
Abstract
PFAPA is an autoinflammatory syndrome characterized by periodic fever, aphthous stomatitis, sterile pharingitis, and adenitis, with an onset usually before the age of five. While the condition is most commonly sporadic, a few cases are familial and are usually compatible with an autosomal dominant (AD) transmission pattern, with reduced penetrance in some pedigrees. We performed exome analysis in a family where PFAPA was present in three relatives in two generations showing apparent AD segregation, identifying several rare and/or novel heterozygous variants in genes involved in the autoinflammatory pathway. Following segregation analysis of candidate variants, only one, c. 2770T>C p.(S924P) in the ALPK1 gene, was found to be consistently present in affected family members. ALPK1 is broadly expressed in different tissues and its protein is the intracellular kinase activated by the bacterial ADP-heptose bisphosphate that phosphorylates and activates TRAF-Interacting protein with Forkhead-Associated domain (TIFA) and triggers the immediate response to Gram-negative bacterial invasion. Sequencing analysis of 13 additional sporadic cases and 10 familial PFAPA cases identified two additional heterozygous missense variants c.1024G>C p.(D342H) and c.710C>T p.(T237M) in two sporadic patients, suggesting that rare variants in ALPK1 may represent a predisposing factor for recurrent periodic fever in a pediatric population.
Similar content being viewed by others

Introduction
PFAPA syndrome was described for the first time in 1987 in 12 children showing the association of recurrent periodic fever along with aphthous stomatitis, pharyngitis and adenitis, in the absence of evidence for infection [1]. The disease usually starts before the age of 5 years and it tends to disappear before reaching adulthood, although occasionally onset in adulthood has been described [2]. Recurrent fever is the main symptom. It tends to recur weekly or monthly, lasts up to 5 days and can reach up to 39.5 °C [3]. It usually responds to low doses of corticosteroids, although this treatment does not prevent its recurrence [4]. The condition does not have an impact on the development of the children with PFAPA. This syndrome has an incidence of 2.3 cases per 10000 children per year [4]. The main burden for patients and their families is represented by the difficulty of excluding other potentially treatable causes of fever, such as infections, autoimmune diseases or cancer. PFAPA should be considered as an autoinflammatory disease with a complex pathogenic architecture, and predisposing genetic factors along with environmental triggers could contribute to onset of the disease [5]. For this reason, PFAPA, unlike other mendelian autoinflammatory syndromes, has been considered a sporadic, non-hereditary condition [6]. Recently a series of familial cases have been published highlighting the possibility that a mendelian transmission may underlie also some sporadic occurrences [7,8,9]. Several confounding factors, including penetrance defect (up to 50%) in some families [10], non-specific symptoms that can overlap with other common pediatric illnesses, and the disappearance of manifestations before adulthood, which can easily induce to overlook familial recurrence, complicate the assessment of genetic mechanisms. Recurrent fever represents a symptom common to other autoinflammatory conditions with known genetic bases and therefore it may be difficult to distinguish PFAPA from the other periodic fever syndromes especially for Familial Mediterranean Fever in endemic countries. For this reason, PFAPA patients are routinely tested for variants in genes already implicated in mendelian periodic fever syndromes [11, 12]. However, so far no clear-cut pathogenic variants (PVs) in known genes have been found in PFAPA cases either sporadic or with family history. A de novo translocation interrupting the SPAG7 gene was found in a sporadic patient, but no additional PVs have been found in other patients [13]. A common polymorphism in CARD8 has been shown to be associated with the disease in a single study [14], but this result needs validation in other cohorts of patients. An exome study on ten familial patients failed to identify common PVs among them [10], while a panel of genes involved in auto inflammation in a large patient series failed to identify indisputably pathogenic variants [14].
For this reason, we decided to perform an exome analysis in a family where PFAPA segregates with an autosomal dominant pattern. After filtering, all novel/rare heterozygous variants were prioritized based on their involvement in the autoinflammatory pathway. All variants, but one in the ALPK1 gene, were excluded by segregation analysis. Sanger sequencing of the coding region of ALPK1 in 23 additional suspected PFAPA patients identified two additional variants.
This study suggests that rare heterozygous variants in the ALPK1 gene represent a predisposing factor that along with environmental triggers can cause recurrent periodic fever in a pediatric population.
Materials and methods
Patients
All patients were referred to the Medical Genetics Clinics of Fondazione Policlinico A. Gemelli at the Università Cattolica del Sacro Cuore with a suspicion of periodic recurrent fever and/or autoinflammatory systemic disorder. A total number of 200 patients were recruited from 2014 until present. Bioethical approval for this study and informed consent was obtained to extend the molecular analysis to other genes for research purposes, if the diagnostic test failed to show pathogenic variants.
We selected from this cohort of 200 patients, cases with a clinical suspicion of PFAPA. Our clinical criteria for PFAPA were not very stringent, as we included in this study all patients with a pediatric onset of recurrent fever, and decided that, at this stage, all the other symptoms associated with PFAPA (such as aphtae and adenitis) should not be considered strong enough criteria to make a clinical selection. Using these relaxed clinical criteria, we selected 23 patients. Clinical synopsis of our patients is described in Supplementary Table 1. For each patient a blood sample was obtained, and the DNA was extracted (using standard procedures), coded and stored at 4 °C until the analysis was performed. All PFAPA patients were tested for 10 genes, described below, previously found involved in periodic recurrent fever. None of them carried pathogenic variants or variants of uncertain significance in any of these genes. Ten of our PFAPA patients were familial cases with one or more first-degree affected relatives showing a possible apparent dominant inheritance, the remaining had a negative family history or there were no information available.
Next generation sequencing (NGS) analysis of genes involved in autoinflammatory diseases
Ten genes were selected based on their previous association with periodic recurrent fever, IL1RN(OMIM 147679), LPIN2(OMIM605519), MEFV(OMIM608107), MVK(OMIM251170), NLRP3(OMIM606416), NLRP12(OMIM609648), NOD2(OMIM605956), PSMB8(OMIM177046), TNFRSF1A(OMIM191190) and PSTPIP1(OMIM606347). Some of those genes are involved in syndromic forms of periodic fever, most with an autosomal dominant inheritance. Libraries, emulsion PCR, sample enrichment and sequencing were performed on an Ion Torrent platform, using Life Technologies reagents (Thermo Fisher Scientific), following manufacturer recommendations. For each sample an average coverage of 600X for 98% of the exons was obtained. Sanger sequencing filled up regions with no or poor coverage. Reads alignment, variant calling and annotation were done using the Ion Reporter software. For most variants a confirmation by Sanger sequencing was not deemed necessary, however some variants were confirmed when the segregation analysis was performed in other family members. Sanger sequencing was done also on a few variants where the low coverage did not allow to immediately confirm their presence.
Exome sequencing and analysis
Whole exome sequencing analysis was done on service at Galseq SRL (Bresso, Milan) requesting an average coverage of 60X on an Illumina platform. For each patient a pair of fastq files was delivered. These were subsequently mapped and filtered using the online platform Galaxy [15]. We customized the following pipeline: we mapped paired reads using Burrows Wheeler Aligner for Illumina [16] then removed duplicates and performed variant calling using FreeBayes. The ensuing vcf file was then annotated using wAnnovar [17]. All bioinformatic analyses were done following best practices recommendations [18, 19]. To evaluate the frequency of each variant we searched the gnomAD database, while the in-silico analysis of each missense variants was done using the website Varsome.com.
The variant annotated file was then filtered using functions on Microsoft Excel. All the variants were then prioritized using Pubmed Fetcher and Genie online software, interrogating Pubmed with the list of genes coming out from our analysis and the following keywords (“innate immunity”, “autoinflammatory disease”, “inflammasome”, “periodic fever”, “recurrent fever”). From each search, a list of genes was generated. All the genes and their variants were manually inspected loading all the reads as a custom track on the UCSC genome browser to confirm their presence and their frequency on gnomAD database.
Sanger sequencing of ALPK1
Intronic primers flanking coding exons were designed using Primer3 application on the UCSC genome browser. Primer sequences will be provided upon request. Each amplicon was PCR amplified using the following standard cycles: 95 °C 5 min, 95 °C 30 s, 60 °C 30 s, 72 °C 15 s, for 34 cycles, final extension at 72 °C for 15 min. 2.5 microliters of each amplicon were purified with 0.5 µl of a 1:1 mixture of Exomicnuclease III and Shrimp Alkaline Phosphatase at 37 °C for 15 min followed by heat inactivation at 80 °C. Cleaned up PCR product was sequenced using BigDye terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) in a final volume of 10 µl and run on a 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA). The electropherograms were analysed by the Sequencing Analysis v5.2 software (Applied Biosystems, Foster City, CA). Variants c.2770T>C, c.1024G>C, c.710C>T were submitted to LOVD database (http://www.lovd.nl/ALPK1 patient ID #00218417, #00218656, #00218657). The accession number for the ALPK1 sequence was NM_025144.3. Exons were counted on the NM_ 025144.3 transcript using the UCSC genome Browser.
Results
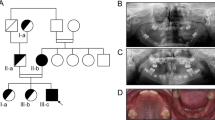
Patient #205 had a clinical diagnosis of PFAPA. In his family recurrent fever was present in his father and paternal aunt and was absent in his brother and sister (Fig. 1a). Panel testing of 10 genes involved in recurrent fever syndromes did not reveal any PV (see Supplemental Material and Methods). We performed exome analysis and included in the subsequent analyses all the variants identified within the coding/splicing regions, with a good coverage, heterozygous, absent in duplicated regions and with a minor allele frequency < 0.001 in the gnomAD population. About 1000 variants were left to be scrutinized. To prioritize them we submitted each gene to several software programs to interrogate Pubmed about proteins that have already been involved in the autoinflammatory pathways, using as keywords “autoinflammatory”, “innate immune system”, “inflammasome”, “periodic fever”, and “recurrent fever”. From each search, a list of candidate genes was generated, usually between 50 and 100. Each variant was manually inspected to confirm that it had a good coverage and was not a common polymorphism in gnomAD. After this search, a list of nine variants was generated (Table 1). These were validated by Sanger sequencing and their segregation analyzed in the remaining individuals of the family. The only variant showing full co-segregation with the phenotype according to an autosomal dominant model, fully penetrant, was the variant c.2770T>C p.(S924P) in the ALPK1 gene (Fig. 1a). The frequency of this variant in gnomAD is 2/246066 (all alleles were in the non-Finnish European population for a frequency of 2/111532): considering the rarity of this variant compared to the estimated prevalence of PFAPA in the general population, we considered this gene a good candidate to be tested in other PFAPA patients. Therefore, we investigated the coding sequence of ALPK1 in a cohort of 23 additional PFAPA patients to identify additional potentially pathogenic variants. All these patients have already been tested with negative results for variants in genes known to be involved in other recurrent fever syndromes (see Material and Methods). We found in a sporadic case (#167) one additional variant, c.1024G>C p.(D342H). This variant has a frequency of 18/276930, with a frequency of 1/126488 in the non-Finnish European population in the gnomAD dataset (Fig. 1b). We re-contacted the family to investigate the parental origin, but the parents were not available for further testing or clinical evaluation for PFAPA symptoms. A third variant, c.710C>T p.(T237M), was found in a sporadic case (#197) and it is absent in gnomAD (Fig. 1c). When we re-contacted this patient to investigate family history, we discovered that he had a more complex phenotype with anhidrosis, blood pancytopenia, splenomegaly, optic neuritis and panuveitis, in addition to PFAPA symptoms. A detailed clinical description is provided in Supplementary Table 2. His parents were also first-degree cousins. Searching the web, looking for additional data on ALPK1, we came across a poster [20] describing a Utahn family, where an almost identical phenotype was present in a mother and her two daughters, who were half-sisters [21]. All three individuals were heterozygotes of the ALPK1 c.710C>T p.(T237M) variant, that had occurred de novo in the affected mother. Segregation analysis in our family showed that the p.(T237M) variant was also de novo in our proband. In the light of parental consanguinity, we decided to rule out the potential contribution of other recessive homozygous alleles to the phenotype. To this aim, we performed exome analysis on patient #197. For variant priorization we used a scheme similar to that described above, but no clear rare pathogenic homozygous alleles that could explain some of the manifestations (see Supplementary Table 2) were present in our patient and not shared by the Utahan family were.

Pedigrees and electropherograms showing ALPK1 variants. a Pedigree of individual #205 (arrowhead), showing an autosomal dominant transmission. Variant C>T predicts a p.(S924P) missense substitution, segregating with the phenotype. b Variant p.(D342H) in patient #167. c Variant p.(T237M) is present only in the proband while is absent in his first-degree cousins parents
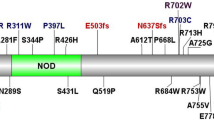
The potential pathogenic role of the three variants in ALPK1 was evaluated using common in silico analyses (Table 2). All different software were concordant in defining a strong predictive damaging effect for c. 710C>T p.(T237M), while for the other two variants the predictions were conflicting. However, the two mutated residues are highly conserved, based on the GERP and DANN scores.
Discussion
In this study we identified through exome sequencing a variant in the ALPK1 gene in a patient with a clinical diagnosis of PFAPA. Segregation analysis showed that the variant was present in all affected individuals, while it was absent in unaffected relatives. Two additional missense ALPK1 variants were found in 2 out of 23 patients with a clinical diagnosis of PFAPA in whom ALPK1 was tested by Sanger sequencing. Given the extreme low frequency of these variants, carriers of those variants in the gnomAD population could represent individuals with PFAPA, especially because in one of our cases periodic fever disappeared during adolescence. The other possible explanation is a potential penetrance defect of those variants, or the complex pathogenic architecture of autoinflammatory diseases requiring the presence of environmental triggers to cause the disease, or the disease could have gone into remission in adulthood and therefore could have been “forgotten”. All these three missense variants were in different regions of the protein; according to common software analysis, all three variants were present in highly conserved genomic positions with a potential effect on the function of the protein.
Compared to the other two ALPK1 variants identified in this study, p.(T237M) shows peculiar characteristics. The patient who carried p.(T237M) had additional clinical manifestations that are usually not associated with PFAPA. p.(T237M) has been reported in an Utahan family with a complex phenotype strikingly similar to that displayed by our proband. The clinical manifestations in the Utahan family [21] and the comparison with our patient are described in Supplementary Table 1. The main difference between the American patients and our patient is the lack of the recurrent fever, or other signs of PFAPA in the former, despite extensive search for manifestations of autoimmune reactivity, since anhidrosis, uveitis, optic nerve neuritis, pancytopenia and splenomegaly were suspected to be the consequence of an abnormal activation of the immune system. Our patient also had keratoconus, enamel defects and hyper-intense lesions in the brain white matter suggesting a mild form of leukoencephalopathy. It is interesting to note that although patient #197 in this study does not have a classical PFAPA phenotype, most of his symptoms are part of the autoinflammatory spectrum, including the dyserythropoietic anemia that is described in Majeed syndrome [22].
Moreover, we excluded, by exome sequencing, the presence of other recessive alleles that could explain the recurrent fever, keratoconus and leukoencephalopathy. There is a possibility that our patient and the Utahan patient show just an accidental resemblance, but we found this hypothesis extremely unlikely given the unique series of symptoms present in both cases. All those symptoms are per se not common and their combination has never been described before. Different approaches for the identification of the ALPK1 variants were used in this case and in the Utahan pedigree. In this study, ALPK1 has been tested as a candidate gene in a cohort of PFAPA patients, while in the Utahan family exome testing was performed. Our finding may provide independent evidence for a causative role of ALPK1, and specifically of variant p.(T237M), in patients with this phenotypic condition.
We are aware that variants with a low frequency do not imply immediately pathogenicity, but the approach that led to the identification of this gene and the three variants in our cohort of PFAPA patients is noteworthy. We did not use a biased candidate gene approach, but we started from variants derived from the whole exome and prioritized them using candidate keywords. In the short list presented (Table 1), there were apparently more suitable candidates, i.e. TLR4 or NLRP9, but the subsequent segregation analysis, that could provide unbiased evidence for candidate genes, excluded them all, but ALPK1. Then, the two additional variants found in a limited cohort of PFAPA patients made this finding stronger.
The role of the ALPK1 protein just recently emerged in the literature [23,24,25,26,27]. It represents the intracytoplasmic kinase activated upon contact with the ADP-heptose biphosphate present in Gram-negative bacteria. This event activates ALPK1 kinase that phosphorylates TIFA protein leading to its oligomerization and to the activation of the NLRP3 inflammasome, thus turning on several cytokines involved in innate immunity [28]. We hypothesize that all three variants could have an activating effect on the kinase; this could happen in the absence of bacterial infections or it could be triggered by a bacterial infection that causes uncontrolled activation followed by fever and other sterile inflammatory manifestations of PFAPA. The variant p.(T237M) could represent an extreme phenotype of the same spectrum, where NLRP3 inflammasome activation mediated by an overactive ALPK1 kinase could trigger all the phenotypic aspects described in carriers of this variant. ALPK1 has never been implicated before in the periodic fever syndromes, but its role in the innate immunity is well established and previous genomic studies found an association, in some populations, between specific ALPK1 variants with gout [23, 29, 30], that is considered a polygenic autoinflammatory disease. Future analyses on additional patients, even with an extended autoinflammatory phenotypic spectrum and functional tests on mutated ALPK1 proteins, will help to identify a precise role for this gene in the recurrent fever phenotype.
Web resources
Galaxy server https://usegalaxy.org/
Galaxy Biomina Server https://galaxyproject.org/public-galaxy-servers/biomina/
Pubmed Fetcher http://biominavm-galaxy.biomina.be/
Genie http://cbdm-01.zdv.uni-mainz.de/~jfontain/cms/
wAnnovar http://wannovar.wglab.org/
gnomAD http://gnomad.broadinstitute.org/
Varsome https://varsome.com/
Genome browser https://genome.ucsc.edu
Accession numbers
The accession number for the ALPK1 sequence reported in this paper is GENBANK: NM_025144.3.
References
Marshall GS, Edwards KM, Butler J, Lawton AR. Syndrome of periodic fever, pharyngitis, and aphthous stomatitis. J Pedia. 1987;110:43–6.
Cantarini L, Vitale A, Bartolomei B, Galeazzi M, Rigante D. Diagnosis of PFAPA syndrome applied to a cohort of 17 adults with unexplained recurrent fevers. Clin Exp Rheuma. 2012;30:269–71.
Thomas KT, Feder HM, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pedia. 1999;135:15–21.
ter Haar N, Lachmann H, Özen S, Woo P, Uziel Y, Modesto C, et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis. 2013;72:678–85.
Manthiram K, Lapidus S, Edwards K. Unraveling the pathogenesis of periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis through genetic, immunologic, and microbiologic discoveries. Curr Opin Rheuma. 2017;29:493–9.
Gattorno M, Caorsi R, Meini A, Cattalini M, Federici S, Zulian F, et al. Differentiating PFAPA syndrome from monogenic periodic fevers. Pediatrics. 2009;124:e721–e728.
Cochard M, Clet J, Le L, Pillet P, Onrubia X, Gueron T, et al. PFAPA syndrome is not a sporadic disease. Rheumatology. 2010;49:1984–7.
Akelma AZ, Cizmeci MN, Kanburoglu MK, Mete E, Bozkaya D, Tufan N, et al. Is PFAPA syndrome really a sporadic disorder or is it genetic? Med Hypotheses. 2013;81:279–81.
Valenzuela PM, Majerson D, Tapia JL, Talesnik E. Syndrome of periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) in siblings. Clin Rheuma. 2009;28:1235–7.
Di GioiaSA, Bedoni N, von Scheven-Gête A, Vanoni F, Superti-Furga A, Hofer M, et al. Analysis of the genetic basis of periodic fever with aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome. Sci Rep. 2015;5:10200.
Adrovic A, Sahin S, Barut K, Kasapcopur O. Familial Mediterranean fever and periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome: shared features and main differences. Rheuma Int. 2019;39:29–36.
Pehlivan E, Adrovic A, Sahin S, Barut K, Kul Cınar O, Kasapcopur O. PFAPA syndrome in a population with endemic familial mediterranean fever. J Pedia. 2018;192:253–5.
Bens S, Zichner T, Stütz AM, Caliebe A, Wagener R, Hoff K, et al. SPAG7 is a candidate gene for the periodic fever, aphthous stomatitis, pharyngitis and adenopathy (PFAPA) syndrome. Genes Immun. 2014;15:190–4.
Cheung MS, Theodoropoulou K, Lugrin J, Martinon F, Busso N, Hofer M. Periodic fever with aphthous stomatitis, pharyngitis, and cervical adenitis syndrome is associated with a CARD8 variant unable to bind the NLRP3 inflammasome. J Immunol. 2017;198:2063–9.
Goecks J, Nekrutenko A, Taylor J, Galaxy Team T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–66.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Current protocols in bioinformatics. John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013, p 11.10.1–11.10.33.
Williams LB. Poster. Whole exome sequencing identifies a mutation in alpk1 responsible for a novel autosomal dominant disorder of vision loss splenomegaly and pancytopenia. 2013. https://webvision.med.utah.edu/2013/05/whole-exome-sequencing-wes-identifies-a-mutation-in-alpk1-responsible-for-a-novel-autosomal-dominant-disorder-of-vision-loss-splenomegaly-and-pancytopenia/. Accessed 30 April 2019.
Tantravahi SK, Williams LB, Digre KB, Creel DJ, Smock KJ, DeAngelis MM, et al. An inherited disorder with splenomegaly, cytopenias, and vision loss. Am J Med Genet Part A. 2012;158A:475–81.
Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet. 2005;42:551–7.
Lee C-P, Chiang S-L, Ko AM-S, Liu Y-F, Ma C, Lu C-Y, et al. ALPK1 phosphorylates myosin IIA modulating TNF-α trafficking in gout flares. Sci Rep. 2016;6:25740.
Milivojevic M, Dangeard A-S, Kasper CA, Tschon T, Emmenlauer M, Pique C, et al. ALPK1 controls TIFA/TRAF6-dependent innate immunity against heptose-1,7-bisphosphate of gram-negative bacteria. PLoS Pathog. 2017;13:e1006224.
Zimmermann S, Pfannkuch L, Al-Zeer MA, Bartfeld S, Koch M, Liu J, et al. ALPK1 and TIFA-dependent innate immune response triggered by the Helicobacter pylori type IV secretion system. Cell Rep. 2017;20:2384–95.
Ryzhakov G, West NR, Franchini F, Clare S, Ilott NE, Sansom SN, et al. Alpha kinase 1 controls intestinal inflammation by suppressing the IL-12/Th1 axis. Nat Commun. 2018;9:3797.
Zhou P, She Y, Dong N, Li P, He H, Borio A, et al. Alpha-kinase 1 is a cytosolic innate immune receptor for bacterial ADP-heptose. Nature. 2018;561:122–6.
Lin T-Y, Wei T-YW, Li S, Wang S-C, He M, Martin M, et al. TIFA as a crucial mediator for NLRP3 inflammasome. Proc Natl Acad Sci USA. 2016;113:15078–83.
Kuo T-M, Huang C-M, Tu H-P, Min-Shan Ko A, Wang S-J, Lee C-P, et al. URAT1 inhibition by ALPK1 is associated with uric acid homeostasis. Rheumatology. 2017;56:654–9.
Tu H-P, Min-Shan KoA, Lee S-S, Lee C-P, Kuo T-M, Huang C-M, et al. Variants of ALPK1 with ABCG2, SLC2A9, and SLC22A12 increased the positive predictive value for gout. J Hum Genet. 2018;63:63–70.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sangiorgi, E., Azzarà, A., Molinario, C. et al. Rare missense variants in the ALPK1 gene may predispose to periodic fever, aphthous stomatitis, pharyngitis and adenitis (PFAPA) syndrome. Eur J Hum Genet 27, 1361–1368 (2019). https://doi.org/10.1038/s41431-019-0421-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0421-6
