
Abstract
Despite the nationwide availability of pharmacogenomic (PGx) guidelines in electronic medication surveillance systems in The Netherlands, PGx guided prescribing is still uncommon in primary care. We set out to investigate the adoption of pharmacist initiated PGx testing in primary care. Community pharmacists were offered a free PGx test covering 40 variants in 8 genes to test patients receiving an incident prescription (IRx) of a selection of 10 drugs. Results of the PGx test along with predicted phenotypes and a therapeutic recommendation based on the Dutch Pharmacogenetics Working Group (DPWG) guidelines were transferred to the pharmacist and physician. Adoption was defined as the percentage of eligible patients that received genotyping. From November 2014–July 2016, 200 patients were included with an adoption of 18.0%. Of the included patients 57.5% received an IRx for atorvastatin, 14.5% started with simvastatin and 28.0% received an IRx for amitriptyline, (es)citalopram, nortriptyline, or venlafaxine. 90% of the patients carried at least one actionable PGx test result in the selected PGx-panel. In 31.0% of the incident prescriptions a combination between a drug with a known gene-drug interaction and an actionable genotype was present and a therapeutic recommendation was provided. The provided recommendations were accepted by the clinicians in 88.7% of the patients. Pharmacist initiated implementation of PGx in primary care is feasible, and the frequency of actionable gene-drug interactions for the selected drugs is high.
Similar content being viewed by others

Introduction
In recent years many of the hurdles hindering the successful application of pharmacogenomics (PGx) have been addressed [1, 2]. The Clinical Pharmacogenetics Implementation Consortium (CPIC) and the Dutch Pharmacogenetics Working Group (DPWG) both have published a range of guidelines on relevant gene-drug interactions providing PGx-test interpretation and therapeutic recommendations for patients with a known genotype [2,3,4].
Several pharmacogenomic implementation projects have been initiated in specialized and secondary care centers. Results have not only shown the feasibility of clinical pharmacogenomics but also highlighted the high frequency of clinically actionable genotypes in the general population underscoring the potential for the use PGx to improve pharmacotherapy [5,6,7,8,9,10,11]. Currently, PGx testing is not routinely implemented in primary care [12]. However, studies have shown that of the drugs with a known gene-drug interactions 25 are among the most frequently used medications in the USA by patients in a home setting. Examples of these drug include statins, TCA’s, SSRI and SNRI’s [13]. Therefore, PGx-informed decision making has a high potential to lead to optimized pharmacotherapy in primary care. Indeed, preliminary studies in primary care setting have shown that PGx informed prescribing leads to a reduction in hospitalization, appears to be cost-effective within the cost-structures currently present in the US healthcare and that gene-drug interactions may appear to be independent risk for hospitalization [14,15,16,17].
In the Netherlands the infrastructure for using PGx information to guide pharmacotherapy is well developed. The DPWG guidelines are fully integrated into available clinical decision support (CDS) nationwide through the G-standard (https://www.knmp.nl/producten-en-diensten/gebruiksrecht-g-standaard/informatie-over-de-g-standaard/the-gstandaard-the-medicines-standard-in-healthcare), and readily available as pop-up alerts (see Fig. 1) during drug prescribing and dispensing [3, 4]. With their dedication to safe and effective pharmacotherapy pharmacists are leading candidates to act on a patients’ genotype. Patients also appear to be positive towards PGx-testing initiated within a community pharmacy setting [18]. Based on the availability of PGx guidelines integrated in the workflow of healthcare professionals and the positive attitude to pharmacy delivered PGx we set out to investigate whether genotype guided dosing in primary care using a PGx test panel in the Netherlands is feasible. Additionally, we wanted to investigate the frequency of actionable genotypes in a panel of the genes CYP2C9, CYP2C19, CYP2D6, CYP3A5, DPYD, SLCO1B1, TPMT and VKORC1 and to study the frequency of gene-drug interactions in primary care.

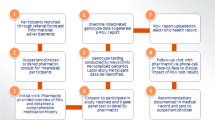
Example of an alert generated through Clinical Decision Support
Methods
Study design and setting
The Implementation of Pharmacogenomics into Primary care Project (IP3) was designed as a prospective multicenter observational study of routine clinical practice with the objective to test the feasibility of pharmacist initiated pharmacogenomics testing in primary care. Secondary aims were to investigate the frequency of actionable phenotypes, the frequency of incident prescriptions with a combination between a drug with a known gene-drug interaction and the associated actionable genotype, and the effects of gene-drug interactions on pharmacotherapy in both the period of empirical treatment prior to inclusion (i.e. without the knowledge of the patients’ genetic make-up) and during genotype-guided dosing in separate exploratory analyses. Actionable phenotypes were defined as a genetically predicted poor, intermediate or ultra-rapid metabolizer phenotype for the genes CYP2C9, CYP2C19, CYP2D6, DPYD and TPMT; a genetically predicted phenotype of heterozygous or homozygous expressor for the gene CYP3A5, a genetically predicted phenotype of lowered transporter activity for the gene SLCO1B1 or a genetically predicted phenotype of high sensitivity for the gene VKORC1 (see supplementary document 1 for associated genetic variants). The central Medical Ethics Review board of the Leiden University Medical Center (LUMC) in Leiden, The Netherlands approved the protocol.
Endpoints
The primary endpoint was the adoption rate of PGx testing. This was defined as the number of genotyped patients divided by the total number of eligible patients in the timeframe of the study. Secondary endpoints included the frequency of actionable genotypes, the number of patients with a combination of an actionable genotype in combination with a drug with a known gene-drug interaction, the number of drug related interventions (defined as stopping of the medication, switching to another drug, switching to a different dose), the number of visits to the pharmacy, and the drug dose. The medication history of the included patients was collected from the electronic patient records in the pharmacy. In order to compare drug dosages of different drugs defined daily dosages (DDD) were used. To account for changing dosages over time, a time-weighted average was calculated for each of the drugs of inclusion.
To study the effects of genotype guided dosing on the secondary endpoints follow-up on all secondary outcomes was prospectively collected from the point of inclusion until the end of the study in which general practitioner and pharmacist had knowledge of actionable PGx test results. In a similar analysis the effects of actionable genotypes on secondary outcomes during the period where individuals had been treated empirically by general practitioners and pharmacists without the knowledge of the patients’ genotype was studied. The follow-up concerning the secondary endpoints was collected prospectively for the period in which general practitioner and pharmacist had knowledge of actionable PGx test results. For the retrospective analysis the DDD, number of visits to the pharmacy and drug related interventions were collected. Patients (see below) were identified through weekly searches in the electronic drug dispensing systems. Treating pharmacists informed eligible patients about the project. When the patient agreed to participate, written informed consent was obtained and a saliva sample for DNA extraction was collected.
Patients
Adult (18 years or older) patients with an incident prescription (defined as no prescription for the incident drug within the preceding 12 months) for at least 28 days for amitriptyline, atomoxetine, atorvastatin, (es)citalopram, clomipramine, doxepin, nortriptyline, simvastatin or venlafaxine in participating primary healthcare centers in the vicinity of Leiden were invited to participate in the study and offered free genetic testing. To investigate the impact of PGx informed prescribing historical data were used. Therefore, patients needed to have at least one prescription for one of the 10 selected drugs for a period of at least 28 days other than the incident prescription in the preceding year. After identification of the patients through automated queries the participating pharmacists manually checked whether patients fulfilled the in- and exclusion criteria. Finally, patients not recruited within 14 days after dispensing the incident prescription were excluded.
Genotyping
Saliva was collected using the Oragene DNA OG-250 (DNA Genotek Inc, Ottawa, Ontario, Canada) and sent to the department of Clinical Pharmacy & Toxicology at the LUMC. DNA was isolated according to the protocol of the manufacturer. Genotypes of CYP2C9, CYP2C19, CYP2D6, CYP3A5, DPYD, SLCO1B1, TPMT and VKORC1 (see supplementary document 1) were determined using the Drug Metabolizing and Transporters (DMET) Plus Array (Affymetrix, Santa Clara, CA). CYP2D6 copy-number variation was detected with qPCR (Thermo Fisher Scientific, Massachusetts, USA). The DMET method was validated for the genes CYP2C9, CYP2C19, CYP3A5, DPYD, SLCO1B1, TPMT and VKORC1 by reanalyzing DNA samples from 94 anonymized blood-donors obtained from Sanquin (Sanquin, Amsterdam, NL) previously analyzed using Sanger Sequencing used for the validation of assays used in the clinical setting (see supplementary document 1 for the associated variants). Validation of the DMET for the detection of genetic variants in the gene CYP2D6 (see supplementary document 1) and qPCR used for the detection of copy-number variants occurred through reanalyzing DNA samples from the CYPTAM study (CYPTAM: NTR1509) previously analyzed using the AmpliChip® (Roche, Indianapolis, US) [19]. The DMET test was further supplemented with the DPYD variants NM_000110.3:c.1236 G > A (rs56038477) and NM_000110.3:c.2846 A > T (rs67376798) which were routinely tested in the LUMC at the time of study execution [20]. The genotypes of the 8 genes in the pharmacogenomic profile were translated into genotype predicted phenotypes using the DPWG guidelines [3, 4]. Finally, a report for the general practitioner and pharmacists was generated which contained the genotypes, the genetic predicted phenotypes along with a therapeutic recommendation to continue current therapy in case of a wildtype genotype or to adjust pharmacotherapy in case of a combination between an actionable genotype and the prescribed drug. The report was transferred to the treating clinicians with the request to record the genetic profile in automated electronic drug prescribing and dispensing systems for active medication surveillance based on the patients’ genotype (see also Introduction). The database containing the genotypes and genetic predicted phenotypes is available at https://databases.lovd.nl/shared/individuals (patient IDs 184080-184279) [21].
Statistical analysis
Primary endpoint
The primary endpoint of this study was the extent of adoption of PGx-testing expressed as the percentage of eligible patients that received genotyping in a percentage.
Secondary endpoints
The statistical analysis of the secondary endpoints was not powered to detect a significant effect and was of exploratory design. To explore whether gene-drug interactions resulted in a difference in the time-weighed DDD’s a multinomial generalized estimating equation (GEE) was carried out using the categorized DDD’s from the prescribed drug at inclusion (amitriptyline, atomoxetine, atorvastatin, (es)citalopram, clomipramine, doxepin, nortriptyline, simvastatin or venlafaxine) as dependent variable, and the occurrence of a gene-drug interaction as an independent variable. The GEE model was chosen because of the hierarchical structure in the data for the prospective collected DDD’s of the drug prescribed at the time of inclusion with genotyped guided dosing. In addition, a multilevel log-linear Poisson analysis was carried out to determine whether a relationship was present between the actionable phenotype and number of switches in dose after inclusion. Furthermore, a multilevel log-linear Poisson analysis was carried out to determine between actionable phenotype and the number of visits to the pharmacy the prospective collected follow-up.
In three separate analyses, the retrospectively collected follow-up from the period before inclusion was analyzed in a similar manner. The categorized DDD’s from one of the ten selected drugs used before inclusion were analyzed using a multinomial GEE with the categorized DDD’s as dependent variable and presence of a gene-drug interaction as an independent variable. Multilevel log-linear Poisson analyses were carried out to analyze the effect gene-drug interactions on the number of switches in dose applied and the number of visits before inclusion. Sensitivity analyses for all the multilevel models were performed to investigate whether factors such as gender, age, weight, height and automated dose dispensing were confounding factors. Model specifications included an exchangeable correlation structure and a robust estimator. In all models the patient was used as the lowest level clustered within prescribing physician.
Results
Characterization of included patients & incident prescriptions
In total 200 patients were included in this study. The characteristics of the patients are presented in Table 1. The included study population contained 103 (51.5%) females and 97 (48.5%) males with mean age was 62 ± 11 years ranging from 27 up to 87. The majority of patients (92.5%) reported that both parents were of Caucasian descent (see Table 1). The majority of the patients received an incident prescription for a statin i.e. atorvastatin (n = 115) or simvastatin (n = 29). The remainder of the patients were included on amitriptyline (n = 15), citalopram (n = 7), escitalopram (n = 3), nortriptyline (n = 17) or venlafaxine (n = 14) (see Table 1). The average turnaround time from drug prescription to returning the results (including time for shipping of the sample to LUMC) was 23,4 ± 8,3 days.
Adoption of PGx
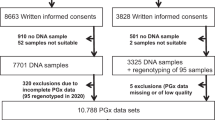
The adoption of PGx testing by healthcare professionals (primary endpoint) was 18.0% (see also Fig. 2). In the 20 month recruitment period, 1864 patients with an incident prescription for one of the 10 drugs and historical use of one of the other nine drugs were identified. Of those patients 483 were not eligible for inclusion. Reasons included duration of prescription < 28 days, use of the incident drug in the preceding 12 months, no use of one of the other nine selected drugs in the previous year, previously included in the study, or was too young (<18 years). Furthermore 267 patients were not considered eligible by the pharmacists. Reasons included palliative treatment, no permanent address, not capable of providing written informed consent. Of the 1,114 eligible patients 652 were not contacted by the pharmacist within two weeks after the patient picked up the incident prescription. Of the 462 patients that were contacted 207 consented to the study, whereas 255 patients did not agree to participate. Of the 207 patients that agreed to participate in the study 7 were excluded because they did not sign informed consent, failed to provide a saliva sample or the saliva contained too little DNA and the patient refused to provide a new sample.

Overview of patient flow
Occurrence of actionable genotypes & gene-drug interactions
Ninety percent of the genotyped patients carried at least one actionable genotype with one and two actionable genotypes per patient being the most frequent with an occurrence of 28.5 and 32.5%, respectively. The remainder of the patients carried up to five actionable genotypes out of the panel of eight tested genes (See Table 2). The frequencies of different genotype predicted phenotypes for each of the eight genes are presented in Table 3. For 31.0% of the patients a combination between a drug with a gene-drug interaction and actionable genotype was present requiring action from the healthcare professionals. The majority of the combinations of drugs with known gene-drug interactions and actionable genotypes consisted of a gene-drug pair between the SLCO1B1 gene with the drug atorvastatin (n = 35). The frequencies of other were CYP2C19 with (es)citalopram (n = 3), CYP2D6 with amitriptyline (n = 6), CYP2D6 with nortriptyline (n = 7), CYP2D6 with venlafaxine (n = 8) and SLCO1B1 with simvastatin (n = 3). Patients with combination of an actionable genotype and associated drug were not significantly different from patients without this gene-drug pair regarding gender, age, length and weight. In an exploratory analysis medication surveillance based a pharmacogenomic profile did not result in a significant change in time weighed DDD’s (OR 1.14, CI 0.68–1.92, p = 0.614), interventions performed on the medication (OR 1.33, CI 0.92–1.93, p = 0.128) or the number of visits to the pharmacy (OR = 0.93, CI 0.63–1.36, p = 0.695). Medication surveillance based on genomic information resulted to 32 additional alerts for drugs other than the drugs of inclusion in 25 patients during the follow-up period.
Medication use during genotype-guided dosing & guideline adherence
An average follow-up of 15.6 months (ranging from 6 to 25 months) of medication use with electronic medication surveillance using genetic predicted phenotypes from the determined panel was prospectively collected. The mean treatment period for the drug of inclusion was 357 days ± 221 (defined as the period between the projected end date of the last prescription and the start date). In 40.5% of the patients the medication of inclusion was stopped before the end of the follow-up period which was higher but not significantly different between patients with and without a gene-drug interaction combination between an actionable genotype and associated drug (48.4 vs. 37.0%. p = 0.128). Follow-up on the patients with such as gene-drug pair upon inclusion showed that in 55 (89%) of the cases with a relevant combination of an actionable genotype with a relevant drug the healthcare professionals adhered to the DPWG guidelines by switching the patient to another drug, adjusting the dose, guarded a maximum dose or performed additional monitoring as instructed per the DPWG guidelines. In 1 case (1.6%) the healthcare professionals did not adhere to the guidelines which was motivated by the fact that patient had tolerated the treatment in the period between inclusion and the moment where the genetic profile in combination with the therapeutic recommendations was provided to the healthcare professionals. In 5 cases (7.8%) the gene-drug pair between CYP2D6 and venlafaxine was present while treatment continued and 1 case (1.6%) the dose-maximum for escitalopram as recommended by the DPWG was surpassed.
Impact of gene-drug interactions on pharmacotherapy in empirical dosing
One of the goals of this study was to explore the effect of gene-drug interactions on pharmacotherapy during the period where healthcare professionals did not have access to the patients’ pharmacogenomic profile. To study this research question patients were required to have received at least one prescription of one of the ten selected drugs in the year before inclusion (other than the drug of the incident prescription). The majority of patients (63.0%) had received a prescription for simvastatin before inclusion in the study which in 108 patients was stopped for a switch to atorvastatin. 57 patients received a prescription for an antidepressant in the year preceding inclusion, most frequently citalopram (n = 19). In 33 cases the drug was still prescribed during follow-up (for example a patient who was included because of an incident prescription for an antidepressant continued statin treatment for hypercholesterolemia). In the retrospective analyses of the mediation use in the period during empirically dosing the occurrence of gene-drug interactions did not lead to a significant difference in time weighted DDD’s (OR = 0.67, CI 0.35–1.29, p = 0.235), interventions (OR = 1.27, CI 0.93–1.75, p = 0.133) or visits to pharmacies (OR = 1.08, CI 0.64–1.83 p = 0.764).
Discussion
This study demonstrates that implementation of genotype guided dosing in primary care by community pharmacists willing to participate in an implementation project is feasible. The IP3 project allowed healthcare professionals to gain experience with individualizing pharmacotherapy based on genetic variation in the setting of their own practice. During this implementation study healthcare professionals adopted PGx testing in 18.0% of all eligible patients. The results of this study further show that actionable genotypes in the tested panel of eight genes are quite common in our group of primary care patients. The majority of patient’s caries at least one actionable genotype. Moreover, in 31% of the patients a combination of drug with a known gene-drug interaction with an actionable genotype was present at the time of inclusion.
Overall, the IP3 study is the first pharmacist initiated pre-emptive pharmacogenomics projects outside the US to evaluate a PGx panel approach to guide individualized pharmacotherapy using CDS in a primary care setting. Similar initiatives implementing PGx panel testing in a primary care setting in the US have been reported by research groups including Brixner et al., Elliot et al., Finkelstein et al. and Perez et al. [14,15,16,17]. In the study performed by Brixner et al. the effect of panel-based genomic screening with 6 genes on the health resource utilization after starting pharmacotherapy with a selection of 61 drugs was studied. The research team compared a cohort of 205 patients who received pharmacogenomic screening with 820 untested patients. Results showed that the screened cohort had a lower rate of hospitalizations (relative risk (RR) of 0.61, 95% confidence interval (CI) = 0.39–0.95, p = 0.027), a lower rate of visits to the emergency department (RR = 0.29, 95% CI = 0.15–0.55, p = 0.0002) and a higher rate of visits to the outpatient clinic (RR = 1.97, 95% CI = 1.74–2.23, p < 0.0001). With this decrease in hospitalizations and visits to emergency departments the authors concluded that prescribing using genomic information could potentially lead to cost-savings [14]. Elliot et al. showed in a prospective, open-label, randomized clinical trial in a hospital-based home health agency that a pharmacogenomic screening in patients aged 50 years or older results in a lower rate of re-hospitalizations per patient at 60 days (RR = 0.48, 95% CI = 0.27-0.82, p = 0.007) and a lower rate of emergency visits per patient at 60 days (RR = 0.58, 95% CI = 0.34-0.99, p = 0.045). In a pilot study using a nested case-control approach Finkelstein et al. showed that frequently hospitalized older adults have more major drug-gene interactions compared to adults who are rarely hospitalized (p < 0.05). Finally, Perez et al. showed in a double-blind, parallel, multi-center randomized controlled trial in which 316 adult patients diagnosed with major depressive disorder were included that genotyping using a PGx panel that the response rate to anti-depressant treatment in the PGx-guided group was significantly higher compared to conventional therapy (Odd Ratio (OR) = 1.62, 95% CI 1.00-2.61, p = 0.0476). Additionally, the side-effect burden measured by the Frequency, Intensity and Burden of Side Effects Rating Burden sub score was significantly higher in to the control group compared to the PGx-guided group (OR = 2.06, 95% CI 1.09-3.89, p = 0.026). These studies indicate that a panel approach can reduce adverse drug reactions, improve drug efficacy and is probably cost-effective within the US healthcare system. When the current study is compared to the frequencies obtained in the pilot of Elliot et al. similarities in the frequencies of actionable genotypes can be observed for CYP2C9 (37.5 vs. 29.8%) and CYP2D6 (47.0 vs. 42.9%) [15].
Additionally, when the minor allele frequencies of variants found in the current study are compared with variant frequencies from a sequencing project of 250 Dutch parent-offspring families (Genome Of the NetherLands), allele frequencies are similar (see Table 4) [22,23,24]. The same is true for the minor allele frequency of variants reported for the European non-Finnish population (n = 185) in the gnomAD database (see Table 4) [25].
Unlike the other four studies no significant effects of gene-drug interactions on dosages, interventions and visits were found in this study. This can be explained by the fact that this study was not powered to detect effects of gene-drug interactions on the secondary endpoints. An additional explanation for the lack of an effect of genotyping on the secondary endpoints can be found in considerable number of patients with an incident prescription for atorvastatin. According to the DPWG guidelines a patient should be switched to an alternate cholesterol lowering therapy in case the patient is a heterozygous carrier of the NM_006446.4:c.521 T > C (rs4149056) variant of the SLCO1B1 gene and additionally uses co-medication that inhibits the enzyme CYP3A4 [3, 4]. For 35 out of the 62 patients with a gene-drug interaction observed in this study, the drug-gene interaction was a combination between the SLCO1B1 521 TC or CC genotype and the drug atorvastatin. As per the guidelines of the DPWG a check on drug-drug interactions mediated through the enzyme CYP3A4 was executed by the pharmacist in case these 35 patients. However, only in seven cases inhibition of activity of CYP3A4 by another drug was found, whereas in the remaining 28 cases no action was required. This dilution of the overall impact by the SLCO1B1 and atorvastatin gene-drug interaction could have been restricted by applying a cap on the number of individuals included for each drug such as currently applied in the U-PGx protocol [26].
The most important limitation of this study is that a substantial number of eligible patients was not invited to participate in the study by the participating pharmacists. As can be seen in Fig. 2, the initial searches identified 1,864 patients that met the requirements regarding type of drug use, while in the end, only 200 patients have been enrolled in the study. For 469 patients the duration of the prescription was less than 28 days (the majority of excluded patients had a prescription with a duration of 14 days) and these patients were not eligible for inclusion. The protocol also allowed pharmacists to exclude patients based on their professional insight (n = 267; use of a Medirol, received end-of-life care, not able to provide informed consent, no known home address or unknown reason) resulting in 1114 patients eligible for inclusion (and used to calculate the percentage of adoption for the primary endpoint). As our study was performed in a naturalistic patient care setting and patients had to be enrolled within 14 days after the first prescription, many patients were unfortunately missed and never invited to participate. We believe that this was the result of several issues: First, our study setting aimed to resemble routine clinical practice, while for the study informed consent procedure was required. This resulted in a substantial time investment from the participating pharmacists compared to ordering a PGx test within routine care. Secondly, the inclusion criterion requiring historical use of an additional drug of the prespecified list made the inclusion procedure complex, resulting in a low amount of patients enrolled at the time of dispensing the drug of inclusion and requiring weekly queries of the drug dispensing records to identify eligible patients. Thirdly, the healthcare professionals did not receive any financial compensation for the time required for the inclusion of patients and the inclusion of patients had to be performed alongside their regular work in clinical practice. As a result of these factors we believe that the adoption of PGx testing in this study is likely an underestimate of adoption in clinical practice.
The substantial number of patients that were not invited by participating pharmacists could potentially introduce bias to our results. Selection bias on provider level could have occurred as highly motivated pharmacists significantly enrolled more patients compared to other colleagues (p < 0.001 in a pearson-chi2 analysis of the different centers vs. percentage of patients contacted). Primarily, this impacts the adoption and may thus lead to an underestimation of adoption in clinical practice. More importantly, selection bias on a provider level may have led to a selective inclusion of specific subpopulations and as a result may have decreased the external validity of the study population. Additionally, it may have had impact on the secondary endpoints including the frequencies of actionable genotypes, the percentage of patients with a combination of an actionable genotype in combination with a drug with a known gene-drug interaction, the number of drug related interventions, the number of visits to the pharmacy and the drug dose.
To evaluate the potential impact of selection bias on the frequencies of actionable genotypes, minor allele frequencies obtained in this study were compared with the Genome Of the NetherLands consortium and showed similar results. Additionally, the minor allele frequencies from patients who reported the descendance of both parents as Caucasian was comparable with the European non-Finnish population in the GnomAD database. Based on the high concordance of our results with results from GoNL, GnomAd and reports in literature of similar distributions of numbers of actionable phenotypes, we feel that the potential selection bias as result from the high number of patients not invited did not impact the reported predicted phenotypes in this study [5,6,7,8,9,10,11, 14,15,16,17, 22,23,24,25].
We also evaluated the potential impact of bias on provider level on the secondary endpoint “percentage of patients with a combination of an actionable genotype and a drug with a clinical recommendation”. As described in the previous paragraph, the minor allele frequencies in this study are comparable with data from large public databases and so are the derived phenotypes. The second factor which influences this endpoint and might be impacted by a selection bias on provider level is the type of drug on which a patient was enrolled in the study. For example, if a pharmacy close to a psychiatric center includes patients at a higher rate than the other centers participating in the project this may affect the distribution of drugs in the study population and as result may impact this endpoint. However, an additional analysis investigating whether a pharmacist contacted the patient vs. the kind of prescribed drug was not significant (p = 0.337), showing that the selection bias on provider level did not have an impact on the drug of inclusion and therefore no impact on the reported percentage of gene-drug interactions found.
Finally, the remaining secondary endpoints (the number of drug related interventions, the number of visits to the pharmacy and the mean drug dose) were studied using GEE and multilevel log-linear Poisson models for the statistical analyses. These statistical models are designed to perform statistical analyses on hierarchical data. Furthermore, because we believe that the percentage of patients with a combination of a actionable phenotype and associate drug with a known gene-drug interaction is accurately estimated and is used as the dependent variable along with the use of the covariates gender, age, weight, height and the use of an automated dose dispensing as confounding factors in the statistical analysis, we believe that the impact of a bias on provider levels on the effect on the number of interventions, mean dose and number of visits to the pharmacy is minimal.
Consequently, we believe that an impact of a possible selection bias on provider level may have led to an underestimation of the adoption. However, the effect of bias on the external validity of the study results appears to be limited. For a larger study investigating implementation of pharmacist-initiated implementation of panel-based PGx screening in routine clinical care the enrollment process should be simplified so that PGx guided therapy can be initiated at the moment of dispensing the first prescription thereby reducing the number of non-routine actions to a minimum.
Similar to previous studies this project has shown that a significant portion of patients in primary care is open to pharmacogenomic testing [18, 27, 28]. Additionally, this study has shown that individualizing medication based on genetic make-up is possible. However, the body of evidence supporting large scale implementation of a preemptive PGx panel approach in primary care within a European healthcare system is still lacking. Large implementation projects such as the U-PGx program are designed to provide an answer to this question [26].
In conclusion, implementation of PGx in primary care by ambitious pharmacists willing to participate in an implementation project is feasible, and the frequency of actionable gene-drug interactions is high.
References
Swen JJ, Huizinga TW, Gelderblom H, de Vries EG, Assendelft WJ, Kirchheiner J, et al. Translating pharmacogenomics: challenges on the road to the clinic. PLoS Med. 2007;4:e209.
Relling MV, Klein TE. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther. 2011;89:464–7.
Swen JJ, Wilting I, de Goede AL, Grandia L, Mulder H, Touw DJ, et al. Pharmacogenetics: from bench to byte. Clin Pharmacol Ther. 2008;83:781–7.
Swen JJ, Nijenhuis M, de Boer A, Grandia L, Maitland-van der Zee AH, Mulder H, et al. Pharmacogenetics: from bench to byte--an update of guidelines. Clin Pharmacol Ther. 2011;89:662–73.
Hoffman JM, Haidar CE, Wilkinson MR, Crews KR, Baker DK, Kornegay NM, et al. PG4KDS: a model for the clinical implementation of pre-emptive pharmacogenetics. Am J Med Genet Part C Semin Med Genet. 2014;166C:45–55.
van Driest SL, Shi Y, Bowton EA, Schildcrout JS, Peterson JF, Pulley J, et al. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin Pharmacol Ther. 2014;95:423–31.
O’Donnell PH, Bush A, Spitz J, Danahey K, Saner D, Das S, et al. The 1200 patients project: creating a new medical model system for clinical implementation of pharmacogenomics. Clin Pharmacol Ther. 2012;92:446–9.
O’Donnell PH, Danahey K, Jacobs M, Wadhwa NR, Yuen S, Bush A, et al. Adoption of a clinical pharmacogenomics implementation program during outpatient care—initial results of the University of Chicago “1200 Patients Project”. Am J Med Genet Part C Semin Med Genet. 2014;166C:68–75.
Bielinski SJ, Olson JE, Pathak J, Weinshilboum RM, Wang L, Lyke KJ, et al. Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time-using genomic data to individualize treatment protocol. Mayo Clin Proc. 2014;89:25–33.
Johnson JA, Elsey AR, Clare-Salzler MJ, Nessl D, Conlon M, Nelson DR. Institutional profile: University of Florida and Shands Hospital Personalized Medicine Program: clinical implementation of pharmacogenetics. Pharmacogenomics. 2013;14:723–6.
Dunnenberger HM, Crews KR, Hoffman JM, Caudle KE, Broeckel U, Howard SC, et al. Preemptive clinical pharmacogenetics implementation: current programs in five US medical centers. Annu Rev Pharm Toxicol. 2015;55:89–106.
Bank PC, Swen JJ, Guchelaar HJ. A nationwide survey of pharmacists’ perception of pharmacogenetics in the context of a clinical decision support system containing pharmacogenetics dosing recommendations. Pharmacogenomics. 2017;18:215–25.
Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in Prescription Drug Use Among Adults in the United States From 1999-2012. J Am Med Assoc. 2015;314(17):1818–31.
Brixner D, Biltaji E, Bress A, Unni S, Ye X, Mamiya T, et al. The effect of pharmacogenetic profiling with a clinical decision support tool on healthcare resource utilization and estimated costs in the elderly exposed to polypharmacy. J Med Econ. 2016;19:213–28.
Elliott LS, Henderson JC, Neradilek MB, Moyer NA, Ashcraft KC, Thirumaran RK. Clinical impact of pharmacogenetic profiling with a clinical decision support tool in polypharmacy home health patients: A prospective pilot randomized controlled trial. PLoS One. 2017;12:e0170905.
Finkelstein J, Friedman C, Hripcsak G, Cabrera M. Pharmacogenetic polymorphism as an independent risk factor for frequent hospitalizations in older adults with polypharmacy: a pilot study. Pharm Pers Med. 2016;9:107–16.
Perez V, Salavert A, Espadaler J, Tuson M, Saiz-Ruiz J, Saez-Navarro C, et al. Efficacy of prospective pharmacogenetic testing in the treatment of major depressive disorder: results of a randomized, double-blind clinical trial. BMC Psychiatry. 2017;17:250.
Swen JJ, van der Straaten T, Wessels JA, Bouvy ML, Vlassak EE, Assendelft WJ, et al. Feasibility of pharmacy-initiated pharmacogenetic screening for CYP2D6 and CYP2C19. Eur J Clin Pharmacol. 2012;68:363–70.
Dezentje VO, Opdam FL, Gelderblom H, Hartigh den J, Van der Straaten T, Vree R, et al. CYP2D6 genotype- and endoxifen-guided tamoxifen dose escalation increases endoxifen serum concentrations without increasing side effects. Breast Cancer Res Treat. 2015;153:583–90.
Lunenburg CA, van Staveren MC, Gelderblom H, Guchelaar HJ, Swen JJ. Evaluation of clinical implementation of prospective DPYD genotyping in 5-fluorouracil- or capecitabine-treated patients. Pharmacogenomics. 2016;17:721–9.
Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–63.
Boomsma DI, Wijmenga C, Slagboom EP, Swertz MA, Karssen LC, Abdellaoui A, et al. The Genome of the Netherlands: design, and project goals. Eur J Hum Genet: Ejhg. 2014;22:221–7.
Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46:818–25.
Francioli LC, Polak PP, Koren A, Menelaou A, Chun S, Renkens I, et al. Genome-wide patterns and properties of de novo mutations in humans. Nat Genet. 2015;47:822–6.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
van der Wouden CH, Cambon-Thomsen A, Cecchin E, Cheung KC, Lucia Davila-Fajardo C, Deneer VH, et al. Implementing pharmacogenomics in Europe: design and implementation strategy of the ubiquitous pharmacogenomics consortium. Clin Pharmacol Ther. 2017;101:341–58.
Haga SB, O’Daniel JM, Tindall GM, Lipkus IR, Agans R. Survey of US public attitudes toward pharmacogenetic testing. Pharm J. 2012;12:197–204.
O’Daniel J, Lucas J, Deverka P, Ermentrout D, Silvey G, Lobach DF, et al. Factors influencing uptake of pharmacogenetic testing in a diverse patient population. Public Health Genom. 2010;13:48–54.
Acknowledgements
We would like to thank V. Rabiea for her assistance in the collection of the follow-up data. This study makes use of data generated by the Genome of the Netherlands (GoNl) Project and the Genome Aggregation Database (gnomAD). A full list of the investigators of the Genome of the Netherlands Project is available from http://www.nlgenome.nl. Funding for the GoNl project was provided by the Netherlands Organization for Scientific Research under award number 184021007, dated 9 July 2009 and made available as a Rainbow Project of the Biobanking and Biomolecular Research Infrastructure Netherlands (BBMRI-NL). The sequencing of samples included in the GoNl project was carried out in collaboration with the Beijing Institute for Genomics (BGI). Additionally, the authors would like to thank the Genome Aggregation Database (gnomAD) and the groups that provided exome and genome variant data to this resource. A full list of contributing groups can be found at https://gnomad.broadinstitute.org/about. This work was supported by unrestricted grants from The Royal Dutch Pharmacist’s Association, The Hague (KNMP), The Netherlands and Zorg & Zekerheid, Leiden, The Netherlands.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Bank, P.C.D., Swen, J.J., Schaap, R.D. et al. A pilot study of the implementation of pharmacogenomic pharmacist initiated pre-emptive testing in primary care. Eur J Hum Genet 27, 1532–1541 (2019). https://doi.org/10.1038/s41431-019-0454-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0454-x
This article is cited by
-
Pharmacogenetic interventions to improve outcomes in patients with multimorbidity or prescribed polypharmacy: a systematic review
The Pharmacogenomics Journal (2022)
-
Clinical implementation of drug metabolizing gene-based therapeutic interventions worldwide
Human Genetics (2022)
-
Appraisal and development of evidence-based clinical decision support to enable perioperative pharmacogenomic application
The Pharmacogenomics Journal (2021)
-
Estimated nationwide impact of implementing a preemptive pharmacogenetic panel approach to guide drug prescribing in primary care in The Netherlands
BMC Medicine (2019)
