
Abstract
Neonatal progeroid syndrome, also known as Wiedemann–Rautenstrauch syndrome, is a rare condition characterized by severe growth retardation, apparent macrocephaly with prominent scalp veins, and lipodystrophy. It is caused by biallelic variants in POLR3A, a gene encoding for a subunit of RNA polymerase III. All variants reported in the literature lead to at least a partial loss-of-function (when considering both alleles together). Here, we describe an individual with several clinical features of neonatal progeroid syndrome in whom exome sequencing revealed a homozygous nonsense variant in POLR3GL (NM_032305.2:c.358C>T; p.(Arg120Ter)). POLR3GL also encodes a subunit of RNA polymerase III and has recently been associated with endosteal hyperostosis and oligodontia in three patients with a phenotype distinct from the patient described here. Given the important role of POLR3GL in the same complex as the protein implicated in neonatal progeroid syndrome, the nature of the variant identified, our RNA studies suggesting nonsense-mediated decay, and the clinical overlap, we propose POLR3GL as a gene causing a variant of neonatal progeroid syndrome and therefore expand the phenotype associated with POLR3GL variants.
Similar content being viewed by others
Introduction
Neonatal progeroid syndrome (MIM 264090), also known as Wiedemann–Rautenstrauch syndrome, is a condition characterized mainly by a progeroid appearance with apparent macrocephaly, prominent scalp veins and a triangular face, sparse hair, lipodystrophy, and intrauterine growth restriction (IUGR) [1, 2]. Because of the recurrence in three unrelated sibships [3, 4], this disorder was thought to be an autosomal recessive condition, but no molecular basis had been identified until relatively recently. Jay et al. first reported an individual with a clinical diagnosis of Wiedemann–Rautenstrauch syndrome in whom exome sequencing revealed biallelic truncating variants in POLR3A (MIM 614258) [5]. This finding was confirmed recently by independent studies. Paolacci et al. found biallelic variants in POLR3A in ten individuals with clinical features consistent with a diagnosis of Wiedemann–Rautenstrauch syndrome. They described five additional individuals in whom a single variant in POLR3A was identified and for whom they hypothesized that large deletions/insertions, or intronic variants in the second allele might explain the phenotype [1]. Furthermore, Wambach et al. and Lessel et al. reported a total of ten additional individuals with biallelic variants in POLR3A associated with Wiedemann–Rautenstrauch syndrome [6, 7]. POLR3A encodes the largest subunit of RNA polymerase III, required for transcription of a subset of nonprotein coding RNAs [8]. Other POLR-related disorders include Treacher-Collins syndrome type 2 (MIM 613717) and type 3 (MIM 248390). Both conditions are characterized by craniofacial anomalies and are associated with variants in POLR1D (MIM 613715) and POLR1C (MIM 610060), respectively [9]. There are also POLR3-related leukodystrophies, which are associated with hypomyelination, hypodontia, and hypogonadotrophic hypogonadism and are caused by variants in POLR3A, POLR3B (MIM 614366), POLR1C, and POLR3K [10,11,12,13,14,15]. POLR3GL (MIM 617457) variants have newly been described in three patients with a phenotype mainly characterized by endosteal hyperostosis, oligodontia and short stature [16]. Here, we describe an individual with a phenotype consistent with a variant of neonatal progeroid syndrome who is homozygous for a nonsense variant in POLR3GL.
Methods
Clinical samples
Written informed consent for research was obtained from the parents of the individual for their own participation and for their child’s participation in the study. This project was approved by the institutional review board of CHU Sainte-Justine. The individual included in the present study was part of a clinical research exome project ongoing at CHU Sainte-Justine.
Array CGH
Array Comparative Genomic Comparation (aCGH) was done using Agilent’s CGXTM HD microarray (from PerkinElmer) with 180,000 probes, according to the manufacturer’s instructions.
Sequencing panels
The panel for RASopathies included the genes BRAF, HRAS, KRAS, MAP2K1, MAP2K2, NRAS, PTPN11, RAF1, RIT1, SOS1, and part of exon 2 of SHOC2. Sequencing was performed at GeneDx laboratory on a clinical basis. The panel did not include deletion and duplication analysis.
ASXL1 sequence analysis was performed at the Genetic Services Laboratory of the University of Chicago on a clinical basis. It did not include deletion and duplication analysis.
Whole-exome sequencing
Exome sequencing was performed using HiSeq 4000 and SeqCap EZ MedExome kit (Roche) at the Génome Québec/CHU Sainte-Justine Integrated Center for Pediatric Clinical Genomics (Montreal, Canada). Alignment was performed using a Burrows-Wheeler algorithm (BWA-mem v0.7.5a), variant calling was done using the Genome Analysis Toolkit (GATK) UnifiedGenotyper v2.6–4 tool, and variant annotation was done using Annovar v2014–11–12 and an in-house pipeline. The average coverage with RefSeq was 53.60 reads. 95.6% of the bases were covered with ≥10 reads and 90.7% were covered with ≥20 reads. The POLR3GL variant was confirmed by Sanger sequencing in the child and parents using standard techniques.
3D protein structural modeling
We used PDB file 5fj8 (https://www.ebi.ac.uk/pdbe/) reported by Hoffmann et al. [17] and the figure was generated using Pymol. [18]
Quantitative reverse transcription PCR analysis
Total RNA from affected individual and two controls were isolated from whole blood using PureLink RNA mini kit (Life Technologies, cat# 12183025). Equal amounts of RNA ware used to synthesize cDNA using qScript cDNA synthesis kit (Quanta Biosciences). cDNA was quantified using PowerUp SYBR green Master Mix (Applied Biosystems) on a LightCycler® 96 system (Roche) using primers 5′-TTCATCCGGCCAGCTGTCC-3′ and 5′-AGCAGAATTGTAATCCGTTCC-3′. The primers were selected for their specificity for POLR3GL and not POLR3G. Amplicons were resolved by agarose gel to determine the size. Relative gene expression levels were analyzed by 2−ΔΔCT method using TBP as the reference gene. Statistical significance was determined using an unpaired two-tailed Student’s t test.
Results
Clinical report
The individual is a 39-month-old female. She is the first child of non-consanguineous French-Canadian parents, and the family history revealed no relatives with issues similar to hers (Fig. 1a). Both parents are healthy. On the first-trimester ultrasound, increased nuchal translucency was noted at 5.69 mm, and the mother was then referred to our tertiary center for follow-up and investigations. Subsequent ultrasounds revealed severe intrauterine growth retardation and hyperechogenic fetal bowel. Chorionic villus sampling was performed and both karyotype and array comparative genomic hybridization (aCGH) were normal. RASopathies panel, performed due to increased nuchal translucency, and ASXL1 sequencing were negative. Maternal serology for common infections was also negative. No abnormality was found on fetal echocardiography. Fetal magnetic resonance imaging (MRI) showed microcephaly, cerebellar hypoplasia, hypertelorism, and dilatation of the lateral ventricles.

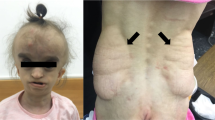
a Pedigree of the family. b Photograph of the individual at 2 years. c Images obtained from a CT scan of the head at the age of 3 months, showing from left to right the frontal, lateral, back and top views. Note the prominent forehead, large patent fontanels (anterior, posterior, mastoid), and triangular face. d Schematic representation of POLR3GL, showing all seven exons and above, the variant identified in the individual. Below are the variants identified in POLR3GL hyperostosis-oligodontia. Introns not drawn to scale. e Screenshot from IGV viewer showing the homozygous variant on the aligned exome (i.e. the BAM file). f Sanger sequencing chromatograms from the proband and her parents. g Total RNA from the affected individual and two unrelated controls was extracted from whole blood and equal amounts were reverse transcribed. POLR3GL relative mRNA levels were measured using qRT-PCR compared to the TBP control gene. Error bars represent standard deviations. h Yeast RNA polymerase III structure and conditions associated with the human orthologs, based on PDB file 5FJ8. Note the DNA in red, RNA in blue, and the POLR3GL/POLR3G ortholog in violet
She was delivered by caesarian section at 33 1/7 weeks of gestation for progressive IUGR. Her birth weight was 910 g (−2.75 SD), her length was 32.0 cm (−4 SD), her head circumference was 25.5 cm (−3 SD) and her Apgar scores were 3, 3 and 7 at 1, 5 and 10 min respectively. She was admitted to the neonatal intensive care unit. She developed severe bronchopulmonary dysplasia and pulmonary hypertension. Physical examination in the neonatal period showed a large anterior fontanel, hypertelorism, short palpebral fissures, proptosis, and midfacial hypoplasia with hypoplasia of the nasal bridge and alae (Fig. 1c). Her ears were low set and posteriorly rotated. She had a small mouth with a high-arched palate. There were no obvious limb malformations, but her fingers were long and ulnarly deviated, and her toes appeared short. Distal finger contractures and axial hypotonia were noted. Genitalia were normal except for hypoplastic labia minora. On skin examination, two infantile hemangiomas were noted, one localized in the frontonasal region and the other on the scapula.
Postnatal echocardiography revealed a bicuspid aortic valve and atrial septal defect type II. Mild ventriculomegaly was noted on cerebral MRI, which was consistent with prenatal findings, however no cerebellar hypoplasia was found. Rhinopharyngolaryngoscopy identified choanal atresia and stenosis of the aperture of the pyriform sinus. An ultrasound revealed left shoulder subluxation. No abnormality was found on abdominal, hip, or spinal ultrasound. Skeletal survey, including limb radiographs, was normal. Audiology and ophthalmology examinations were within normal limits. Transferrin isoelectric focusing and very long chain fatty acid analysis were both normal. Gastrostomy was performed because of feeding difficulties. She was discharged at five months of life with home oxygen for her bronchopulmonary dysplasia.
She was readmitted at 25 months of age for marked failure to thrive. Endocrinological evaluation revealed normal levels of growth hormone and thyroid-stimulating hormone. Follow-up cerebral MRI at 33 months revealed findings suggestive of communicating hydrocephalus. No lesion was identified to explain hydrocephalus. The white matter was normal, there was no abnormal enhancement with gadolinium, and the hypophysis was normal. Home oxygen was no longer necessary at 32 months and was thus discontinued. Examination at 34 months of age revealed a head circumference of 45 cm (1st percentile, −2.2 SD); a weight of 6.9 kg (<1st percentile, −5.1 SD), and a height of 68 cm (<1st percentile, −6.7 SD). She exhibited global developmental delay. She was able to sit without support but did not walk. She said two words and understood simple commands. Examination revealed mostly the same findings identified during the neonatal period (Fig. 1b). There was an infantile hemangioma of <1 cm in diameter on the left scapula. In addition, a protruding forehead with prominent scalp veins were noted and the anterior fontanel was still patent. No lipodystrophy, nor even a tendency towards lipodystrophy, was noted on examination, notably on the arms, legs, abdomen, and buttocks. Adiposity was perfectly normal on the whole body. The triceps skinfold measurement was 10 mm (+1 SD) on both arms. She had two teeth at the age of 7 months and lost them around the age of 23 months, and four other teeth grew between the age of 30 and 34 months. Marked axial hypotonia persisted.
Whole-exome sequencing
Data from whole exome sequencing were filtered for rare variants with minor allele frequencies below 0.5%. A total of 451 rare or novel variants were thus identified. Among the genes affected by these variants, 132 were previously associated with genetic disorders, in many cases it was a single heterozygous variant in a gene causing a recessive disease. None of the disorders associated with these 132 genes caused phenotypes resembling that presented by the individual described here. Further filtering for variants based on gnomAD frequencies narrowed the list to 73 variants (see supplementary file for homozygous or potential biallelic variants without homozygotes in gnomAD, heterozygous variants absent from gnomAD, and X-linked variants without hemizygotes in gnomAD. Human and mouse phenotypes associated with these genes are also listed).
We then considered whether variants in genes not previously associated with disorders were present (at the time the other patients with POLR3GL variants were not yet reported). A homozygous variant in exon 5 of POLR3GL (MIM 617457), NM_032305.2:c.358C>T; p.(Arg120Ter) was identified. Both parents are heterozygous for this variant (Fig. 1e, f). The variant creates a stop codon. We performed qRT-PCR, which showed an 84% decrease in POLR3GL mRNA level compared with controls (Fig. 1g), suggesting nonsense-mediated decay resulting in loss of function.
Discussion
POLR3GL encodes one of the 17 subunits of the RNA polymerase III, which transcribes small non-coding RNAs [17] (Fig. 1d, g). Another subunit of the RNA polymerase III named POLR3G is closely related to POLR3GL, and RNA polymerase III can contain either subunit. The POLR3GL and POLR3G genes arose from the duplication of a common ancestor and share 46% of their residues but have distinct expression patterns [19]. All RNA polymerase III subunits’ expression can be activated by MYC, except for POLR3GL, which is constitutively expressed. POLR3G is expressed in pluripotent cells but its expression decreases during differentiation, while POLR3GL is expressed ubiquitously, at stable levels in both pluripotent cells and throughout differentiation [20, 21]. In the GTEx database, expression levels are slightly less than half in the brain, colon, esophagus, liver, pancreas, and blood, compared with other tissues. Haurie et al. observed that suppression of POLR3GL expression leads to cell death [21], and Renaud et al. suggested that cell death would occur solely in cells with low or absent expression of POLR3G [19]. While the expression levels of the subunits differ for reasons which remain to be determined, RNA polymerase III occupies the same targets whether it includes POLR3G or POLR3GL. Together with two other subunits (POLR1C and POLR3F), POLR3G, or POLR3GL form a subcomplex which plays an active role in the initiation of transcription and which can be selectively detached from RNA polymerase III [22, 23]. The POLR3GL gene is found in the intronic region of NBPF10. The function of NBPF10 remains unclear and variants in this gene are not known to cause any recognizable condition.
Variants in POLR3GL have recently been associated with endosteal hyperostosis and oligodontia. Terhal et al. described monozygotic twins and a third unrelated individual with biallelic POLR3GL splice site variants. Their phenotype was characterized by endosteal hyperostosis, oligodontia, short stature, and mild facial dysmorphisms including upslanting palpebral fissures, thin lips, and long columella. Other features included motor retardation without intellectual disability, club feet, and delayed puberty. One patient also presented non-progressive spastic paraparesis, microcephaly, and hypoplasia of corpus callosum [16]. Thus, as highlighted in Table 1, the phenotype of these individuals is markedly different from the individual we describe. They mention that the patients they describe have a phenotype along the spectrum of other POLR3A/B/K and 1C-related disorders, such as 4H leukodystrophy (hypomyelination, hypodontia, hypogonadotropic hypogonadism; MIM 612440). No Polr3gl knockout mouse model has been reported. For Polr3a and Polr3g, homozygous knockout mice are embryonic lethal [24], similarly to mice homozygous for the variant equivalent to the human leukodystrophy variant POLR3B NM_018082.6:c.308 G>A, p.(Arg103His) [25], whereas mice homozygous for the variant equivalent to the human hypomyelinating leukodystrophy variant POLR3A NM_007055.4:c.2015G>A, p.(Gly672Glu) manifest slightly perturbed gait [26]. In human cell lines with the POLR3A NM_007055.4:c.2554 A>G, p.(Met852Val) variant, reductions in tRNAs and in myelin basic protein mRNA were observed [27].
The variant identified in the individual we describe is present in the gnomAD database in three individuals (see https://gnomad.broadinstitute.org/variant/1-145457572-G-A) but it has never been reported in the homozygous state in this or other population variant databases. The nucleotide substitution creates a stop codon and we therefore hypothesize that it would lead to loss of function by a nonsense-mediated decay mechanism. This hypothesis is supported by qRT-PCR performed on the individual’s blood, which demonstrated an 84% decrease in POLR3GL mRNA level. These elements advocate for pathogenicity of the variant.
The phenotype of the individual we describe appears to be fairly different compared with the other individuals with POLR3GL variants. While they share a few characteristics, namely short stature and oligodontia, the individual we report here exhibits a distinctive facies with prominent forehead and apparent scalp veins. Her phenotype is more consistent with neonatal progeroid syndrome, which has been associated with specific variants in POLR3A. POLR3A gene also encodes for a subunit of the RNA polymerase III. Jay et al., Paolacci et al., Wambach et al., and Lessel et al. recently described a total of 21 individuals with a clinical diagnosis of Wiedemann–Rautenstrauch syndrome in whom whole exome sequencing revealed biallelic variants in POLR3A [1, 5,6,7]. Table 1 details the clinical phenotype of these individuals with POLR3A variants who have been reported in the literature and compares it with the phenotype of the individual described here.
There are no definitive criteria for neonatal progeroid syndrome, but the individual we describe shares most of the core features of the syndrome. She had marked IUGR followed by severe growth retardation. Her head, with relative macrocephaly and prominent forehead and scalp veins, was characteristic. Natal teeth, a common finding in neonatal progeroid syndrome, were not present, but she had early eruption of two teeth that she lost briefly thereafter, similarly to other descriptions of neonatal progeroid syndrome. Furthermore, the presence of biallelic variants in the individual we describe is consistent with the autosomal recessive inheritance of neonatal progeroid syndrome. However, we observed neither abnormal fat distribution nor decreased subcutaneous fat tissue in this individual, whereas lipodystrophy is typically present in neonatal progeroid syndrome [2, 6]. This patient was born prematurely and had chronic respiratory illness, both of which may negatively affect fat mass. Changes in fat distribution may yet appear with age. Although this patient did not conform to all features of the progeroid syndrome, there are striking similarities, as is shown in the figures and tables.
Hence, we suggest that she has a variant of neonatal progeroid syndrome and that this is caused by a homozygous deleterious variant in POLR3GL. The distinct phenotype of this individual compared with other individuals with biallelic variants in POLR3GL could be explained by the different effects of the variants on RNA transcription. While variants reported by Terhal et al. are predicted to cause aberrant splicing of POLR3GL [16], while the variant identified in this individual leads to loss of function by nonsense-mediated decay. It is also possible that variants or epigenetic changes in other genes play a role in the phenotype of the individual we describe. Further identification of individuals carrying similar variants in POLR3GL will be helpful to improve our understanding of genotype-phenotype correlations.
Other conditions related to variants in genes encoding for RNA polymerase subunits include POLR3-related leukodystrophy, also called 4H-leukodystrophy, characterized by hypomyelination, hypodontia, and hypogonadotropic hypogonadism, and caused by biallelic variants in POLR3A, POLR3B, POLR1C, or POLR3K [10,11,12,13,14,15]. The individual we describe does not have hypomyelination on brain MRI and does not share most of the clinical features of this syndrome, with the exception of hypodontia and short stature. Treacher-Collins syndrome is mostly associated with variants in TCOF1, but variants in POLR1C and POLR1D have also been identified in a small proportion of individuals [9]. POLR1C and POLR1D encode two subunits shared by RNA polymerases I. The POLR1C subunit is also part of RNA polymerase III. In yeast, it has been shown that variants in either POLR1C or POLR1D affect the assembly of both RNA polymerases I and III, leading to reduced RNA production [28]. Zebrafish models showed an abnormal ribosomal synthesis and consequently, a deficiency in neural crest cells involved in craniofacial development [29]. Treacher-Collins syndrome is characterized by craniofacial abnormalities, including malar and mandibular hypoplasia, ear malformations, and ocular anomalies. The individual we describe did not have the main features of Treacher-Collins syndrome, and there is only a minor overlap with her phenotype as she exhibited midface hypoplasia and choanal atresia.
The present report strongly suggests an association of biallelic loss-of-function variants in POLR3GL with a variant of neonatal progeroid syndrome. To date, POLR3A is the only gene known to cause neonatal progeroid syndrome, while POLR3GL has recently been associated with endosteal hyperostosis and oligodontia. The phenotype of the patient presented here is more consistent with neonatal progeroid syndrome than with the phenotype of the three individuals with biallelic POLR3GL variants reported previously with a phenotype suggestive of POLR3-related leukodystrophy. Therefore, it expands the phenotypic spectrum associated with variants in POLR3GL. Differences in the phenotype of this patient compared with other patients described with POLR3GL variants could be due in part to the different impact of the variants on transcription, but we cannot exclude that other variants or epigenetic changes in other genes could contribute to the phenotype. Future case reports and studies in model organisms will be helpful to determine how deficiencies of POLR3A or POLR3GL affect development. We suggest that sequencing of POLR3GL might be considered in individuals with features compatible with neonatal progeroid syndrome, and no variant in POLR3A.
References
Paolacci S, Li Y, Agolini E, Bellacchio E, Arboleda-Bustos CE, Carrero D, et al. Specific combinations of biallelic POLR3A variants cause Wiedemann–Rautenstrauch syndrome. J Med Genet. 2018;55:837–46.
Paolacci S, Bertola D, Franco J, Mohammed S, Tartaglia M, Wollnik B, et al. Wiedemann-Rautenstrauch syndrome: A phenotype analysis. Am J Med Genet Part A. 2017;173:1763–72.
Pivnick EK, Angle B, Kaufman RA, Hall BD, Pitukcheewanont P, Hersh JH, et al. Neonatal progeroid (Wiedemann–Rautenstrauch) syndrome: report of five new cases and review. Am J Med Genet. 2000;90:131–40.
Arboleda G, Morales LC, Quintero L, Arboleda H. Neonatal progeroid syndrome (Wiedemann-Rautenstrauch syndrome): report of three affected sibs. Am J Med Genet Part A. 2011;155A:1712–5.
Jay AM, Conway RL, Thiffault I, Saunders C, Farrow E, Adams J, et al. Neonatal progeriod syndrome associated with biallelic truncating variants in POLR3A. Am J Med Genet Part A. 2016;170:3343–6.
Wambach JA, Wegner DJ, Patni N, Kircher M, Willing MC, Baldridge D, et al. Bi-allelic POLR3A loss-of-function variants cause autosomal-recessive Wiedemann-Rautenstrauch syndrome. Am J Hum Genet. 2018;103:968–75.
Lessel D, Ozel AB, Campbell SE, Saadi A, Arlt MF, McSweeney KM, et al. Analyses of LMNA-negative juvenile progeroid cases confirms biallelic POLR3A mutations in Wiedemann-Rautenstrauch-like syndrome and expands the phenotypic spectrum of PYCR1 mutations. Hum Genet. 2018;137:921–39.
Sepehri S, Hernandez N. The largest subunit of human RNA polymerase III is closely related to the largest subunit of yeast and trypanosome RNA polymerase III. Genome Res. 1997;7:1006–19.
Dauwerse JG, Dixon J, Seland S, Ruivenkamp CA, van Haeringen A, Hoefsloot LH, et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet. 2011;43:20–22.
Bernard G, Vanderver A. POLR3-related leukodystrophy; in: Adam MP, Ardinger HH, Pagon RA. et al. (eds): Gene Reviews ((R)). Seattle (WA), 1993.
Dorboz I, Dumay-Odelot H, Boussaid K, Bouyacoub Y, Barreau P, Samaan S, et al. Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation. Neurol Genet. 2018;4:e289.
Bernard G, Chouery E, Putorti ML, Tetreault M, Takanohashi A, Carosso G, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:415–23.
Tetreault M, Choquet K, Orcesi S, Tonduti D, Balottin U, Teichmann M, et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:652–5.
Wolf NI, Vanderver A, van Spaendonk RM, Schiffmann R, Brais B, Bugiani M, et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology. 2014;83:1898–905.
Thiffault I, Wolf NI, Forget D, Guerrero K, Tran LT, Choquet K, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun. 2015;6:7623.
Terhal PA, Vlaar JM, Middelkamp S, Nievelstein RAJ, Nikkels PGJ, Ross J, et al. Biallelic variants in POLR3GL cause endosteal hyperostosis and oligodontia. Eur J Hum Genet. 2019. [Epub ahead of print].
Hoffmann NA, Jakobi AJ, Moreno-Morcillo M, Glatt S, Kosinski J, Hagen WJ, et al. Molecular structures of unbound and transcribing RNA polymerase III. Nature. 2015;528:231–6.
Schrodinger, LLC. The PyMOL molecular graphics system, Version 1.8, 2015.
Renaud M, Praz V, Vieu E, Florens L, Washburn MP, l’Hote P, et al. Gene duplication and neofunctionalization: POLR3G and POLR3GL. Genome Res. 2014;24:37–51.
Lund RJ, Rahkonen N, Malonzo M, Kauko L, Emani MR, Kivinen V, et al. RNA polymerase III subunit POLR3G regulates specific subsets of polyA(+) and small RNA transcriptomes and splicing in human pluripotent stem cells. Stem Cell Rep. 2017;8:1442–54.
Haurie V, Durrieu-Gaillard S, Dumay-Odelot H, Da Silva D, Rey C, Prochazkova M, et al. Two isoforms of human RNA polymerase III with specific functions in cell growth and transformation. Proc Natl Acad Sci USA. 2010;107:4176–81.
Wang Z, Roeder RG. Three human RNA polymerase III-specific subunits form a subcomplex with a selective function in specific transcription initiation. Genes Dev. 1997;11:1315–26.
Geiduschek EP, Kassavetis GA. The RNA polymerase III transcription apparatus. J Mol Biol. 2001;310:1–26.
Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, et al. High-throughput discovery of novel developmental phenotypes. Nature. 2016;537:508–14.
Choquet K, Pinard M, Yang S, Moir RD, Poitras C, Dicaire MJ, et al. The leukodystrophy mutation Polr3b R103H causes homozygote mouse embryonic lethality and impairs RNA polymerase III biogenesis. Mol Brain. 2019;12:59.
Choquet K, Yang S, Moir RD, Forget D, Lariviere R, Bouchard A, et al. Absence of neurological abnormalities in mice homozygous for the Polr3a G672E hypomyelinating leukodystrophy mutation. Mol Brain. 2017;10:13.
Choquet K, Forget D, Meloche E, Dicaire MJ, Bernard G, Vanderver A, et al. Leukodystrophy-associated POLR3A mutations down-regulate the RNA polymerase III transcript and important regulatory RNA BC200. J Biol Chem. 2019;294:7445–59.
Walker-Kopp N, Jackobel AJ, Pannafino GN, Morocho PA, Xu X, Knutson BA. Treacher Collins syndrome mutations in Saccharomyces cerevisiae destabilize RNA polymerase I and III complex integrity. Hum Mol Genet. 2017;26:4290–4300.
Noack Watt KE, Achilleos A, Neben CL, Merrill AE, Trainor PA. The roles of RNA polymerase I and III subunits Polr1c and Polr1d in craniofacial development and in zebrafish models of Treacher Collins syndrome. PLoS Genet. 2016;12:e1006187.
Acknowledgements
We would like to thank the family for participating in this study and all clinicians involved in the care of this child. We thank the funding support from the Canadian Institutes of Health Research (RN315908) and the Fonds de Recherche du Québec - Santé (30647). GB has received the New Investigator Salary Award from the Canadian Institutes of Health Research (2017–2022).
Funding
We acknowledge support from the Canadian Institutes of Health Research of Canada and the Fonds de Recherche Santé Québec to PMC. GB has received the New Investigator Salary Award from the Canadian Institutes of Health Research (2017–2022).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This project was approved by the institutional review board of CHU Sainte-Justine. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Written informed consent was obtained from all participants included in the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Accession Numbers Variant entry was deposited to: https://databases.lovd.nl/shared/genes/POLR3GL (ID: #0000498533)
Supplementary information
Rights and permissions
About this article

Cite this article
Beauregard-Lacroix, E., Salian, S., Kim, H. et al. A variant of neonatal progeroid syndrome, or Wiedemann–Rautenstrauch syndrome, is associated with a nonsense variant in POLR3GL. Eur J Hum Genet 28, 461–468 (2020). https://doi.org/10.1038/s41431-019-0539-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0539-6
This article is cited by
-
tRNA dysregulation and disease
Nature Reviews Genetics (2022)
-
Whole-exome sequencing reveals POLR3B variants associated with progeria-related Wiedemann-Rautenstrauch syndrome
Italian Journal of Pediatrics (2021)
-
Novel Targets in Glucose Homeostasis and Obesity—Lesson from Rare Mutations
Current Diabetes Reports (2020)
