant decreases protein flexibility. Interestingly, several papers have highlighted that the TRP channel lower pore opening and closing depends on movement within the S4-S5 linker, the TRP helix, and the pore helices [22, 23]. In addition, a new group of transient potential ankyrin type 1 (TRPA1) antagonists, a channel that is structurally related to TRPV5, have been shown to antagonize transitional movement in the S4-S5 linker and TRP helix, which are required for channel opening [24]. Taken together, these studies suggest that reduced flexibility in the TRP helix could have detrimental effects on protein function. As such, we investigated whether the channel function of the TRPV5 p.(Val598Met) variant is altered. To this end, TRPV5 wildtype and p.(Val598Met) plasmids were transfected in HEK293 cells and TRPV5-dependent Ca2+ transport was quantified using a 45Ca2+ uptake assay. Uptake is significantly increased in HEK293 cells that express the wildtype TRPV5 channel, compared to mock-transfected HEK293 cells (p = 0.0026) (Fig. 3C). In contrast, transfection with the TRPV5 p.(Val598Met) mutant channel produces 45Ca2+ uptake similar to mock-transfected cells (p = 0.6), suggesting that the TRPV5 p.(Val598Met) channel is non-functional. Functional TRPV5 channels consist of four subunits. To see whether TRPV5 p.(Val598Met) could have a dominant negative effect on the channel activity (simulating the heterozygous state), we co-transfected TRPV5 wildtype and p.(Val598Met) plasmids (1:1 ratio) in HEK293 cells. While there is a slight decrease in 45Ca2+ uptake compared to the TRPV5 wildtype condition, this is likely due to less TRPV5 wildtype DNA used in the co-transfection setup than in the condition that contains only TRPV5 wildtype DNA (Fig. 3C). In support of this, there is no significant difference (p = 0.2119) compared to the co-transfection of TRPV5 and mock (condition added to control for DNA amount) (Fig. 3C), indicating that TRPV5 p.(Val598Met) does not produce a dominant negative effect.

A Zoom of the wildtype TRPV5 channel structure (model obtained from the protein data bank (PDB) database with accession number: 6O1P) with p.(Val598Met) and its surroundings highlighted. Helix-loop-helix (HLH), transmembrane helices 1 and 6 (S1/S6), pre helix before first transmembrane helix (pre-S1). B Zoom of the mutated TRPV5 channel with a focus on the interactions that p.Met598 makes with residues in the pre-S1 and HLH domains. The mutated protein model was made by DynaMut2 prediction software. C Relative 45Ca2+ uptake in HEK293 cells transiently transfected with either mock, TRPV5 wildtype (wt), TRPV5 p.(Val598Met) (V598M), co-transfection of TRPV5 wildtype with TRPV5 p.Val598Met (wt/V598M), co-transfection of mock with TRPV5 wildtype (mock/wt) and transfection of TRPV5 wildtype treated with the TRPV5 blocker ruthenium red (RR). Uptake is normalized to the TRPV5 wt condition. The experiment consists of 4 independent biological replicates (N = 4) and individual data points corresponding to 3 technical replicates per condition. RR=ruthenium red. TRPV5 wt vs TRPV5 p.(Val598Met): p = 0.0026. D Representative immunoblot showing expression of TRPV5 (via HA-tag) and β-actin from one of the biological replicates of the 45Ca2+ uptake experiment shown in A. The triangle on the right side of the blot serves as an indicator for the complex glycosylated band of TRPV5.
We further investigated the underlying cause of the loss of function seen in the TRPV5 p.(Val598Met) channel by measuring protein expression via immunoblotting in TRPV5 wildtype and p.(Val598Met) transfected HEK293 cells. Interestingly, the TRPV5 p.(Val598Met) mutant channel lacks the complex-glycosylation band typically seen when visualizing TRPV5 (Fig. 3D). The condition where TRPV5 wildtype and TRPV5 p.(Val598Met) are co-transfected also showed reduced complex-glycosylation.
TRPV5 p.(Val598Met) mutant channel plasma membrane insertion is unaffected but proteasomal degradation is enhanced
The lack of complex-glycosylation on the TRPV5 p.(Val598Met) channel could point to a trafficking or folding defect that prevents TRPV5 p.(Val598Met) from reaching the plasma membrane. As such, the abundance of both channels at the plasma membrane was assessed with a cell-surface biotinylation assay. The abundance of TRPV5 p.(Val598Met) is clearly diminished in both the plasma membrane and whole-cell fractions, compared to the wildtype channel (Fig. 4A). However, the plasma membrane to whole-cell TRPV5 expression ratio is similar between all of the conditions (Fig. 4B), indicating no trafficking defect of TRPV5 p.(Val598Met) to the plasma membrane. The fact that TRPV5 p.(Val598Met) expression levels are lower in both cellular compartments, suggests a more upstream effect. To test this further, we treated TRPV5 wildtype and p.(Val598Met) transfected HEK293 cells with inhibitors of lysosomal (bafilomycin A1) and proteasomal (MG-132) protein degradation. Interestingly, inhibition of the proteasomal degradation rescued non-glycosylated TRPV5 p.(Val598Met) protein expression levels, while inhibition of the lysosomal protein degradation had no effect (Fig. 4C, D). Neither inhibitor changed the expression level of the TRPV5 wildtype, indicating that the rescue observed in the TRPV5 p.(Val598Met) condition is due to targeting of this defective protein to the proteasome.

A Representative immunoblot of a cell-surface biotinylation experiment in HEK293 cells transfected with TRPV5 wildtype (wt) and TRPV5 p.(Val598Met). The top panel shows the membrane fraction whereas the bottom panel corresponds to the whole-cell lysate fraction. No biotin control, where biotin is not added to the cells, confirms that there is no non-specific binding of unbiotinylated TRPV5 to the neutravidin beads. B Semi-quantification of expression signal of TRPV5 wt and TRPV5 p.(Val598Met) protein, plotted as the area under the curve (AUC), normalized to the TRPV5 wt expression. The top two panels show the whole-cell lysate and membrane fraction expression respectively, while the bottom panel depicts the trafficking efficiency as the ratio of membrane fraction expression divided by whole-cell lysate expression. The individual data points indicate the 3 independent biological replicates (N = 3). C Representative immunoblot of HEK293 cells transiently transfected with TRPV5 wt and TRPV5 p.(Val598Met) (TRPV5 V598M), treated with inhibitors of proteasomal and lysosomal protein degradation pathways. MG = MG-132 and baflo = Bafilomycin A1. D Semi-quantification of TRPV5 wt and TRPV5 p.(Val598Met) expression levels in 3 independent experiments where MG-132 and Bafilomycin A1 were added. Expression levels are normalized to the untreated TRPV5 wt condition. The asterisk denotes the significant difference between the conditions TRPV5 p.(Val598Met) without any inhibitors and TRPV5 p.(Val598Met) with Bafilomycin A1 (p < 0.05).
Discussion
Renal Ca2+ reabsorption by DCT and CNT is an important factor in determining the blood Ca2+ level and is under strict endocrine control [8]. The apical membranes of cells lining these tubules harbor TRPV5 as the central protein controlling calcium permeability. Herein, using a combination of homozygosity mapping and WES, we describe the homozygous missense variant (c.1792G>A; p.(Val598Met)) in TRPV5 that causes renal Ca2+-wasting hypercalciuria (RCWH). Functional characterization of the TRPV5 p.(Val598Met) mutant in HEK293 cells revealed that the channel is incapable of transporting significant amounts of Ca2+. TRPV5 p.(Val598Met) mutant had a complete absence of complex N-linked glycosylation and was also subject to increased proteasomal degradation.
Since its discovery in 1999, TRPV5 has been in the spotlight as a candidate gene causing IH. Despite the expectation, extensive GWASs failed to demonstrate any association between common variants in TRPV5 and nephrolithiasis/IH. However, in a large case-control study from Iceland, the rare missense TRPV5 variant (rs757494578, c.1589T>G; p.(Leu530Arg)) was associated with recurrent kidney stones [14]. The Ca2+ transport defect and the absence of complex-glycosylation that were observed in the TRPV5 p.(Leu530Arg) variant are remarkably similar to our observations for the p.(Val598Met) mutant channel [25]. Each TRPV5 monomer is synthesized as a 6-pass transmembrane protein in the endoplasmic reticulum (ER), undergoes N-linked glycosylation at p.Asn358 which is completed as complex-glycosylation in Golgi apparatus, and finally reaches the plasma membrane [11, 26, 27]. While it is noteworthy that both of these variants lose their complex-glycosylation, it does not fully explain the loss of transport function. Studies have shown that disruption of TRPV5 complex-glycosylation, via a p.(Asn358Gln) mutation, does not drastically reduce 45Ca2+ uptake and trafficking to plasma membrane in HEK293 cells [28].
We initially speculated, based on the decreased flexibility predicted by structural modeling and the observed loss-of-function effect, that channel pore opening and closing may be affected in the TRPV5 p.(Val598Met) mutant. However, the follow-up experiments showed that the loss-of-function effect could be attributed to increased breakdown of the mutant channel and a potential folding defect. Furthermore, a loss of complex-glycosylation can point to a folding defect in the mutated protein. It is known that proteins that repeatedly fail their folding checkpoints in the ER are de-mannosylated and transported to the cytosol, where they are broken down by the proteasome [29]. A well-known example is the cystic fibrosis transmembrane conductance regulator (CFTR) channel. Studies have shown that mutated versions of CFTR lose their complex-glycosylation tree, fail to fold correctly, and are subsequently broken down [30]. To check if this also applies to TRPV5, we inhibited the two main pathways of protein degradation. Inhibition of the proteasomal degradation by MG132, which reduces the degradation of mutant CFTR, resulted in the rescue of TRPV5 p.(Val598Met) protein, showing that the mutant protein is recognized as misfolded and targeted for destruction.
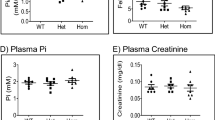
Although a well-defined TRPV5-related phenotype has not been previously described in humans, the critical role of Trpv5 in renal Ca2+ handling has long been demonstrated in mice [12, 31]. Trpv5-knockout mice (Trpv5−/−) present with marked hypercalciuria as a result of renal Ca2+ wasting, which is compensated by an increase of 1,25-dihydroxy vitamin D3 leading to normocalcemia. Additionally, these mice have PTH levels comparable to control mice [12]. This phenotypic picture is strikingly similar to the clinical phenotype observed in the individuals with homozygous TRPV5 mutations presented here (II-1, II-2, II-6), except for hyperparathyroidism in the proband. On the other hand, the heterozygous mice (Trpv5 + /−) do not have any significant hypercalciuria compared to wildtypes, which is in line with our observations that only show an insignificant reduction of TRPV5 function in double-transfected HEK293 cells [12]. These are in parallel with the phenotype of the heterozygotes (I-1, I-2, II-4, II-5) who either are not hypercalciuric or exhibit non-persistent mild hypercalciuria (Supplementary Table S1). It is known that dietary factors have a significant impact on calcium excretion [32]. Thus, it is likely that the non-persistent hypercalciuria in the unaffected family members is due to temporary environmental factors. Even so, any possible contribution of pathogenic heterozygous TRPV5 variants to urinary calcium excretion in humans is yet to be explored.
The severe skeletal findings and unexpectedly high PTH levels which are neither observed in the proband’s siblings nor the Trpv5-knockout mice are highly indicative of an additional intrinsic bone disorder in individual II-2. In fact, the mice lacking Trpv5 show no major bone deformation even at long-term follow-up (up to 78 weeks) [33, 34]. However, these mice demonstrate an accelerated reduction in trabecular and cortical bone thickness [12, 35]. Similar to the proband presented here, knockout mice do not manifest any increase in the bone resorption marker urinary deoxypyridinoline, while the bone formation marker, serum osteocalcin, is significantly higher compared to wild-type mice. In contrast to observations in the knockout mice, another bone formation marker, ALP, is strikingly higher in the proband, which also differs from his siblings with homozygous p.(Val598Met) [12].
All in all, the proband’s severe bone phenotype accompanied by increased PTH and ALP cannot be explained only by the TRPV5 defect. In recent years, with the widespread diagnostic utilization of WES, previously underestimated blended phenotypes have frequently been discovered, reaching up to 7.5% in certain populations [36]. Based on this observation, the search for an additional variant in the proband using a virtual hypercalciuria/hyperparathyroidism panel for disease-causing variants did not uncover any other germline variant (Supplementary Table S2). Nevertheless, a search for somatic variants revealed the mosaic pathogenic GNAS variant (p.(Arg201Cys)) which is a well-known cause of fibrous dysplasia. Considering the involvement of multiple long bones, increased serum ALP levels, and absence of GNAS-related McCune-Albright-Syndrome-associated extraskeletal findings, the skeletal deformities seen in the proband are compatible with POFD [37, 38]. Clinical presentation in POFD becomes apparent during early childhood and progresses until adulthood despite normal in-utero skeletal development similar to the clinical course seen in the proband [38]. However, hypercalciuria is inexplicable solely by fibrous dysplasia, because hypercalciuria has only been mentioned in a few cases of fibrous dysplasia that are complicated by Cushing’s Syndrome [39, 40]. Thus, the hypercalciuria is a consequence of the TRPV5 mutation, but not the GNAS mutation, in the proband.
Hyperparathyroidism in the proband can be explained neither by GNAS nor TRPV5 mutations alone. However, it is possible that hyperparathyroidism may arise as a result of an additive effect of these mutations [36]. Some individuals with fibrous dysplasia can have hyperparathyroidism due to accompanying vitamin D deficiency or primary hyperparathyroidism (parathyroid adenoma, hyperplasia), which are both ruled out in the proband [41,42,43,44]. The observation that elder Trpv5−/− mice demonstrate severely elevated serum PTH in contrast to young knockout mice may provide some explanation for the hyperparathyroidism seen in the proband [45]. It has been proposed that the elevated PTH level is a necessary compensatory mechanism to sustain normocalcemia in elder Trpv5−/− mice that cannot tolerate the age-related changes in intestinal and bone Ca2+ metabolism due to the already strained Ca2+ homeostasis [45]. The POFD in the proband undoubtedly burdens the Ca2+ metabolism and decreases the bone mineral density, which can presumably be compensated by increased PTH [42]. Normal PTH levels in proband’s affected siblings without the somatic GNAS mutation support the additive effect hypothesis. However, it is yet to be seen if hyperparathyroidism would arise in these siblings with advancing age.
Blended phenotypes caused by a combination of germline and somatic variants are quite rare and primarily observed in hematological disorders that involve a germline disorder ameliorated by indirect somatic genetic rescue [46, 47]. A previous study showed that osteoclast activity is decreased in Trpv5−/− mice, whereas fibrous dysplasia lesions demonstrate excessive osteoclast activation due to increased Gsα signaling [35, 48, 49]. Considering the contrasting impact of TRPV5 and GNAS mutations on osteoclastic activity, it is reasonable to postulate that the acquisition of a somatic GNAS mutation would lead to a transient and partial rescue of defective osteoclastic activity due to TRPV5 mutation.
In conclusion, comprehensive genetic and functional studies performed here demonstrate that TRPV5 is involved in autosomal recessively inherited IH through increased renal calcium excretion, which we name “Renal Calcium-Wasting Hypercalciuria” (RCWH). The anticipated relation between TRPV5 and IH is now demonstrated in humans after >20 years. The complete loss of function seen in the TRPV5 p.(Val598Met) channel, coupled with the unexpected autosomal recessive inheritance pattern suggests that a complete absence of TRPV5 function may be required to cause RCWH in childhood. The recessive inheritance pattern and extremely low allele frequency of the TRPV5 variant described in this work suggest that RCWH could be found most often in highly consanguineous populations and emphasize the importance of exploration of this disorder within those communities.
Data availability
Disease-causing TRPV5 variants have been submitted to ClinVar with the accession number SCV003852754. All data for cell culture studies were collected and saved in accordance with the FAIR principles. The whole-exome sequencing, SNP microarray, and other raw data are available upon reasonable request.
References
Kruse K, Kracht U, Kruse U. Reference values for urinary calcium excretion and screening for hypercalciuria in children and adolescents. Eur J Pediatr. 1984;143:25–31.
Robertson W, Morgan D. The distribution of urinary calcium excretions in normal persons and stone-formers. Clin Chim Acta. 1972;37:503–8.
Bollerslev J, Rejnmark L, Zahn A, Heck A, Appelman-Dijkstra NM, Cardoso L, et al. European expert consensus on practical management of specific aspects of parathyroid disorders in adults and in pregnancy: recommendations of the ESE Educational Program of Parathyroid Disorders (PARAT 2021). Eur J Endocrinol. 2022;186:R33–63.
So NP, Osorio AV, Simon SD, Alon US. Normal urinary calcium/creatinine ratios in African-American and Caucasian children. Pediatr Nephrol. 2001;16:133–9.
Alon US, Berenbom A. Idiopathic hypercalciuria of childhood: 4-to 11-year outcome. Pediatr Nephrol. 2000;14:1011–5.
Milart J, Lewicka A, Jobs K, Wawrzyniak A, Majder-Łopatka M, Kalicki B. Effect of vitamin D treatment on dynamics of stones formation in the urinary tract and bone density in children with idiopathic hypercalciuria. Nutrients. 2020;12:2521.
Hunter DJ, de Lange M, Snieder H, Mac Gregor AJ, Swaminathan R, Thakker RV, et al. Genetic contribution to renal function and electrolyte balance: a twin study. Clin Sci. 2002;103:259–65.
Downie ML, Alexander RT. Molecular mechanisms altering tubular calcium reabsorption. Pediatr Nephrol. 2022;37:707–18.
Albright F, Henneman P, Benedict PH, Forbes AP. Idiopathic hypercalciuria (a preliminary report). Proc R Soc Med. 1953;46:1077–81.
Pak CY, Kaplan R, Bone H, Townsend J, Waters O. A simple test for the diagnosis of absorptive, resorptive and renal hypercalciurias. N Engl J Med. 1975;292:497–500.
Hoenderop JG, van der Kemp AW, Hartog A, van de Graaf SF, van Os CH, Willems PH, et al. Molecular identification of the apical Ca2+ channel in 1, 25-dihydroxyvitamin D3-responsive epithelia. J Biol Chem. 1999;274:8375–8.
Hoenderop JG, van Leeuwen JP, van der Eerden BC, Kersten FF, WCM van derKemp A, Mérillat A-M, et al. Renal Ca 2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Investig. 2003;112:1906–14.
Khaleel A, Wu M-S, Wong HS-C, Hsu Y-W, Chou Y-H, Chen H-Y. A single nucleotide polymorphism (rs4236480) in TRPV5 calcium channel gene is associated with stone multiplicity in calcium nephrolithiasis patients. Mediators Inflamm. 2015;2015:375427.
Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, et al. Common and rare variants associated with kidney stones and biochemical traits. Nat Commun. 2015;6:7975.
de Groot T, Lee K, Langeslag M, Xi Q, Jalink K, Bindels RJ, et al. Parathyroid hormone activates TRPV5 via PKA-dependent phosphorylation. J Am Soc Nephrol. 2009;20:1693–704.
Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–5.
Groza T, Gomez FL, Mashhadi HH, Muñoz-Fuentes V, Gunes O, Wilson R, et al. The International Mouse Phenotyping Consortium: comprehensive knockout phenotyping underpinning the study of human disease. Nucleic Acids Res. 2023;51:D1038–45.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Dang S, van Goor MK, Asarnow D, Wang Y, Julius D, Cheng Y, et al. Structural insight into TRPV5 channel function and modulation. Proc Natl Acad Sci USA. 2019;116:8869–78.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
Rodrigues CHM, Pires DEV, Ascher DB. DynaMut2: assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021;30:60–9.
Cao E. Structural mechanisms of transient receptor potential ion channels. J Gen Physiol. 2020;152:e201811998.
Pumroy RA, Fluck EC 3rd, Ahmed T, Moiseenkova-Bell VY. Structural insights into the gating mechanisms of TRPV channels. Cell Calcium. 2020;87:102168.
Terrett JA, Chen H, Shore DG, Villemure E, Larouche-Gauthier R, Déry M, et al. Tetrahydrofuran-based transient receptor potential ankyrin 1 (TRPA1) antagonists: ligand-based discovery, activity in a rodent asthma model, and mechanism-of-action via cryogenic electron microscopy. J Med Chem. 2021;64:3843–69.
Wang L, Holmes RP, Peng J-B. The L530R variation associated with recurrent kidney stones impairs the structure and function of TRPV5. Biochem Biophys Res Commun. 2017;492:362–7.
Chang Q, Hoefs S, Van Der Kemp A, Topala C, Bindels R, Hoenderop J. The ß-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310:490–3.
de Groot T, van der Hagen EA, Verkaart S, Te Boekhorst VA, Bindels RJ, Hoenderop JG. Role of the transient receptor potential vanilloid 5 (TRPV5) protein N terminus in channel activity, tetramerization, and trafficking. J Biol Chem. 2011;286:32132–9.
Leunissen EH, Nair AV, Büll C, Lefeber DJ, van Delft FL, Bindels RJ, et al. The epithelial calcium channel TRPV5 is regulated differentially by klotho and sialidase. J Biol Chem. 2013;288:29238–46.
Molinari M. N-glycan structure dictates extension of protein folding or onset of disposal. Nat Chem Biol. 2007;3:313–20.
Xiong X, Chong E, Skach WR. Evidence that endoplasmic reticulum (ER)-associated degradation of cystic fibrosis transmembrane conductance regulator is linked to retrograde translocation from the ER membrane. J Biol Chem. 1999;274:2616–24.
Müller D, Hoenderop JG, Vennekens R, Eggert P, Harangi F, Méhes K, et al. Epithelial Ca2+ channel (ECAC1) in autosomal dominant idiopathic hypercalciuria. Nephrol Dial Transplant. 2002;17:1614–20.
Heller HJ, Doerner MF, Brinkley LJ, Adams-Huet B, Pak CY. Effect of dietary calcium on stone forming propensity. J Urol. 2003;169:470–4.
Loh NY, Bentley L, Dimke H, Verkaart S, Tammaro P, Gorvin CM, et al. Autosomal dominant hypercalciuria in a mouse model due to a mutation of the epithelial calcium channel, TRPV5. PLoS ONE. 2013;8:e55412.
van der Eerden BC, Koek WNH, Roschger P, Zillikens MC, Waarsing JH, van der Kemp A, et al. Lifelong challenge of calcium homeostasis in male mice lacking TRPV5 leads to changes in bone and calcium metabolism. Oncotarget. 2016;7:24928–41.
van der Eerden BC, Hoenderop JG, de Vries TJ, Schoenmaker T, Buurman CJ, Uitterlinden AG, et al. The epithelial Ca2+ channel TRPV5 is essential for proper osteoclastic bone resorption. Proc Natl Acad Sci USA. 2005;102:17507–12.
Rosina E, Pezzani L, Pezzoli L, Marchetti D, Bellini M, Pilotta A, et al. Atypical, composite, or blended phenotypes: how different molecular mechanisms could associate in double-diagnosed patients. Genes. 2022;13:1275.
Javaid MK, Boyce A, Appelman-Dijkstra N, Ong J, Defabianis P, Offiah A, et al. Best practice management guidelines for fibrous dysplasia/McCune-Albright syndrome: a consensus statement from the FD/MAS international consortium. Orphanet J Rare Dis. 2019;14:139.
Robinson C, Collins MT, Boyce AM. Fibrous dysplasia/McCune-Albright syndrome: clinical and translational perspectives. Curr Osteoporos Rep. 2016;14:178–86.
Kessel D, Hall C, Shaw D. Two unusual cases of nephrocalcinosis in infancy. Pediatr Radiol. 1992;22:470–1.
Kirk JM, Brain CE, Carson DJ, Hyde JC, Grant DB. Cushing’s syndrome caused by nodular adrenal hyperplasia in children with McCune-Albright syndrome. J Pediatr. 1999;134:789–92.
Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis. 2012;7:S4.
Corsi A, Collins MT, Riminucci M, Howell PG, Boyde A, Robey PG, et al. Osteomalacic and hyperparathyroid changes in fibrous dysplasia of bone: core biopsy studies and clinical correlations. J Bone Miner Res. 2003;18:1235–46.
Turcu AF, Clarke BL. Fibrous dysplasia of bone associated with primary hyperparathyroidism. Endocr Pract. 2013;19:226–30.
Vasilev V, Daly AF, Thiry A, Petrossians P, Fina F, Rostomyan L, et al. McCune-Albright syndrome: a detailed pathological and genetic analysis of disease effects in an adult patient. J Clin Endocrinol Metab. 2014;99:E2029–38.
van Abel M, Huybers S, Hoenderop JG, van der Kemp AW, van Leeuwen JP, Bindels RJ. Age-dependent alterations in Ca2+ homeostasis: role of TRPV5 and TRPV6. Am J Physiol Renal Physiol. 2006;291:F1177–83.
Choijilsuren HB, Park Y, Jung M. Mechanisms of somatic transformation in inherited bone marrow failure syndromes. Hematol Am Soc Hemato. Educ Program. 2021;2021:390–8.
Döcker D, Schubach M, Menzel M, Spaich C, Gabriel H-D, Zenker M, et al. Germline PTPN11 and somatic PIK3CA variant in a boy with megalencephaly-capillary malformation syndrome (MCAP)-pure coincidence? Eur J Hum Genet. 2015;23:409–12.
Piersanti S, Remoli C, Saggio I, Funari A, Michienzi S, Sacchetti B, et al. Transfer, analysis, and reversion of the fibrous dysplasia cellular phenotype in human skeletal progenitors. J Bone Miner Res. 2010;25:1103–16.
Riminucci M, Robey PG, Saggio I, Bianco P. Skeletal progenitors and the GNAS gene: fibrous dysplasia of bone read through stem cells. J Mol Endocrinol. 2010;45:355-64.
Acknowledgements
We thank all members of the family for participating in this study. The authors would like to thank Aranka Gerritsen for the technical assistance that she provided during the biochemical characterization of the TRPV5 p.(Val598Met) variant, Can Holyavkin from Gen Era Diagnostics for his assistance in obtaining SNP data from microarrays, Merve Ulak from Technohealth for facilitating WES studies.
Funding
This work is supported by the Hacettepe University Scientific Research Projects Coordination Unit grant (Grant Number: TUK-2022-19957) and the Radboudumc BMS PhD grant. Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK).
Author information
Authors and Affiliations
Contributions
Conceptualization: NGL, MvG, JvW, AC, JH. Methodology: NGL, MvG, JvW, AC, JH. Software: MA. Formal analysis: NGL, MvG, MA, AC. Investigation: NGL, MvG, SC. Resources: NGL, SC, JvW, FKC, JH. Writing—original draft: NGL, MvG, SC, MA, AC. Writing—review & editing: NGL, MvG, SC JvW, FKC, AC, JH. Visualization: MvG, AC. Supervision: JvW, AC, JH. Project administration: AC, JH. Funding acquisition: NGL, JvW, AC, JH.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study protocol was approved by the local ethics committees of Hacettepe University and Dr. Sami Ulus Obstetrics and Gynecology, Children Health and Disease Training and Research Hospital (GO21/446, 30.03.2021, and 2012-KAEK-15/2030, 22.01.2020). The study was conducted in accordance with the Declaration of Helsinki and written informed consent including publication of individual details was obtained from all participants and/or their parents. Additionally, written informed consent for publication of medical images was obtained for the proband.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guleray Lafci, N., van Goor, M., Cetinkaya, S. et al. Decreased calcium permeability caused by biallelic TRPV5 mutation leads to autosomal recessive renal calcium-wasting hypercalciuria. Eur J Hum Genet (2024). https://doi.org/10.1038/s41431-024-01589-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41431-024-01589-9
