
Abstract
Split-hand/foot malformation (SHFM) is a clinically and genetically heterogeneous condition. We sequentially performed screening of the previously identified Japanese founder 17p13.3 duplication/triplication involving BHLHA9, array comparative genomic hybridization, and whole exome sequencing (WES) in newly recruited 41 Japanese families with non-syndromic and syndromic SHFM. We also carried out WES in seven families with nonsyndromic and syndromic SHFM in which underlying genetic causes including pathogenic copy-number variants (CNVs) remained undetected in our previous studies of 56 families. Consequently, we identified not only known pathogenic CNVs (17p13.3 duplications/triplications [n = 21], 2q31 deletion [n = 1], and 10q24 duplications [n = 3]) and rare variants in known causative genes (TP63 [n = 3], DLX5 [n = 1], IGF2 [n = 1], WNT10B [n = 3], WNT10B/PORCN [n = 1], and PORCN [n = 1]), but also a de novo 19q13.11 deletion disrupting UBA2 (n = 1) and variants that probably affect function in LRP6 (n = 1) and UBA2 (n = 1). Thus, together with our previous data based on testing of 56 families, molecular studies for a total of 97 families with SHFM revealed underlying genetic causes in 75 families, and clinical studies for the 75 families indicated a certain degree of correlation between genetic causes and phenotypes. The results imply that SHFM primarily occurs as a genetic disorder with genotype–phenotype correlations. Furthermore, the results together with previous data such as the development of SHFM in Lrp6 knockout mice, the presence of SHFM in two subjects with 19q13 deletions involving UBA2, and strong mouse Uba2 expression in the developing limb buds, imply that LRP6 and UBA2 represent plausible candidate genes for SHFM.
Similar content being viewed by others

Introduction
Split-hand/foot malformation (SHFM) is a rare limb anomaly caused by failure to maintain the central portion of the apical ectodermal ridge (AER) in the developing autopod [1, 2]. It is characterized by median clefts of the hands and feet and aplasia/hypoplasia of the phalanges, metacarpals, and metatarsals, and is often accompanied by other digital anomalies such as syndactyly [1, 2].
SHFM is a clinically heterogeneous condition. Indeed, SHFM occurs as a nonsyndromic form with limb-confined manifestations or as a syndromic form with extra-limb manifestations, and both nonsyndromic and syndromic forms take place as digit-confined abnormalities or in association with long bone defects as observed in SHFM with long bone deficiency (SHFLD) primarily affecting the tibia and in Gallop–Wolfgang complex (GWC) characterized by femoral bifurcation [1, 2]. Furthermore, SHFM is associated with variable expressivity and reduced penetrance [1, 2].
SHFM is also a genetically heterogeneous condition. Indeed, SHFM has been identified in patients with heterozygous copy-number variants (CNVs) such as: (1) deletions affecting DLX5 and/or DLX6 at 7q21.3 (SHFM1; MIM: 183600), (2) duplications around an LBX1–FGF8 region at 10q24 (SHFM3; MIM: 246560), (3) deletions involving the HOXD cluster at 2q31 (SHFM5; MIM: 606708), and (4) duplications or triplications involving BHLHA9 at 17p13.3 (SHFLD3; MIM: 612576) [1,2,3]. Furthermore, SHFM has also been found in patients with (1) heterozygous variants of TP63 at 3q28 (SHFM4; MIM: 605289), FGFR1 at 8p11.2, and PORCN at Xp11.23, (2) heterozygous or biallelic variants of DLX5 or DLX6 at 7q21.3, (3) monoallelic variants of paternally expressed IGF2 at 11p15.5, and (4) biallelic variants of WNT10B at 12q31 (SHFM6; MIM: 225300), FGFR2 at 10q26.13, CDH3 at 16q22.1, and MAP3K20 at 2q31.1 [1,2,3]. In addition, several candidate genes/loci have been suggested for SHFM [1,2,3].
To date, we have identified underlying genetic causes in 37 of 56 Japanese families with SHFM, while such genetic causes could not be detected in the remaining 19 families. Actually, we revealed heterozygous tandem duplications/triplications at 17p13.3 in 28 families (43 patients) with SHFM, SHFLD, or GWC, heterozygous 10q24 duplications in three families (five patients) with SHFM, biallelic WNT10B variants in a single family (one patient) with SHFM, heterozygous FGFR1 variants in four families (four patients) with SHFM and hypogonadotropism, and a de novo IGF2 variant on the paternally inherited allele in a single family (one patient) with SHFM, Silver–Russell syndrome, and disorder of sex development (Fig. 1) [4,5,6,7].

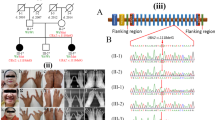
Molecular diagnostic flowchart. This study consists of group 1 and group 2, and both groups comprise subjects with non-syndromic and syndromic SHFM. Molecular analyses employed in this study are highlighted with blue, and those performed in our previous studies are highlighted with orange. Asterisk represents including family #26 in which 26-II-3 had a de novo 19q13.11 deletion of uncertain significance. Daggar represents in two of three families, the variant was identified in only one allele (simple heterozygosity)
Thus, tandem duplications/triplications involving BHLHA9 at 17p13.3 constitute the major underlying factor for SHFM in Japanese patients (Fig. S1) [4]. In this regard, it is notable that the 17p13.3 duplications/triplications are associated with highly variable expressivity and obviously low penetrance. This would primarily be due to the relevance of multiple minor genetic and environmental factors, because a major modifier(s) has not been identified despite extensive studies [4]. It is also notable that an identical 210,050 bp segment has been duplicated/triplicated in all the patients, with a genomic breakage and fusion between nonhomologous sequences in association with a 4-bp microhomology at the fusion point. This suggests that a founder duplication was produced through a microhomology-mediated replication error and underwent subsequent triplication through an unequal interchromatid exchange [4]. It is likely, therefore, that the founder duplications/triplications are widely spread in Japan primarily via clinically normal carriers and are identified via manifesting patients [4].
However, underlying genetic causes remain to be determined in a substantial fraction of patients with SHFM, and characteristic clinical findings related to each underlying genetic cause also remain to be clarified. Here, we report comprehensive clinical and molecular findings in Japanese families with SHFM, and suggest that LRP6 and UBA2 represent plausible candidate genes for SHFM.
Materials and methods
Families and subjects
We performed molecular and clinical studies in subjects from two groups of families: group 1, newly recruited 41 families (families #1–36 and #39–43; 41 probands, seven affected relatives, and 75 nonaffected relatives including two relatives with microform in family #28); and group 2, seven of the 19 families in which the underlying genetic causes remained undetected in our previous studies (families #37–38 and #44–48; seven probands, one affected relative, and eight nonaffected relatives) [4,5,6,7] (Figs. 1 and 2). For group 2, we had not obtained IC for whole exome sequencing (WES) from most families examined in our previous studies, and we could obtain IC for WES from seven families. All the 48 probands had a normal karyotype. For convenience, each subject is indicated by a family-generation-person style (e.g., the proband of family 1 is described as subject 1-II-1).

The pedigrees of 48 families examined in this study. Arrows indicate probands. Asterisk represents although not studied molecularly, 32-I-1 is considered as a carrier, because 32-II-1 and 32-II-2 are compound heterozygotes with maternally and nonmaternally (paternally) transmitted variants. Class 1, nonsyndromic SHFM confined to digits; class 2, nonsyndromic SHFM involving long bones; class 3, syndromic SHFM confined to digits; and class 4, syndromic SHFM involving long bones
Clinical classification
We divided SHFM into four classes: class 1, nonsyndromic SHFM confined to digits (SHFM-C1); class 2, nonsyndromic SHFM involving long bones (SHFM-C2); class 3, syndromic SHFM confined to digits (SHFM-C3); and class 4, syndromic SHFM involving long bones (SHFM-C4). Of the 48 probands and eight affected relatives from the 48 families, 40 had SHFM-C1, five had SHFM-C2, eight had SHFM-C3, and three had SHFM-C4.
Genetic studies
We sequentially performed screening of the Japanese founder duplication/triplication involving BHLHA9, genomewide array comparative genomic hybridization (aCGH), and WES in group 1, and carried out WES in group 2 (Fig. 1). We also performed additional analyses required in the above approach, such as DNA sequencing, microsatellite analysis, parental origin analysis, and X-inactivation analysis, by the standard methods. These studies were performed with leukocyte genomic DNA samples. The primers utilized in this study are shown in Table S1.
The Japanese founder 17p13.3 duplication/triplication was screened by conventional PCR analysis for the fusion point (Fig. S1) [4]. When PCR products were obtained, we carried out quantitative PCR (q-PCR) analysis, to determine the copy number (duplication or triplication).
Genomewide aCGH was performed with a catalog human array (4 × 180 K format, ID G4449A) (Agilent Technologies). Obtained data were analyzed with the Agilent Genomic Workbench 7.0. We searched for known pathogenic CNVs for SHFM and CNVs of unknown significance absent from Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/).
WES was carried out with SureSelect Human All Exon V6 (Agilent Technologies). Captured libraries were sequenced by NextSeq 500 (Illumina) with 150 bp paired-end reads. Exome data processing, variant calling, and variant annotation were performed, as described previously [8], using Human GRCh37 (ftp://ftp.broadinstitute.org/bundle/b37/) as the reference genome. We extracted rare variants with minor allele frequencies of ≤0.01 in all the public and in-house databases described in Table 1, and performed in silico pathogenicity predictions for extracted rare variants by several methods shown in Table 1. Subsequently, we searched for rare variants identified in causative genes for SHFM reported in the literature [1,2,3], and those detected in candidate genes for SHFM such as genes located on previously reported pathogenic CNVs for SHFM [1,2,3] and those reported to have produced SHFM in knockout mice (Mouse Genome Informatics; http://www.informatics.jax.org/). A rare variant-positive gene(s) shared by ≥two families or found to reside on a significance-unknown CNV(s) was also regarded as a candidate gene(s). We further examined inheritance patterns of rare variants in families examined by ≥trio analysis (proband and parents, as well as other available relatives), tissue expression patterns of rare variant-positive genes, occurrence of nonsense-mediated mRNA decay (NMD) for truncating variants [9], the probability of being loss-of-function intolerant (pLI) for candidate genes [10], and a cryptic exonic deletion(s) using WES-based CNV calling developed by Nord et al. [11]. The variants were evaluated by the methods described in Sequence Variant Nomenclature (http://varnomen.hgvs.org/) and the ACMG standards and guidelines [12].
Results
The results are summarized in Figs. 1 and 2. Representative photos and roentgenograms are shown in Fig. 3, and clinical findings are summarized in Table S2.

Representative photos and roentgenograms. U upper, and L lower
Screening of the Japanese founder 17p13.3 duplication/triplication
The duplications and triplications containing BHLHA9 were identified in probands from families #1–19 and families #20–21 of group 1, respectively. They were also detected in all the three affected relatives and in 15 of 31 nonaffected relatives examined in families #1–21. Notably, when both clinically normal parents were examined, either of them was invariably found to be a nonmanifesting carrier. Of the 24 affected subjects with the duplications/triplications, 18 had SHFM-C1, five had SHFM-C2, and one had SHFM-C4 with hypospadias and imperforate anus.
Genomewide aCGH
Genomewide aCGH was carried out for the remaining 20 families in group 1, revealing known pathogenic CNVs in five subjects from four families, i.e., a de novo 6.5–6.6 Mb deletion at 2q31 in 22-II-1, a de novo 440–470 kb duplication at 10q24 in 23-II-1, a familial 500–540 kb duplication at 10q24 shared by 24-II-1 and 24-I-2, and a de novo 520–550 kb duplication at 10q24 in 25-II-2 (Fig. S2). The 2q31 deletion (SHFM5) involved the HOXD cluster and was associated with SHFM-C4 with growth failure, developmental delay, and severe limb abnormalities. The 10q24 duplications (SHFM3) involved LBX1, BTRC, POLL, and DPCD in common, and were accompanied by SHFM-C1.
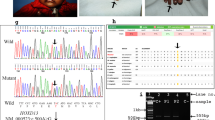
Furthermore, a de novo ~1.35 Mb deletion at 19q13.11 absent from the public databases was identified on a paternally derived chromosome of 26-II-3 with SHFM-C4 accompanied by sparse hair and eyebrows and hypodontia (Fig. 4a). The proximal and distal breakpoints were determined to reside at an intergenic sequence between RHPN2 and GPATCH1 and at an intragenic sequence between exon 5 and exon 6 of UBA2, respectively.

19q13.11 deletions and UBA2 variants. a Molecular findings in family #26. Genomewide aCGH analysis shows a de novo ~1.35 Mb deletion, Chr19:g.(33,555,825_33,571,785)_(34,925,874_34,929,549)del (according to the human genome reference build hg19), at 19q13.11 in 26-II-3, and microsatellite analysis for D19S416 confirms the deletion and its presence on the paternally inherited chromosome. The proximal and the distal breakpoints are localized between RHPN2 and GPATCH1 and between exon 5 and exon 6 of UBA2, respectively, by aCGH, the WES-based CNV calling, and q-PCR analysis for exons 4–6 of UBA2. The deletion contains 13 protein-coding genes and disrupts UBA2. b Summary of deletions involving UBA2 and UBA2 variants. The gray and blue bars indicate previously reported deletions identified in SHFM-negative and SHFM-positive subjects respectively, and the red bar denotes the deletion identified in 26-II-3. The NM_005499.2:c.71 G > T (p.(Gly24Val)) reported previously and NM_005499.2:c.1324dupT (p.(Tyr442Leufs*17)) identified in this study have been found in SHFM-negative and SHFM-positive subjects respectively
WES
WES was performed for 23 families (#26–36 and 39–43 in group 1, and #37–38 and 44–48 in group 2); family #26 was included because of uncertain significance of the 19q13.11 deletion. We carried out ≥ trio analyses for families #26–45, and singleton analyses for probands #46–48. At least, 98.8% of RefSeq coding sequences were covered by ≥10 reads.
WES revealed following novel or recurrent rare variants in known causative genes (Table 1 and Fig. S3): (1) a de novo TP63 variant affecting function in 27-II-1 with SHFM-C1; (2) a TP63 variant affecting function, shared by 28-II-1 with SHFM-C1 and 28-I-2 and 28-II-2 with microphenotype (left second to fourth finger nail dysplasia in 28-I-2 and syndactyly of the second to third toes in 28-II-2); (3) a TP63 variant probably affecting function, common to 29-II-1 and 29-II-2 with SHFM-C1 and 29-I-2 with apparently normal phenotype; (4) a de novo DLX5 variant affecting function in 30-II-1 with SHFM-C1; (5) a de novo IGF2 variant affecting function in 31-II-1 with SHFM-C3 accompanied by Silver–Russell syndrome (the IGF2 variant resided on the paternally inherited allele, and was predicted to drastically impair the protein structure by disrupting the S–S binding) (Fig. S4); (6) compound heterozygosity for a WNT10B variant of unknown function derived from 32-I-2 with normal phenotype and a WNT10B variant probably affecting function, which was absent from 32-I-2 and thus assumed to be inherited from 32-I-1 with normal phenotype, in 32-II-1 and 32-II-2 with SHFM-C1; (7) simple heterozygosity for a WNT10B variant probably affecting function shared by 33-II-1 with SHFM-C1, 34-II-1 with SHFM-C3 accompanied by ventricular septal defect, and clinically normal 33-I-2, 34-I-1, and 34-II-2; (8) coexistence of a WNT10B variant probably affecting function and a PORCN variant of unknown function common to 35-II-1 with SHFM-C3 accompanied by microphthalmia, cryptorchidism, pituitary hypoplasia, and developmental delay, and clinically normal 35-I-2; and (9) a de novo PORCN variant probably affecting function in 36-II-1 with SHFM-C3 accompanied by clinical features suggestive of mild focal dermal hypoplasia (FDH) (the PORCN variant appeared to have occurred in the postzygotic period, because of the low variant allele frequency in WES (25/109 of the read counts) and the small peak for the variant allele on Sanger sequencing). The eight missense variants were completely absent from the databases and were assessed to have high pathogenicity by in silico analyses. The WNT10B frameshift variant common to families #32–35 have been registered as a rare variant in gnomAD (exome_EAS) and HGVD, and was predicted to undergo NMD. In addition, no variant was identified in the intronic sequences including the pyrimidine-rich sequences and the branch sites of the apparently normal WNT10B alleles in the probands of families #33–35. Two females with PORCN variants (35-I-2 and 36-II-1) had random X-inactivation (31%:69% and 47%:53%, respectively).
Furthermore, WES revealed two frameshift variants, which were completely absent in the databases and were predicted to undergo NMD, in candidate genes (Fig. S3). First, a c.3984_3987del (p.(Asp1329Serfs*51)) variant in LRP6 (low density lipoprotein receptor-related protein 6), which was assessed as probably affecting function, was identified in 37-II-1 with SHFM-C1 and 37-II-2 with SHFM-C3 associated with hypospadias and tethered cord syndrome. This variant was also revealed in their father (37-I-1) with apparently normal phenotype, although digital roentgenograms were not obtained in the father. LRP6 was regarded as a candidate gene because of SHFM phenotype in homozygous Lrp6 knockout mice [13]. Second, a de novo c.1324dupT (p.(Tyr442Leufs*17)) variant in UBA2 (ubiquitin-like modifier-activating enzyme 2), which as assessed as probably affecting function, was found in 38-II-1 with SHFM-C4 accompanied by undermasculinized external genitalia. UBA2 was regarded as a candidate gene, because of its disruption by the 19q13.11 deletion in 26-II-3, positive SHFM in two previously reported patients with 19q13 deletions involving UBA2 (Fig. 4b) [14, 15], and strong Uba2 expression in the developing mouse limb buds [16]. UBA2 and LRP6 had the pLI value of 1.00. In both families #37 and #38, no other rare variant was identified in candidate genes for SHFM under assumption of a Mendelian inheritance pattern with complete penetrance.
Neither rare variant nor cryptic exonic CNV was detected in a causative or candidate gene in the remaining families #39–48 and in 26-II-3 with the 19q13.11 deletion.
Data depository
All the CNVs and sequence variants identified in this study were submitted to the Leiden Open Variant Database (https://databases.lovd.nl/shared/genes), together with phenotypes (Table S3).
Discussion
We performed comprehensive clinical and molecular studies in two groups of families with SHFM-C1–4. To our knowledge, such detailed studies have not been carried out previously.
We identified the Japanese founder duplication/triplication in families #1–21 (roughly half of families in group 1). No other CNVs involving BHLHA9 was detected by aCGH as described in our previous report [4, 5], although various types of duplications have been revealed in non-Japanese patients [17]. Thus, we identified the Japanese founder duplication/triplication in 49 of 97 families (50.5%) with SHFM examined to date (Fig. 1). Notably, we analyzed both of the clinically normal parents in 30 of the 49 families, invariably showing heterozygosity for the duplication/triplication in either of them. We also revealed the duplication/triplication in 37 of 78 clinically normal relatives (47.4%) in the 30 families. Furthermore, we have detected the duplication in two of 1000 Japanese general population [4]. These findings imply that the duplication/triplication is widely spread in Japan and cause SHFM in subjects with high susceptibility, with an estimated penetrance of ~1.7% and an odds ratio of ~500 (for details, see Supplementary Document).
We found a de novo 2q31 deletion (SHFM5) in 22-II-1 with SHFM-C4. Since the deletion encompassed the HOXD cluster involved in the limb development [18], loss of the HOXD cluster would be the major cause for the SHFM-C4 in 22-II-1. Indeed, several types of 2q31 deletions involving the HOXD cluster and balanced chromosomal rearrangements with the breakpoints around the HOXD cluster have been identified in patients with variably expressed growth and developmental delay, craniofacial anomalies, undermasculinized genitalia, and/or limb abnormalities including SHFM [19].
We also found 10q24 duplications (SHFM3) in families #23–25 with SHFM-C1. For SHFM3, several types of duplications have been identified in multiple patients with SHFM [3, 20]. In this context, it is notable that (1) a region spanning LBX1, BTRC, POLL, and FBXW4 is duplicated in most patients, although there appears to be no gene that is involved in all the duplications [20]; (2) FGF8, whose protein product play an important role in the AER maintenance and functions as a ligand of FGFR1 relevant to SHFM with hypogonadotropism [21], resides adjacent to FBXW4, although FGF8 is not duplicated in most patients [20]; (3) FGF8 enhancers are interspersed around the duplicated region [22]; and (4) mouse Lbx1, Fbxw4, and Fgf8 are expressed in the developing limbs [22]. Thus, the 10q24 duplications may have altered the expression of FGF8, and possibly LBX1 and/or FBXW4 as well, leading to the development of SHFM.
A de novo 19q13.11 deletion was identified in 26-II-3 with SHFM-C3. This deletion is discussed with the UBA2 variant, because it disrupted UBA2.
Sequence variants were primarily identified in the known causative genes. In this regard, several matters would be worth pointing out. First, this study provides support for variable expressivity and reduced penetrance of TP63 variants [1, 2, 23, 24]. Furthermore, TP63 variants detected in families #27–28 with SHFM-C1 have also been identified in patients with ectrodactyly, ectodermal dysplasia, and cleft lip/palate (EEC) syndrome [23, 24]. Since TP63 variants have predominantly been reported in patients with SHFM-C3 such as EEC syndrome rather than in those with SHFM-C1 [23, 24], it is likely that TP63 variants usually result in SHFM-C3 and occasionally lead to SHFM-C1 as well as apparently normal phenotype, depending on other multiple genetic and environmental factors. Second, this study argues for the important role of heterozygous DLX5 variants in the development of SHFM-C1, as reported previously [25]. Third, this study supports the development of SHFM by apparently amorphic rather than hypomorphic IGF2 variants [7]. Indeed, SHFM has occurred in patients with apparently null IGF2 variants (nonsense and frameshift variants subject to NMD [7, 26] and the missense variant disrupting the S–S binding of IGF2 protein identified in this study). By contrast, SHFM has never taken place in patients with epimutations (hypomethylations) of the H19/IGF2:IG-DMR (differentially methylated region) that occur in a postzygotic period with mosaicism for cells with epimutated DMR silencing IGF2 expression and those with normally methylated DMR permitting IGF2 expression [7, 27]. In addition, mouse Igf2 is strongly expressed in the developing limbs [28], and Igf2 knockout mice often manifest abnormal long bone morphology [29]. Fourth, the WNT10B frameshift variant common to families #32–35 appears to have spread in Japan. Since WNT10B variants have autosomal recessive effects [1, 2], it is assumed that another variant is hidden in a noncoding region(s) of WNT10B or in another gene for SHFM of 33-II-1 and 34-II-1 with apparently simple heterozygosity for the frameshift variant. Alternatively, it might be possible that simple heterozygosity leads to SHFM in patients with markedly high susceptibility because of multiple other genetic and environmental factors. Fifth, coexistence of a WNT10B frameshift variant and a PORCN variant in 35-II-1 might have exerted an additive or synergic effect on the development of SHFM. Indeed, since PORCN variants usually lead to FDH in an X-linked dominant manner with male lethality [30], the deleterious effect of the PORCN variant is assumed to be considerably mild in 35-II-1 as well as in 35-I-2 with a random X-inactivation. Thus, while phenotype of 35-II-1 such as SHFM and microphthalmia could be explained by the PORCN variant [30], it is uncertain whether such phenotype is caused by the PORCN variant alone. Lastly, the apparent mosaicism would explain the mild FDH phenotype in 36-II-1 with a PORCN variant probably affecting function and a random X-inactivation.
Notably, we identified an LRP6 variant subject to NMD in family #37. LRP6 has been regarded not only as a causative gene for autosomal dominant neural tube defects, tooth agenesis, and cleft lip/palate with incomplete penetrance [31,32,33], but also as a candidate gene for SHFM because of the development of SHFM in homozygous Lrp6 knockout mice [13]. Furthermore, the knockout mice also exhibit urogenital abnormalities, caudal to lumbar region anomalies such as spina bifida, and neural tube defect [13], and 37-II-2 showed SHFM-C3 with hypospadias and tethered cord syndrome which appears to be developmentally linked with spina bifida. Such phenotypic similarity between Lrp6 knockout mice and 37-II-2, together with SHFM-C1 in 37-II-1, would argue for LRP6 being a candidate gene for SHFM-C1 and SHFM-C3. Furthermore, there are several findings in support of the relevance of LRP6 variants to the development of SHFM: (1) heterozygous LRP6 variants are associated with minor digital anomalies such as clinodactyly of the fifth fingers and underdeveloped thumbs [32, 33]; (2) LRP6 acts as a coreceptor with Frizzled protein family members in the canonical WNT/β-catenin signaling pathway involved in limb development [31,32,33]; and (3) heterozygous variants in LRP4, which inhibits LRP5/LRP6 signaling, are associated with Cenani-Lenz syndactyly syndrome characterized by complex syndactyly of hands and feet, oligodactyly, radial and ulnar shortening, and teeth abnormalities [34]. Notably, the LRP6 variant was also detected in the father with apparently normal phenotype. This could be due to incomplete penetrance of the LRP6 variant with a dominant effect. Alternatively, another CNV or sequence variant for SHFM might be transmitted from the mother to the affected children, although such a variant was not identified by aCGH and WES.
We also identified a de novo UBA2 frameshift variant subject to NMD in 38-II-1 with SHFM-C4 with undermasculinized genitalia. UBA2 is regarded as a candidate gene for SHFM, because UBA2 was disrupted by the 19q13.11 deletion in 26-II-3 with SHFM-C3 accompanied by sparse hair and eyebrows and hypodontia. In this regard, a single patient with a de novo missense variant in UBA2 and 16 patients with different types of 19q13 deletions involving UBA2 have been reported (Fig. 4b) [14, 15, 35,36,37]. Notably, two patients with 19q13 deletions have SHFM [14, 15], and most patients of both sex have aplasia cutis congenita reminiscent of sparse hair in 26-II-3 and most male patients manifest underdeveloped genitalia as observed in 38-II-1 [14, 15, 35,36,37]. Furthermore, it is worth pointing out that (1) UBA2 plays an important role in sumoylation as a subunit of dimeric E1-activating enzyme [14, 15, 35,36,37]; (2) TP63 is sumoylated by SUMO (small ubiquitin-like modifier) such as SUMO-1 and SUMO-2 [38, 39], and altered sumoylation of TAp63α (an isoform of TP63) has been suggested to underlie the development of SHFM in patients with TP63 variants on the sterile α motif domain [39]; and (3) a 17q25 deletion including SUMO-2 has been identified in a patient with SHFM [3]. These findings, together with a strong expression of mouse Uba2 in the developing limb buds [16], would argue that UBA2 represents a novel gene for SHFM, with a low penetrance. In addition, altered sumoylation has also been implicated as an underlying factor for aplasia cutis congenita possibly via a modified activity of ΔNp63α (another isoform of TP63) involved in skin terminal differentiation and that for undermasculinized genitalia possibly via a modified activity of the androgen receptor essential for genital masculinization [15, 35,36,37]. However, the 19q13.11 deleted region in 26-II-3, which is also deleted in the two previously reported patients with SHFM, is associated with haploinsufficiency of 14 genes including UBA2 (Fig. 4). Thus, the relevance of other deleted genes such as LRP3 (the paralog of LRP6) to the development of SHFM might remain possible at present.
We studied a total of 97 Japanese families with SHFM to date. Several findings are noteworthy for the overall results (Table 2). First, we revealed significant CNVs or rare variants in 75 families (99 subjects) of the 97 families (77%), although WES was not performed in previously reported 12 CNVs-negative families (Fig. 1). This implies that SHFM primarily occurs as a genetic disorder, while the inheritance pattern is often complicated by reduced penetrance and variable expressivity [1, 2, 4, 23, 24]. Second, a considerable degree of correlation was observed between underlying genetic causes and phenotypes. For example, it was found that (1) 17p13.3 duplication was primarily associated with SHFM-C1 affecting the upper limbs, and 17p13.3 triplication was predominantly accompanied by SHFM-C2 involving the lower limbs; (2) 17p13.3 duplication/triplication was the sole underlying cause for SHFM-C2, with the dosage effect (the relative frequency of long bone deficiency was significantly higher in triplications than in duplications) (8/12 vs. 12/55, P = 0.0042 by the Fisher′s exact test); (3) 10q24 duplication was invariably accompanied by SHFM-C1 affecting four limbs, whereas 2q31 and 19q13.11 deletions resulted in SHFM3/SHFM-4; and (4) TP63, DLX5, and WNT10B variants were primarily associated with SHFM-C1 involving both upper and lower limbs, whereas PORCN, FGFR1, IGF2, LRP6, and UBA2 variants were primarily accompanied by SHFM-C3/SHFM-4 affecting both upper and lower limbs. Third, the prevalence of SHFM was apparently more frequent in males than in females (61:38). Such male dominance has also been reported in non-Japanese patients with BHLHA9 duplications [17]. This might be due to testosterone effects, because testosterone influences the digital growth pattern as indicated by the lower second to fourth digit length ratio in males than in females [40]. Lastly, genital and caudal region anomalies were observed in five subjects (two with the 17p13.3 duplication, one with the LRP6 variant, one with the UBA2 variant, and one with unknown cause) (Tables S2 and S4). This may imply the operation of similar genetic factors in the formation of limbs and genitalia, as exemplified by HOXA13 for hand-foot-genital syndrome [41].
In summary, this study implies that SHFM primarily occurs as a genetic disorder with some clinical features characteristic of each underlying cause, and that LRP6 and UBA2 represent plausible candidate genes for SHFM.
References
Gurrieri F, Everman DB. Clinical, genetic, and molecular aspects of split-hand/foot malformation: an update. Am J Med Genet A. 2013;161A:2860–72.
Everman DB. Split-hand/foot malformation. In: Stevenson RE, Hall JG, Everman DB, Solomon BD, editors. Human malformations and related anomalies. 3rd ed. Oxford, UK: Oxford University Press; 2016. p. 169–73.
Carter TC, Sicko RJ, Kay DM, Browne ML, Romitti PA, Edmunds ZL, et al. Copy-number variants and candidate gene mutations in isolated split hand/foot malformation. J Hum Genet. 2017;62:877–84.
Nagata E, Kano H, Kato F, Yamaguchi R, Nakashima S, Takayama S, et al. Japanese founder duplications/triplications involving BHLHA9 are associated with split-hand/foot malformation with or without long bone deficiency and Gollop-Wolfgang complex. Orphanet J Rare Dis. 2014;9:125.
Nagata E, Haga N, Fujisawa Y, Fukami M, Nishimura G, Ogata T. Femoral-tibial-digital malformations in a boy with the Japanese founder triplication of BHLHA9. Am J Med Genet A. 2015;167A:3226–8.
Ohtaka K, Fujisawa Y, Takada F, Hasegawa Y, Miyoshi T, Hasegawa T, et al. FGFR1 analyses in four patients with hypogonadotropic hypogonadism with split-hand/foot malformation: implications for the promoter region. Hum Mutat. 2017;38:503–6.
Yamoto K, Saitsu H, Nakagawa N, Nakajima H, Hasegawa T, Fujisawa Y, et al. De novo IGF2 mutation on the paternal allele in a patient with Silver-Russell syndrome and ectrodactyly. Hum Mutat. 2017;38:953–8.
Hiraide T, Nakashima M, Yamoto K, Fukuda T, Kato M, Ikeda H, et al. De novo variants in SETD1B are associated with intellectual disability, epilepsy and autism. Hum Genet. 2018;137:95–104.
Kuzmiak HA, Maquat LE. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med. 2006;12:306–16.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genom. 2011;12:184.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–8.
Chowdhury S, Bandholz AM, Parkash S, Dyack S, Rideout AL, Leppig KA, et al. Phenotypic and molecular characterization of 19q12q13.1 deletions: a report of five patients. Am J Med Genet A. 2014;164A:62–9.
Abe KT, Rizzo IMPO, Coelho ALV, Sakai N Jr, Carvalho DR, Speck-Martins CE. 19q13.11 microdeletion: clinical features overlapping ectrodactyly ectodermal dysplasia-clefting syndrome phenotype. Clin Case Rep. 2018;6:1300–7.
Costa MW, Lee S, Furtado MB, Xin L, Sparrow DB, Martinez CG, et al. Complex SUMO-1 regulation of cardiac transcription factor Nkx2-5. PLoS One. 2011;6:e24812.
Klopocki E, Lohan S, Doelken SC, Stricker S, Ockeloen CW. Soares et al. Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion. J Med Genet. 2012;49:119–25.
Zakany J, Duboule D. The role of Hox genes during vertebrate limb development. Curr Opin Genet Dev. 2007;17:359–66.
Dimitrov B, Balikova I, de Ravel T, Van Esch H, De Smedt M, Baten E, et al. 2q31.1 microdeletion syndrome: redefining the associated clinical phenotype. J Med Genet. 2011;48:98–104.
Li CF, Angione K, Milunsky JM. Identification of critical region responsible for split hand/foot malformation type 3 (SHFM3) phenotype through systematic review of literature and mapping of breakpoints using microarray data. Microarrays. 2015;5:E2.
Becic T, Kero D, Vukojevic K, Mardesic S, Saraga-Babic M. Growth factors FGF8 and FGF2 and their receptor FGFR1, transcriptional factors Msx-1 and MSX-2, and apoptotic factors p19 and RIP5 participate in the early human limb development. Acta Histochem. 2018;120:205–14.
Marinić M, Aktas T, Ruf S, Spitz F. An integrated holo-enhancer unit defines tissue and gene specificity of the Fgf8 regulatory landscape. Dev Cell. 2013;24:530–42.
van Bokhoven H, Brunner HG. Splitting p63. Am J Hum Genet. 2002;71:1–13.
Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999;99:143–53.
Wang X, Xin Q, Li L, Li J, Zhang C, Qiu R, et al. Exome sequencing reveals a heterozygous DLX5 mutation in a Chinese family with autosomal-dominant split-hand/foot malformation. Eur J Hum Genet. 2014;22:1105–10.
Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, et al. Paternally inherited IGF2 mutation and growth restriction. N Engl J Med. 2015;373:349–56.
Eggermann T, Begemann M, Binder G, Spengler S. Silver-Russell syndrome: genetic basis and molecular genetic testing. Orphanet J Rare Dis. 2010;5:19.
Conover CA, Bale LK, Overgaard MT, Johnstone EW, Laursen UH, Füchtbauer EM, et al. Metalloproteinase pregnancy-associated plasma protein A is a critical growth regulatory factor during fetal development. Development. 2004;131:1187–94.
Hardouin SN, Guo R, Romeo PH, Nagy A, Aubin JE. Impaired mesenchymal stem cell differentiation and osteoclastogenesis in mice deficient for Igf2-P2 transcripts. Development. 2011;138:203–13.
Bostwick B, Fang P, Patel A, Sutton VR. Phenotypic and molecular characterization of focal dermal hypoplasia in 18 individuals. Am J Med Genet C Semin Med Genet. 2016;172C:9–20.
Allache R, Lachance S, Guyot MC, De Marco P, Merello E, Justice MJ, et al. Novel mutations in Lrp6 orthologs in mouse and human neural tube defects affect a highly dosage-sensitive Wnt non-canonical planar cell polarity pathway. Hum Mol Genet. 2014;23:1687–99.
Ockeloen CW, Khandelwal KD, Dreesen K, Ludwig KU, Sullivan R. van Rooij IALM, et al. Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet Med. 2016;18:1158–62.
Massink MP, Créton MA, Spanevello F, Fennis WM, Cune MS, Savelberg SM, et al. Loss-of-function mutations in the WNT co-receptor LRP6 cause autosomal-dominant oligodontia. Am J Hum Genet. 2015;97:621–6.
Sukenik Halevy R, Chien HC, Heinz B, Bamshad MJ, Nickerson DA. Mutations in the fourth β-propeller domain of LRP4 are associated with isolated syndactyly with fusion of the third and fourth fingers. Hum Mutat. 2018;39:811–5.
Venegas-Vega C, Nieto-Martínez K, Martínez-Herrera A, Gómez-Laguna L, Berumen J, Cervantes A. et al. 19q13.11 microdeletion concomitant with ins(2;19)(p25.3; q13.1q13.4)dn in a boy: potential role of UBA2 in the associated phenotype. Mol Cytogenet. 2014;7:61.
Melo JB, Estevinho A, Saraiva J, Ramos L, Carreira IM. Cutis Aplasia as a clinical hallmark for the syndrome associated with 19q13.11 deletion: the possible role for UBA2 gene. Mol Cytogenet. 2015;8:21.
Marble M, Guillen Sacoto MJ, Chikarmane R, Gargiulo D, Juusola J. Missense variant in UBA2 associated with aplasia cutis congenita, duane anomaly, hip dysplasia and other anomalies: a possible new disorder involving the SUMOylation pathway. Am J Med Genet A. 2017;173:758–61.
Ghioni P, D’Alessandra Y, Mansueto G, Jaffray E, Hay RT, La Mantia G, et al. The protein stability and transcriptional activity of p63alpha are regulated by SUMO-1 conjugation. Cell Cycle. 2005;4:183–90.
Ranieri M, Vivo M, De Simone M, Guerrini L, Pollice A, La Mantia G, et al. Sumoylation and ubiquitylation crosstalk in the control of ΔNp63α protein stability. Gene. 2018;645:34–40.
Williams TJ, Pepitone ME, Christensen SE, Cooke BM, Huberman AD, Breedlove NJ, et al. Finger-length ratios and sexual orientation. Nature. 2000;404:455–6.
Mortlock DP, Innis JW. Mutation of HOXA13 in hand-foot-genital syndrome. Nat Genet. 1997;15:179–80.
Acknowledgements
We are grateful to all patients and their parents for their cooperation. We thank Ms. Aya Kitamoto and Mr. Naoki Adachi for their technical support. We also thank Drs Toshiro Nagai, Hironao Numabe, Seiji Mizuno, Toshiro Nagai, Keisuke Nagasaki, Hiroaki Yagasaki, You Omura, Noriyuki Namba, Satoshi Okada, Sadao Ito, Hiromi Iwata, Yuri Endo, Kaori Hara, Takahito Wada, and Akira Oishi for providing clinical information and patient samples.
Funding
This work was supported by Grants from the Japan Agency for Medical Research and Development (AMED) (JP17ek0109297 and JP18ek0109301) and by Grant-in-Aid for Scientific Research on Innovative Areas (JP17H06428) from the Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Institutional Review Board Committee of Hamamatsu University School of Medicine, and was performed after obtaining written informed consent (IC) from adult subjects (≥20 years old) or parents of nonadult subjects (<20 years old). We also obtained informed assent from children aged ≥6 years.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yamoto, K., Saitsu, H., Nishimura, G. et al. Comprehensive clinical and molecular studies in split-hand/foot malformation: identification of two plausible candidate genes (LRP6 and UBA2). Eur J Hum Genet 27, 1845–1857 (2019). https://doi.org/10.1038/s41431-019-0473-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0473-7
This article is cited by
-
Rare phenotype: Hand preaxial polydactyly associated with LRP6-related tooth agenesis in humans
npj Genomic Medicine (2021)
-
Genome sequencing in families with congenital limb malformations
Human Genetics (2021)
