
Abstract
Familial adenomatous polyposis (FAP) is characterised by the development of hundreds to thousands of colorectal adenomas and results from inherited or somatic mosaic variants in the APC gene. Index patients with suspected FAP are usually investigated by APC coding region sequence and dosage analysis in a clinical diagnostic setting. The identification of an APC variant which is predicted to alter protein function enables predictive genetic testing to guide the management of family members. This report describes a 4-generation family with a phenotype consistent with FAP, but in which an APC variant had not been identified, despite testing. To explore this further, quantitative PCR (qPCR) was employed to assess APC transcription, demonstrating reduced levels of APC RNA. Next generation sequencing (NGS) identified the APC 5′UTR/ Exon 1 variant, c.-190 G>A, that had been reported previously in an another FAP family with APC allelic imbalance. Quantitative RNA studies and DNA sequencing of the APC promoters/ Exon 1 may be useful diagnostically for patients with suspected FAP when coding region variants cannot be identified.
Similar content being viewed by others

Introduction
Familial adenomatous polyposis (FAP), due to germline or somatic mosaic variants affecting APC, is the second most common cause of inherited colorectal cancer (CRC) after Lynch syndrome. FAP affects ~1 in 8000 individuals (reviewed in [1,2,3]). It is characterised by the development of hundreds to thousands of colorectal adenomas and, if untreated, will progress to CRC. In addition to FAP, there are several other, rarer syndromes which are characterised by multiple, though usually fewer, colorectal adenomas and an increased risk of CRC. These include MUTYH-associated polyposis [4], Polymerase-proofreading associated polyposis [5], NTHL1-associated polyposis [6], and MSH3-associated polyposis [7].
The identification of causative variants in families with inherited polyposis syndromes is important for the prevention of CRC. Good clinical practice includes referral of patients with multiple colorectal adenomas for genetic counselling and consideration of diagnostic testing of APC and other polyposis genes. Well over 90% of patients with a phenotype of classical FAP have a germline APC variant which is predicted to alter protein function identified through sequencing of coding exons and deletion/duplication analysis via multiplex ligation-dependent probe analysis [8]. Of those patients with an attenuated phenotype, with <100 adenomas, APC, MUTYH or other causative germline variants are detected in only 20–50% of cases [8]. The monogenic mechanisms potentially operating in the group who have no APC variant identified (NVI) include promoter and other non-coding variants, somatic mosaicism, the involvement of other genes and epigenetic effects.
This paper describes a 4-generation family with a clinical diagnosis of FAP. Despite genetic diagnostic testing performed in several expert centres, the genetic basis for the disease had not been determined.
Methods
Subjects
A study at Cardiff University, ‘Genetic Mechanisms in Colorectal Polyposis’ (approved by the NHS Research Ethics Committee for Wales: REC 3, study 12/WA/0071) is currently investigating patients with at least ten colorectal polyps who had no protein-altering APC variant identified (NVI) through genetic testing in a clinical diagnostic setting. All patients recruited to the study give written, informed consent.
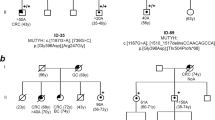
One of the probands (Individual 2.1, Fig. 1) participating in the study was a 44-year-old female who had undergone a colectomy and proctectomy for clinical FAP. At least 11 other family members were also affected (Fig. 1). The family history was provided by recruited family members but detailed clinical information on other family members was not available.

Family Pedigree. Proband is marked by an arrow (2.1)
Quantitative PCR (qPCR)
APC transcript levels in leucocyte RNA were first determined within a healthy control cohort. RNA was prepared from venous blood samples from 40 normal controls, including 7 unaffected adult relatives of NVI patients and 33 healthy individuals without a personal or family history of polyposis or CRC recruited through a local study: Causes of Bowel Polyps: Recruitment of Healthy Controls (approved by Cardiff University School of Medicine Ethics Committee). RNA was converted to cDNA, which underwent qPCR using Taqman technology (details of experimental protocols are in Supplementary 1 and 2, available at the European Journal of Human Genetics webpage). cDNA from Individual 2.1 underwent qPCR using the same methods, along with a positive control with FAP due to a previously characterised APC promoter deletion that abrogated transcription, FAP1. Results were analysed using ThermoFisher Cloud software, and APC expression levels in Individual 2.1 and the FAP-positive control were compared to the healthy cohort to give an Rq value.
Ultra-deep sequencing (UDS), variant calling and validation in DNA
UDS across the whole genomic locus of APC, hg19 chr5:g.112042936-112186350, was undertaken in genomic DNA extracted from whole blood from Individual 2.1. Reference sequence NM_001127511.2 was used, and the first delimited exon, which is untranslated, was assigned as Exon 1.
Target sequence capture was undertaken using the Haloplex assay (Agilent). Sequencing was performed by the Wales Gene Park Genomics Facility using the HiSeq (Illumina). Rare variants, present in ≤1% of the population, according to dbSNP data or The 1000 Genomes Project data, were analysed using CADD software and were assessed using the Integrative Genomics Viewer (IGV). Variants which had a CADD score ≥15 were validated with Sanger sequencing (details in Supplementary 3, available at the European Journal of Human Genetics webpage).
Results
qPCR studies
The APC Rq value for FAP1 was 0.48 and for Individual 2.1 it was 0.56 (Mean delta Ct = 8.238). An assessment for APC allelic imbalance in Individual 2.1 was attempted but homozygosity for the chosen SNPs in her genomic DNA precluded informativity of the analysis.
The qPCR results for all controls and NVI polyposis patients are in Supplementary 4, available at the European Journal of Human Genetics webpage. A further 3 NVI polyposis patients also had apparently reduced APC expression, but the cause in these patients was not identified despite APC ultradeep sequencing, karyotype analysis where possible and APC promoter methylation studies.
APC capture and UDS
The mean depth of coverage across the APC locus for Individual 2.1 was 2458 reads, with 97.7% of the target region covered at a minimum of 1× read and 73.4% at a minimum of 1000× reads. The APC promoter/Exon 1 variant c.-190 G>A (hg19 chr5:g.112043225G>A) was identified in 2845/5546 (51%) reads and confirmed by Sanger sequencing. It had a CADD score of 22.4. The proband’s father and one cousin, who both had a clinical diagnosis of FAP, were subsequently recruited for investigation (Fig. 1 Individuals 1.1 and 2.2). Both were found to carry the c.-190 G>A variant. qPCR studies gave Rq values of 0.56 for the father and 0.63 for the cousin.
The variant has been submitted to the LOVD database (patient ID 00213111).
Discussion
Li et al. [9] previously reported the APC c.-190 G>A variant in another family with FAP and profuse fundic gland polyposis, in which 5 individuals were affected over three generations. The authors performed electromobility shift assays showing that the variant led to reduced protein binding, and that the protein likely to be affected was the transcription factor YY1. They demonstrated allelic imbalance of APC expression in carriers of the variant due to abrogation of transcription.
In the Li’s paper, other specific germline variants in the APC promoter 1B region resulted in a phenotype of gastric polyposis. Individual 2.1 was found to have ‘multiple fundic gland polyps in the body and fundus’ when she attended upper gastrointestinal endoscopic surveillance, but details of upper GI endoscopy could not be obtained for other family members.
The variant has been categorised as ‘disease causing’ (HGMD Accession CR165704) and predictive genetic testing is now being offered to relatives of Individual 2.1.
Our findings suggest that quantification of APC transcription may help to direct the search for an unusual underlying genetic mechanism in NVI patients with colorectal polyposis.
Other studies have also demonstrated the importance of analysing transcripts in the search for unusual underlying mechanisms in NVI polyposis patients. As early as 1993, Powell et al. [10] used an allele-specific expression assay to show that 3/11 APC NVI patients with clinical FAP had significantly reduced expression of one APC allele. Laken et al. [11] used monoallelic mutation analysis to reveal that 7/9 APC NVI patients had reduced/no expression from one of their APC alleles. More recently Yan et al. [12] identified a patient with colorectal tumours and reduced levels of the APC protein. This patient had the expected 50:50 APC allelic ratio in gDNA, but a 66:34 APC allelic ratio in cDNA from lymphoblastoid cells [12]. Early findings regarding APC AI have been supported by Castellsagué et al. [13]. Of 23 APC/ MUTYH NVI polyposis families who were heterozygous for the SNP rs2229992 (hg19 chr5: g.112162854T>C), two were shown to exhibit APC AI. The AI in one family was suggested to result from promoter variants [13]. In 2012 transcript analysis in a sample of 125 NVI polyposis patients found that 8% had a reproducible aberrant transcript pattern, the majority of which reflected insertions between two exons originating from exonised sequences deep within the corresponding intron [8].
Considering APC qPCR studies, a rigorously determined Rq threshold would be required for diagnostic translation of transcription assays. With whole genome sequencing emerging as a realistic basis for genetic diagnosis it is likely that many more potentially regulatory non-coding variants will be identified in future. In this case, transcription studies and/or transcript analysis might form a second line test to provide evidence for or against their pathogenicity.
References
Mishra N, Hall J. Identification of patients at risk for hereditary colorectal cancer. Clin Colon Rectal Surg. 2012;25:67–82.
Fearnhead NS, Wilding JL, Bodmer WF. Genetics of colorectal cancer: hereditary aspects and overview of colorectal tumorigenesis. Br Med Bull. 2002;64:27–43.
Bodmer W. Familial adenomatous polyposis (FAP) and its gene, APC. Cytogenet Cell Genet. 1999;86:99–104.
Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, et al. Inherited variants of MYH associated with somatic G:C–>T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–32.
Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–44.
Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Sprujit L, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet. 2015;47:668–71.
Adam R, Spier I, Zhao B, Kloth M, Marquez J, Hinrichsen I, et al. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am J Hum Genet. 2016;99:337–51.
Spier I, Horpaopan S, Vogt S, Uhlhaas S, Morak M, Stienen D, et al. Deep intronic APC mutations explain a substantial proportion of patients with familial or early-onset adenomatous polyposis. Hum Mutat. 2012;33:1045–50.
Li J, Woods SL, Healey S, Beesley J, Chen X, Lee JS, et al. Point mutations in exon 1B of APC reveal gastric adenocarcinoma and proximal polyposis of the stomach as familial adenomatous polyposis variant. Am J Hum Genet. 2016;98:830–42.
Powell SM, Petersen GM, Krush AJ, Booker S, Jen J, Giardiello FM, et al. Molecular diagnosis of familial adenomatous polyposis. N Engl J Med. 1993;329:1982–7.
Laken SJ, Papadopoulos N, Petersen GM, Gruber SB, Hamilton SR, Giardiello FM, et al. Analysis of masked mutations in familial adenomatous polyposis. Proc Natl Acad Sci USA. 1999;96:2322–6.
Yan H, Dobbie Z, Gruber SB, Marowitz S, Romans K, Giardiello FM, et al. Small changes in expression affect predisposition to tumorigenesis. Nat Genet. 2002;30:25–26.
Castellsagué E, González S, Guinó E, Stevens KN, Borràs E, Raymond VM, et al. Allele-specific expression of APC in adenomatous polyposis families. Gastroenterology. 2010;139:439–47.
Acknowledgements
This work was supported by grants from the Pathological Society of Great Britain and Ireland and from Health and Care Research Wales to the Wales Gene Park and Wales Cancer Research Centre.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Short, E., Thomas, L.E., Davies, A. et al. APC transcription studies and molecular diagnosis of familial adenomatous polyposis. Eur J Hum Genet 28, 118–121 (2020). https://doi.org/10.1038/s41431-019-0486-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0486-2
