
Abstract
Attenuated adenomatous polyposis (AAP) is a heterogeneous syndrome in terms of clinical manifestations, heritability and etiology of the disease. Genetic heterogeneity and low penetrance alleles are probably the best explanation for this variability. Certainly, it is known that APC and MUTYH are high penetrance predisposition genes for adenomatous polyposis, but they only account for 5–10% of AAP. Other new predisposition genes, such as POLE, POLD1, NTHL1, AXIN2 or MSH3, have been recently described and have been associated with AAP, but their relative contribution is still not well defined. In order to evaluate the genetic predisposition to AAP in a hospital based population, germline DNAs from 158 AAP subjects were screened for genetic variants in the coding regions and intron-exon boundaries of seven associated genes through a next-generation sequencing (NGS) custom gene panel. Splicing, segregation studies, somatic mutational screening and RNA quantitative expression assays were conducted for selected variants. In four of the probands the adenoma susceptibility could be explained by actionable mutations in APC or MUTYH, and one other patient was a double carrier of two truncating variants in both POLE and NTHL1. Furthermore, 16 additional patients harbored uncertain significance variants in the remaining tested genes. This report gives information about the contribution of the newly described adenomatous polyposis predisposition genes in a Spanish attenuated polyposis cohort. Our results highly support the convenience of NGS multigene panels for attenuated polyposis genetic screening and reveals POLE frameshift variants as a plausible susceptibility mechanism for AAP.
Similar content being viewed by others

Introduction
Attenuated adenomatous polyposis (AAP) is usually defined by the presence of more than 10 and less than 100 adenomas along the large intestine and/or rectum. It is a highly heterogeneous syndrome in terms of polyposis severity, family history, and lifetime risk of colorectal cancer (CRC)1,2,3. Adenoma burden can be low or mild, it may be accompanied by hyperplastic or serrated polyps, the polyposis diagnosis age is variable and later than classical forms, and CRC risk can range from 40% to 80% depending on the adenoma burden. A positive family history is frequently absent, but horizontal or vertical inheritance patterns are also observed. The diffusion of early detection CRC screening programs, together with the improvement of colonoscopy techniques, has given rise to an increase in the detection of patients with multiple colorectal adenomas. These patients are referred to the oncogenetic counseling units for genetic testing in order to find an explanation to their adenoma susceptibility that helps in the understanding of the disease and makes possible a satisfactory genetic counseling. Nowadays, APC (MIM *611731) and MUTYH (MIM *604933) are the two main clinical actionable predisposition genes for adenomatous polyposis3. That means prevalence and cancer risk estimations are well-defined, allowing an accurate genetic counseling and effective high risk monitoring programs for carriers. They both together explain the vast majority of classical adenomatous polyposis (>100 adenomas), but they are only able to explain a small fraction of AAP2. In fact, in our hospital settings, we are not able to explain more than 5–10% of the patients referred to our laboratory after the routine genetic tests. Therefore, the identification of the underlying susceptibility causes of those unexplained AAP cases is a priority in our laboratory.
Recently, other adenomatous polyposis predisposition genes, such as POLE4, POLD14, NTHL15, MSH36 and AXIN27 (MIM *174762, *174761, *602656, *600887 and *604025, respectively), have been described. However, they are still not well characterized and only a few reports have attempted to estimate the extent to which some of these genes, individually, are involved in different CRC populations8,9,10,11,12,13. Therefore, they are still poorly implemented in the daily clinical practice.
Under this scenario of high genetic heterogeneity, the use of next-generation sequencing (NGS) gene panels for the diagnosis of hereditary AAP seems to be the best choice. Aiming to delve into the genetic study of unexplained AAP, we have screened the whole coding sequences and intron-exon boundaries of APC, MUTYH, POLE, POLD1, NTHL1, MSH3 and AXIN2 in a cohort of AAP collected at our oncogenetic counseling unit. The main objective of this work is to determine the contribution of the AAP associated genes in an unexplained AAP cohort.
Results
Clinical features of the study cohort
A clinical description of the study cohort is summarized in Table 1. The study cohort consisted of 158 AAP patients, coming from the Oncogenetic Counseling Units of Hospital Clínico San Carlos and Hospital 12 de Octubre, in Madrid. The average polyposis diagnosis age was 62.9 (ranged from 33 to 80) and the average polyp burden 31.2 (from 10 to 100). Detailed clinical description of participants is given in Suppl. Table 1.
Germline DNA screening
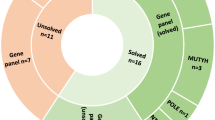
After genetic panel screening, the average read depth per sample was 895 reads, with a minimum count of 326 and a maximum of 4616. However, two regions showed reiterative low coverage, so they were reanalyzed by high resolution melt (HRM) analysis, NM_000038.4: exon 13 (APC) and NM_00128425.1: exon 15 (MUTYH). Once the screening was completed, 28 variants located in six genes were validated in 24 patients (Table 2).
Eight patients harbored pathogenic variants (class-5): Two patients harbored class-5 variants in APC, one patient was a biallelic carrier of two class-5 variants, another was MUTYH biallelic carrier of a class-5 and a class-3 variants, and three patients were monoallelic MUTYH class-5 carriers. Finally, one patient was a double carrier of two deleterious variants, in POLE and NTHL1. The remaining sixteen patients harbored class-3 (Table 2).
Reclassification of variants
After the splicing, segregation and somatic analyses, it was possible to confirm two MUTYH variants (c.739C > G; p.(Arg247Gly) and c.1510_1517delinsCCAACAGCCA; p.Thr504Profs*68) as likely pathogenic (class-4) and pathogenic (class-5), respectively, and to reclassify the variant POLD1: c.2007-5C > T as benign (class-1) and POLE: c.6716C > T p.(Ala2239Val) as likely benign (class-2) (Table 2, Suppl. Table 3A,B). In the end, 11 out of 28 variants were classified as class-5 or -4, two variants were classified as class-1 or -2, discarding their involvement in the AAP predisposition, and 15 variants remained as uncertain significance (class-3) variants.
MUTYH:c.739C > G p.(Arg247Gly)
MUTYH variant c.739C > G; p.(Arg247Gly) was detected in trans (Suppl. Fig. 1a) with c.1187G > A; p.(Gly396Asp) in a male with more than 20 adenomas at the age of 37. One of his brothers was also a carrier of both variants and presented more than 50 adenomas and CRC at the age of 41(Fig. 1a). The variant is located in the hMSH6 binding domain, and missense mutations located in this domain have been shown to affect the A/8-oxoG binding and glycosylase activities14. Adenomatous tissue from the proband was screened for somatic mutations and G > T changes were found in APC, KRAS, TP53 and MAP2K (Table 3, ID 35). As it is well known, adenomas and tumors coming from MUTYH biallelic carriers show a deficiency in the 8-oxo-hidroxyguanine repair system, leading to an increase in the G > T mutation rate, frequently in APC and KRAS15. Thus, c.739C > G; p.(Arg247Gly) was reclassified as class-4 (Suppl. Table 3).

Pedigrees of the explained polyposis cases. (a) MUTYH biallelic mutations carrier families. (b) APC mutations carrier families. †polyposis diagnosis after family mutation identification. Black square = cancer; black circle = >10 adenomas; +=variant carrier; −non carrier; +/+biallelic carrier; GC = gastric cancer; CRC = colorectal cancer; BC = breast cancer; y = years; A = adenomas.
MUTYH:c.1510_1517delinsCCAACAGCCA p.Thr504Profs*68
MUTYH variant c.1510_1517delinsCCAACAGCCA p.(Thr504Profs) was detected in trans (Suppl. Fig. 1b) with c.1187G > A; p.(Gly396Asp) in a male with more than 40 adenomas at the age of 56. In this instance, the subject did not present a family history of polyposis or CRC (Fig. 1a). Blood derived cDNA sequencing revealed an extension corresponding to 22 amino acids after the stop codon (p.Thr504Profs*68). The novel frameshift variation was located at the 3′-end of the coding sequence, altering the amino acid sequence of the whole proliferating cell nuclear antigen (PCNA) binding domain, which is essential for MUTYH’s activity during DNA replication16. Indeed, point mutations at the PCNA-binding domain have been proven to decrease the activity of the enzyme17. Somatic analysis of DNA from affected adenomas showed G > T changes at KRAS locus (Table 3, ID 89). Therefore, c.1510_1517delinsCCAACAGCCA p.Thr504Profs*68 was reclassified as class-5 (Suppl. Table 3).
POLD1:c.2007-5C > T
After cDNA analysis, no splicing alteration was detected in the c.2007-5 carrier. So it was reclassified as class-1.
POLE:c.6716C > T p.(Ala2239Val)
POLE variant c.6716C > T p.(Ala2239Val) was detected in homozygosis in both an affected and a healthy member, and it was absent in another affected member. Therefore, it was reclassified as class-2 variants.
Co-occurrence of truncating variants at POLE and NTHL1 loci
Two heterozygous truncating variants, POLE:c.141delG; p.Phe48Leufs*6 and NTHL1:c.268C > T; p.Gln90*, were detected in the same germline DNA (Fig. 2a). The patient was a woman with full-blown late AAP diagnosed at the age of 70 and without a previous family history of polyposis or CRC. Segregation analyses could only be achieved in two of her daughters, detecting POLE:c.141delG; p.Phe48Leufs*6 in one of them, who had been diagnosed of a low grade dysplastic tubular adenoma at the age of 46 (Fig. 2b).

Identification of POLE and NTHL1 double heterozygote. (a) POLE and NTHL1 protein domains and location of truncating variants detected in patient 83. (b) Pedigree chart; POLE+ = POLE variant carrier; POLE− = POLE wild type genotype; NTHL1+ = NTHL1variant carrier; NTHL1− = NTHL1wild type genotype; LC = lung cancer; A = adenoma. (c) POLE wild type expression in blood and tissue cDNA samples. (d) NTHL1 wild type allele expression in blood and tissue cDNA samples. COLON pool sample has been used as a reference sample in both NTHL1 and POLE assays. Significant ρ-values are indicated with *(ρ < 0.05) and **(ρ < 0.01). Each sample was analyzed in triplicates. AC = adenocarcinoma; AD = adenoma; CRC = colorectal cancer.
In order to investigate the involvement of these variants in the adenoma and CRC formation, four adenomas (83AD1, 83AD3, 83AD4, 83AD5) and one adenocarcinoma (Haggit-0) (83AC1) from the proband were analyzed for somatic mutations (Table 3, ID 83). A total of 13 driver mutations was detected, two of which were indels, four C > A changes and seven were C > T changes. Eleven somatic mutations were located in genes involved in the early adenoma formation (APC, RNF43 and CTNNB1), whereas two mutations were detected in genes involved in later stages (ARID1A, NRAS).
In addition, NTHL1 and POLE specific wild type allele expression was checked at tissue level. Quantification analyses were performed in three adenomas (83AD1, 83AD3, 83AD4), one adenocarcinoma (83AC1) and healthy colon tissue from the proband (83COLON). POLE wild type allele expression in the carrier’s colon tissue (83COLON) did not show a significant change compared to the colon control pool (fold difference (FD) = 0.94 ± 0.27, ρ = 0.82), whereas NTHL1 wild type allele expression in 83COLON did (FD = 0.30 ± 0.08, ρ = 0.004).
Both POLE and NTHL1 transcripts were over-expressed in the CRC control pool (FDPOLE = 2.06 + 0.08, ρ = 0.003; FDNTHL1 = 1.87 + 0.28, ρ = 0.018). However, all adenomas and the adenocarcinoma tested from the carrier showed a decrease in the expression of POLE wild type allele (FD average = 0.46 ± 0.20, ρ < 0.001), whereas NTHL1 wild type allele showed significant over-expression in one adenoma and one adenocarcinoma from the proband when compared to its healthy tissue (ρ = 0.0156 and ρ < 0.001, respectively) (Fig. 2c,d).
Discussion
AAP is becoming one of the largest groups of patients attending our Genetic Counselling Unit. Genetic screening of MUTYH and APC is recommended in those cases with more than 10 adenomas18. The diagnostic approach in our hospital consists in the screening of the four most prevalent MUTYH mutations in Spanish population19, c.536A > G; p.(Tyr179Cys), c.1187G > A; p.(Gly396Asp), c.1227_1228dup; p.(Glu410Glyfs*43) and c.1437_1439delGGA; p.(Glu480del), and the subsequent screening of point mutations and copy number variations (CNVs) in the whole coding sequence of the APC gene in those cases with a higher polyp burden and high suspicion of inheritance pattern. However, by this approach we are only able to explain around 5% of the cases with more than 10 adenomas, or 7% when restricting the criteria to full-blown polyposis (more than 20 adenomas or 10 synchronous adenomas). In order to improve the diagnostic sensitivity and investigate the contribution of currently known high-penetrance adenomatous polyposis genes in our AAP population, we have conducted a genetic screening in an unexplained AAP cohort through a custom gene panel, including APC, MUTYH, POLE, POLD1, NTHL1, AXIN2 and MSH3 coding regions.
Our study cohort was made up of 158 subjects, 132 (83.5%) of which fulfilled the clinical criteria for full-blown polyposis (Table 1). There is only a very recent cross-sectional study analyzing the prevalence of pathogenic variants in genes associated with colorectal polyposis and/or CRC in a cohort of 3789 polyposis patients, of which 2979 presented between 10 and 100 adenomas. All patients underwent panel testing of at least 14 CRC predisposition genes, including APC and MUTYH. However, only POLD1 and POLE were tested on a subset of the adenoma cohort, and the remaining adenomatous polyposis predisposition genes (NTHL1, AXIN2 and MSH3) were not tested20. Therefore, this is the largest multiple polyposis cohort in which joint genetic screening for the full coding sequences of all AAP predisposition genes has been done so far.
Two APC truncating mutations were detected in two subjects diagnosed with late-onset full-blown polyposis and a family history initially suspicious of recessive inheritance (Fig. 1b). Both had been previously tested for MUTYH but not for APC, probably due to the lack of family information at the time of diagnosis, the late polyposis onset of the probands, and also the stricter clinical criteria for the recommendation of full APC mutational screening for AAP in our hospital.
In addition, the screening of the whole coding sequence of MUTYH, allowed the detection of two MUTYH non-recurrent mutations, c.739C > G p.(Arg247Gly) and c.1510_1517delinsCCAACAGCCA p.Thr504Profs*68, both in co-occurrence with the recurrent mutation c.1187G > A; p.(Gly396Asp), being able to explain the polyposis susceptibility in two more subjects (Fig. 1a).
APC/MUTYH mutational rates are low in AAP population2. Conventional genetic screening technologies imply the sequential analysis of each gene amplicon by amplicon, which makes these protocols costly in both time and money. Therefore, those laboratories without high sample volumes are forced to restrict their clinical criteria for AAP diagnosis in order to make the analysis cost-effective. NGS multigene panels reduce the time and cost of these genetic studies, increasing the cost-effectiveness and making the complete screening of samples feasible for small laboratories. The identification of pathogenic mutations in APC and MUTYH with an NGS panel in our study cohort is a clear example of underdiagnoses, supporting the necessity of parallel sequencing for APC and MUTYH routine genetic screening in AAP patients.
None of the patients showed actionable mutations (class-4 and -5) in any of the genes associated with new polyposis syndromes (MSH3, NTHL1, POLD1, POLE or AXIN2). However, 10 uncertain significance (class-3) variants were detected in AXIN2, NTHL1, POLD1 and POLE (Table 2). Three of these variants were located outside the proofreading domain of POLE/POLD1. Although there is no evidence of association between missense variants outside these regions and cancer susceptibility, there is a recent report that describes somatic driver mutations located outside the exonuclease domains, and suggesting that other domains may be responsible for proofreading21. Therefore, we decided to include these variants as class-3 variants for AAP predisposition.
Considering other previous studies that included multiple polyposis cohorts, the frequency of actionable mutations in all of these genes seems to be very low in tested populations, but not insignificant (around 1–2%). Just like it is not irrelevant the number of variants of uncertain significance that have been described in this and other works9,10,12 (1–2% per gene), and whose probable pathogenicity might be proven during the coming years. For this reason it is important to include all these genes in the routine analysis of AAP through NGS panels.
A remarkable finding of this work is the detection of a double heterozygous for two truncating variants, at exon two of NTHL1 and exon two of POLE (Fig. 2a). The carrier was a woman with late full-blown polyposis, who was also diagnosed with endometrial hyperplasia and hypothyroidism (Fig. 2b).
POLE encodes the catalytic subunit of DNA polymerase epsilon, which is responsible for the replication of the leading DNA strand during the S phase; it is an essential protein and biallelic truncating mutations are not viable. POLE, together with POLD1, is the only nuclear polymerase with an intrinsic 3′–5′exonuclease proofreading activity capable of correcting mistakes made during DNA synthesis22. A few years ago, germline mutations located in the proofreading domain of POLE and POLD1 were associated with adenomatous polyposis predisposition4, showing tumors with a very high mutational burden due to the lack of exonuclease but not polymerase activity. According to this, truncating variants at POLE would not be supposed to confer this genetic instability because they would lead to a complete inactivation of the enzyme without any polymerase-exonuclease imbalance. However, it is not clear to what extent the lack of one POLE allele can lead to cancer predisposition in some other way.
On the other hand, NTHL1 encodes the DNA glycosylase NTHL1, which is involved in removing oxidative pyrimidine lesions through the base excision repair (BER) pathway. Resembling to MUTYH-associated polyposis, germline biallelic mutations at NTHL1 have been recently associated with adenomatous polyposis predisposition5, leading to a deficiency in the repair of 5-hydroxycytosine and a consequent increase in the C > T somatic mutation rate. Like other glycosylases involved in the BER pathway, NTHL1’s repair activity can be completed by a short or long BER patch, mainly depending on the proliferative activity of the cell23. Thus, in high division rate tissues NTHL1 is coupled to the DNA synthesis and follows the long-patch BER pathway, dependent of PCNA and where POLE and POLD1 are the polymerases responsible for filling the gap after NTHL1’s action and strand cleavage23.
Therefore, it is plausible that POLE and NTHL1 co-occurring truncating variants may have a synergistic effect leading to a polyposis predisposition in high division tissues such as the colon epithelium. To check this hypothesis, somatic mutation screening and RNA expression analyses were performed in adenomas and CRC from the carrier. The analysis of somatic mutations showed a tendency to C > T changes (Table 3), although no clear definition of the mutational signature could be done due to the low number of mutations. Furthermore, a quantitative analysis of POLE wild type allele showed a significant decrease of POLE expression in all tested carrier’s adenomas and adenocarcinoma (Fig. 2c), which is consistent with a replicative stress due to POLE haploinsufficiency. However and unlike POLE, NTHL1 expression was increased in two of the carrier’s adenomas and adenocarcinoma tissues (Fig. 2d). This over-expression was probably triggered by a greater oxidative DNA damage in tissues with a high division rate, which has been shown to up-regulate BER glycosylases24. Wild type NTHL1 transcript levels in the carrier’s affected tissues reached similar levels than non-carrier’s affected pool, which is not consistent with an NTHL1 haploinsufficiency. Therefore, we discarded the involvement of the NTHL1 monoallelic mutation c.268C > T p.(Gln90*) in the polyposis predisposition of this patient and we considered the possibility that POLE:c.141delG p.Phe48Leufs*6 could confer the adenoma predisposition by itself. Only 0.005% of POLE coding variants described in gnomAD are truncating (frameshift or nonsense); non homozygous have been described and all truncating variants show allelic frequencies lower than 1/10000. Moreover, there are two studies in the literature describing germline frameshift variants at POLE; c.5621_5622delGT was detected in a sporadic CRC patient with a diagnosis age of 2625, and c.1370_1371delAT p.Tyr457fs*9 was later detected in an AAP patient10. In addition, as it has been mentioned above, somatic driver mutations have been recently described in POLE and POLD1 polymerase domains21.
Furthermore, the presence of both variants was checked in two of the proband’s daughters; one harbored the POLE variant, but not the NTHL1 one, and the other did not harbor any of the variants (Fig. 2b). The POLE carrier showed a dysplastic tubular adenoma at the age of 46. Despite the large size of the family, no other relatives had been diagnosed of cancer or polyposis, which would support the hypothesis of a digenic or oligogenic inheritance with other undetected variants or, a de novo POLE mutation in the proband with a dominant effect that is not yet possible to observe due to the young age of the daughter harboring the variant (Fig. 2b).
To our knowledge, this is the first report describing the probable association of a truncating germline POLE variant with the predisposition of AAP by a haploinsufficiency mechanism in adenomatous and colorectal tumor tissues of the carrier. This result highlights not only the genetic heterogeneity and complexity of the syndrome, but also the potential of NGS gene panels for the detection and diagnosis of new inheritance forms of complex diseases. Further efforts should be done for the study and characterization of germline POLE truncating variants in other AAP populations, as well as their co-occurrence with variants in other associated genes.
In accordance with other AAP studies, our results showed a major number of subjects without detection of variants in any of the genes tested. It can be thought the involvement of other unknown predisposition genes in the susceptibility of AAP and the convenience of wider genetic analysis, such as exome sequencing, for the elucidation of new AAP predisposition genes in this unexplained group. However, despite most of the new AAP associated genes have been discovered by exome sequencing approaches, there are other works without such successful results26,27. The failure of detecting new predisposition genes in AAP is probably due to the high clinical and genetic heterogeneity of the condition, as well as the low prevalence of pathogenic mutations in the already associated genes. Probably, polygenic inheritance models in which the susceptibility is explained by the accumulation of multiple low penetrance alleles28, and lifestyle risk factors such as smoking, alcohol, body mass index, diet and physical activity29 play a major role in the unexplained AAP.
In our cohort, all patients with pathogenic mutation detection presented full blown AAP (more than 20 adenomas) and an average diagnosis age lower than the general study cohort (57.2). Other works analyzing AAP cohorts, such as Grover30 who analyzed MUTYH and APC in 4223 patients with 10 to 100 adenomas, or Stanich20 who also analyze POLD1 and POLE and other CRC genes in 2979 patients with 10 to 100 adenomas, showed similar clinical results. These results suggest that the low mutation detection rate in AAP is partially because of the lack of strict clinical criteria for the selection of patients with high probability to detect pathogenic mutation in any known predisposition gene. Therefore, redefinition of stronger clinical criteria and the use of panel gene testing are necessary for the improvement of genetic testing in AAP.
This work is a translational study aiming to analyze the contribution of known and new described adenomatous polyposis predisposition genes and the suitability of their genetic testing in a hospital based cohort that have been referred to the genetic counseling unit. The results shown above lead to the following conclusions:
-
1.
The contribution of new predisposition genes is much smaller than that of the known genes APC and MUTYH. Related pathogenic mutations have not been detected in any gene. However, uncertain significance variants have been detected in all genes but MSH3. Since they are recently described genes, the identification of potential pathogenic variants and further clarification of their pathogenicity is important for the definition of the syndrome.
-
2.
Somatic genetic screening of affected tissues allows the detection of certain mutational signatures associated with DNA repair deficiency, helping the classification of the variants.
-
3.
The expected low mutation detection rate in AAP study cohort point to the necessity of stronger clinical criteria for the improvement of the diagnostic sensitivity in AAP genetic testing.
-
4.
Although the number of POLE/POLD1 truncating mutations detected in AAP cases is very limited, due to the genetic heterogeneity of the disease, the relevance of these genes and the decreased levels of carrier’s tumor and adenoma samples shown in this work, special attention should be paid to those POLE/POLD1 truncating variants in order to determine their pathogenicity.
-
5.
Summarizing, this work highlights the need of multigene panel testing in highly genetic heterogeneous syndromes such as AAP, not only to increase the cost-effectiveness and the diagnostic sensitivity of the analysis, but also to better detect other potentially pathogenic variants or other inherited forms of the disease that would not be detected by other gene directed approaches.
Patients and Methods
Patients
The inclusion criterion was the detection of between 10 and 100 adenomas in the colon and/or rectum. All the samples had been screened, at least, for the most frequent MUTYH (NM_0012222.2) mutations in the Spanish population19: c.536A > G; p.(Tyr179Cys), c.1187G > A; p.(Gly396Asp), c.1227_1228dup; p.(Glu410Glyfs*43) and c.1437_1439delGGA; p.(Glu480del), by HRM and/or Sanger sequencing. The presence of biallelic MUTYH mutations was considered as an exclusion criterion, but not the presence of monoallelic MUTYH mutations. APC screening was not an inclusion criterion, but the previous detection of a pathogenic APC mutation was considered as an exclusion criterion.
A total of 158 unrelated AAP cases was included in the study (Suppl. Table 1), comprising 123 subjects from Hospital Clínico San Carlos (Madrid) and 35 from Hospital 12 de Octubre (Madrid).
Ethical approval was obtained from Hospital Clínico San Carlos’ Ethical Research Committee (approval number: C.I.-14/241-E_BS). A written informed consent was obtained from each participant. Methods were compliant with the relevant guidelines and regulations.
Sample extraction
Peripheral-blood DNA/RNA extraction was performed with the MagNA Pure Compact extractor (Roche), according to the manufacturer’s protocol. Tissue DNA/RNA (from normal epithelium, adenoma or tumor sections) was obtained from formalin-fixed paraffin-embedded (FFPE) tissues with a purity greater than 80% as determined by an experienced pathologist. DNA extractions were performed with the QIAamp DNA FFPE Tissue kit (Qiagen N.V.) and RNA extractions with the RNAeasy FFPE kit (Qiagen N.V.). SuperScript First-Strand Synthesis System for RT-PCR (Thermo Fisher Scientific) was used to synthesize cDNA, either from blood or tissue RNA, using random hexamers and oligo-dT, according to the manufacturer’s instructions.
Germline DNA screening
Germline DNAs were screened for APC, MUTYH, POLE, POLD1, NTHL1, MSH3 and AXIN2 variants in the coding regions and intron-exon boundaries through an NGS Haloplex custom panel (Agilent Technologies). Enriched libraries were obtained according to the manufacturer’s protocol, and subsequently sequenced on a MiSeq System (Illumina, Inc.). Data analysis and variant calling were achieved with the SureCall software (Agilent Technologies) following the recommended pipeline for Haloplex libraries. Low covered regions (read depth <50) were analyzed by HRM. CNVs analyses at APC and MUTYH loci were performed by multiplex ligation and probe amplification (MLPA) (SALSA® MLPA®probemix P043 and P378, MRC-Holland).
All rare (novel or minor allele frequency (MAF) <0.01 according to the gnomAD31 and 1000 genomes project32 databases), deleterious or possible deleterious variants (according to protein and/or splicing alteration prediction tools) were selected for validation by Sanger sequencing. MaxEnt, and human splicing finder (HSF)33 were used to predict splicing alterations, while SIFT34, Polyphen235 and MutationTaster36 predicted protein damage.
Classification of variants
Variants were classified in five different pathogenicity classes (class-5 or pathogenic, class-4 of likely pathogenic, class-3 or uncertain significance, class-2 or likely benign and class-1 or benign) according to the public mutational databases (InSight37 LOVD38, UMD39, ClinVar40) and supporting pathogenic and/or benign evidences, in accordance to the ACMG-SHERLOC criteria41,42.
Characterization of variants
Segregation analyses
Whenever possible, segregation analyses of candidate variants were performed on DNA from available family members by Sanger sequencing (oligonucleotides shown in Suppl. Table 2).
Splicing analyses
Blood derived cDNAs from patients harboring variants either with a positive splicing alteration prediction or located between five nucleotides from the intron-exon boundaries, were subjected to transcript analyses by Sanger sequencing (oligonucleotides shown in Suppl. Table 2).
Adenoma/tumor DNA screening
Adenoma and tumor DNAs were screened for somatic mutations using the commercial amplicon-based TruSight Tumor 26 panel (TST26) (Illumina, Inc.), which includes 174 relevant regions located in 26 genes involved in solid tumors. Sequencing was performed on a MiSeq System (Illumina, Inc.). Data analysis and variant calling were performed through the plug-in specific Amplicon DS workflow for the MiSeq Reporter Software tool (Illumina, Inc.). PASS filter variants, according to the default settings, were first selected. Variants that failed to pass default filters, but that were detected in both pools (i.e. showing a low coverage depth or strand bias) were manually inspected using the Integrative Genome Viewer (IGV) browser43. Known germline polymorphisms, variants detected in the majority of samples tested, variants previously detected in the patient’s germline DNA or present in all tested samples from the same subject were discarded.
By single-molecule molecular inversion probe (smMIP) sequencing, two adenomas (83AD1 and 83AD4) and one adenocarcinoma (83AC1) were also investigated for the occurrence of somatic mutations in the open reading frames and hotspot regions of 57 genes involved in CRC development, as described previously44.
In order to characterize the variant MUTYH:c.1510_1517delinsCCAACAGCCA p.(Thr504Profs*68), three adenomas of patient 89 were analyzed for KRAS somatic mutations at codons 12 and 13 by the Therascreen KRAS RGQ PCR Kit (Qiagen N.V.), according to the manufacturer’s protocol.
cDNA expression analyses
NTHL1 and POLE wild type allele expression levels were evaluated in three adenomas (83AD1, 83AD3 and 83AD4), one adenocarcinoma (83AC1) and one healthy colon tissue (83COLON) from patient 83 and compared to healthy colon and colorectal cancer control pools (COLON and CRC). The healthy colon pool was made up of eight FFPE colon tissue-derived RNA samples from unrelated healthy subjects, while the colon cancer pool was made up of eight FFPE CRC tissue-derived RNA samples from unrelated patients. All samples were previously treated with RNase-free recombinant DNase I (Roche) and the lack of germline DNA traces was checked by specific germline and cDNA amplification (Suppl. Fig. 2, primer sequences on Suppl. Table 2).
For each of the genes to be tested, primer pairs were designed for the specific detection of the wild type allele, and PSMB4 was used as an endogenous gene (Suppl. Table 2). For each sample, 5 ng/μL of RNA was amplified in triplicates with the KAPA SYBR® FAST Universal Kit (Roche) in a Light Cycler® 96 (Roche), according to the manufacturer’s instructions.
Reclassification of variants
Reclassification of variants was achieved according to the supporting pathogenic and/or benign evidences, in accordance to the ACMG-SHERLOC criteria41,42
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Lucci-Cordisco, E., Risio, M., Venesio, T. & Genuardi, M. The growing complexity of the intestinal polyposis syndromes. Am J Med Genet A 161A, 2777–87 (2013).
Mongin, C. et al. Unexplained polyposis: a challenge for geneticists, pathologists and gastroenterologists. Clin Genet 81, 38–46 (2012).
Jasperson, K. W., Tuohy, T. M., Neklason, D. W. & Burt, R. W. Hereditary and familial colon cancer. Gastroenterology 138, 2044–58 (2010).
Palles, C. et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45, 136–44 (2013).
Weren, R. D. et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 47, 668–71 (2015).
Adam, R. et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am J Hum Genet 99, 337–51 (2016).
Rivera, B. et al. A novel AXIN2 germline variant associated with attenuated FAP without signs of oligondontia or ectodermal dysplasia. Eur J Hum Genet 22, 423–6 (2014).
Elsayed, F. A. et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur J Hum Genet 23, 1080–4 (2015).
Bellido, F. et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med 18, 325–32 (2016).
Spier, I. et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer 137, 320–31 (2015).
Belhadj, S. et al. Delineating the Phenotypic Spectrum of the NTHL1-Associated Polyposis. Clin Gastroenterol Hepatol 15, 461–462 (2017).
Esteban-Jurado, C. et al. POLE and POLD1 screening in 155 patients with multiple polyps and early-onset colorectal cancer. Oncotarget 8, 26732–26743 (2017).
Grolleman, J. E. et al. Mutational Signature Analysis Reveals NTHL1 Deficiency to Cause a Multi-tumor Phenotype. Cancer Cell 35, 256–266.e5 (2019).
Bai, H. et al. Functional characterization of two human MutY homolog (hMYH) missense mutations (R227W and V232F) that lie within the putative hMSH6 binding domain and are associated with hMYH polyposis. Nucleic Acids Res. 33, 597–604 (2005).
Al-Tassan, N. et al. Inherited variants of MYH associated with somatic G:C–>T:A mutations in colorectal tumors. Nat Genet 30, 227–32 (2002).
van Loon, B. & Hubscher, U. An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase lambda. Proc Natl Acad Sci USA 106, 18201–6 (2009).
Brinkmeyer, M. K. & David, S. S. Distinct functional consequences of MUTYH variants associated with colorectal cancer: Damaged DNA affinity, glycosylase activity and interaction with PCNA and Hus1. DNA Repair (Amst) 34, 39–51 (2015).
Syngal, S. et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 110, 223–62; quiz 263 (2015).
Gomez-Fernandez, N. et al. Molecular analysis of the APC and MUTYH genes in Galician and Catalonian FAP families: a different spectrum of mutations? BMC Med Genet 10, 57–68 (2009).
Stanich, P. P. et al. Prevalence of Germline Mutations in Polyposis and Colorectal Cancer-Associated Genes in Patients With Multiple Colorectal Polyps. Clin. Gastroenterol. Hepatol, https://doi.org/10.1016/j.cgh.2018.12.008 (2018).
Campbell, B. B. et al. Comprehensive Analysis of Hypermutation in Human. Cancer. Cell 171, 1042–1056 e10 (2017).
Henninger, E. E. & Pursell, Z. F. DNA polymerase epsilon and its roles in genome stability. IUBMB Life 66, 339–51 (2014).
Akbari, M. et al. Extracts of proliferating and non-proliferating human cells display different base excision pathways and repair fidelity. DNA Repair (Amst) 8, 834–43 (2009).
Fleming, A. M., Ding, Y. & Burrows, C. J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc Natl Acad Sci USA 114, 2604–2609 (2017).
Smith, C. G. et al. Exome resequencing identifies potential tumor-suppressor genes that predispose to colorectal cancer. Hum Mutat 34, 1026–34 (2013).
Spier, I. et al. Exome sequencing identifies potential novel candidate genes in patients with unexplained colorectal adenomatous polyposis. Fam. Cancer 15, 281–288 (2016).
Broderick, P. et al. Validation of Recently Proposed Colorectal Cancer Susceptibility Gene Variants in an Analysis of Families and Patients-a Systematic Review. Gastroenterology 152, 75–77.e4 (2017).
Cheng, T. H. T. et al. Common colorectal cancer risk alleles contribute to the multiple colorectal adenoma phenotype, but do not influence colonic polyposis in FAP. Eur. J. Hum. Genet. 23, 260–263 (2015).
Øines, M., Helsingen, L. M., Bretthauer, M. & Emilsson, L. Epidemiology and risk factors of colorectal polyps. Best Pract Res Clin Gastroenterol 31, 419–424 (2017).
Grover, S. et al. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA 308, 485–492 (2012).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–91 (2016).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37, e67 (2009).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–81 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–9 (2010).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11, 361–2 (2014).
Thompson, B. A. et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 46, 107–115 (2014).
Fokkema, I. F. et al. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 32, 557–63 (2011).
Beroud, C., Collod-Beroud, G., Boileau, C., Soussi, T. & Junien, C. UMD (Universal mutation database): a generic software to build and analyze locus-specific databases. Hum Mutat 15, 86–94 (2000).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46, D1062–D1067 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–24 (2015).
Nykamp, K. et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med 19, 1105–1117 (2017).
Robinson, J. T. et al. Integrative genomics viewer. Nat Biotechnol 29, 24–6 (2011).
Eijkelenboom, A. et al. Reliable Next-Generation Sequencing of Formalin-Fixed, Paraffin-Embedded Tissue Using Single Molecule Tags. J Mol Diagn 18, 851–863 (2016).
Acknowledgements
The authors wish to thank the polyposis patients and relatives for their participation. They also thank the donors and the Biobank of the “Instituto de Investigación Sanitaria San Carlos” for the human specimens used in this study. The present study was supported by grants from the Instituto de Salud Carlos III, Spain (www.isciii.es) and European Regional Development FEDER funds (PI14/00929, PI16/01292 and CIBERONC (Centro de Investigación Biomédica en Red en Oncología) network CB16/12/00301).
Author information
Authors and Affiliations
Contributions
V.L. performed the most of experiments and analyses of the results. D.R. and S.T. contributed to the selection of patients from Hospital 12 de Octubre and validation cohort. L.M.M. contributed to the germline DNA gene panel sequencing and data analysis. M.J.F.-A. and J.S. collected and assessed FFPE tissue for DNA and RNA extraction. C.P. and P.P.S. participated in the selection of Hospital Clínico San Carlos’s participants and clinical data curation. J.G. and R.M.V. performed somatic mutational screening by smMIP, P.L. and V.G.B. helped in the analysis of results. M.H., E.D.R. and T.C. gave advice throughout the project, P.G. and T.C. designed the study, P.G. supervised all the study, V.L. and P.G. wrote the manuscript, M.H., E.D.R., T.C., V.G.B. and R.M.V. reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lorca, V., Rueda, D., Martín-Morales, L. et al. Contribution of New Adenomatous Polyposis Predisposition Genes in an Unexplained Attenuated Spanish Cohort by Multigene Panel Testing. Sci Rep 9, 9814 (2019). https://doi.org/10.1038/s41598-019-46403-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46403-5
This article is cited by
-
Genotype–Phenotype Correlations in Autosomal Dominant and Recessive APC Mutation-Negative Colorectal Adenomatous Polyposis
Digestive Diseases and Sciences (2023)
-
Intestinal and extraintestinal neoplasms in patients with NTHL1 tumor syndrome: a systematic review
Familial Cancer (2022)
-
Pathogenic germline variants are associated with poor survival in stage III/IV melanoma patients
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
