
Abstract
We aimed to identify novel deletions and variants of TP63 associated with orofacial clefting (OFC). Copy number variants were assessed in three OFC families using microarray analysis. Subsequently, we analyzed TP63 in a cohort of 1072 individuals affected with OFC and 706 population-based controls using molecular inversion probes (MIPs). We identified partial deletions of TP63 in individuals from three families affected with OFC. In the OFC cohort, we identified several TP63 variants predicting to cause loss-of-function alleles, including a frameshift variant c.569_576del (p.(Ala190Aspfs*5)) and a nonsense variant c.997C>T (p.(Gln333*)) that introduces a premature stop codon in the DNA-binding domain. In addition, we identified the first missense variants in the oligomerization domain c.1213G>A (p.(Val405Met)), which occurred in individuals with OFC. This variant was shown to abrogate oligomerization of mutant p63 protein into oligomeric complexes, and therefore likely represents a loss-of-function allele rather than a dominant-negative. All of these variants were inherited from an unaffected parent, suggesting reduced penetrance of such loss-of-function alleles. Our data indicate that loss-of-function alleles in TP63 can also give rise to OFC as the main phenotype. We have uncovered the dosage-dependent functions of p63, which were previously rejected.
Similar content being viewed by others

Introduction
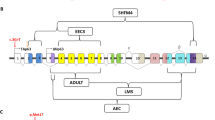
Transcription factor p63 encoded by TP63 is a key regulator in epithelial commitment and development [1, 2]. TP63 encodes a large number of p63 isoforms. The two different transcription start sites give rise to transactivation (TA) isoforms, which contain a TA domain and to ΔN isoforms, which lack the canonical TA domain, but contain an N-terminal TA-competent domain, called TAΔN. Alternative splicing at the 3ʹ end of the gene adds to the complex variety of p63 isoforms by generating at least five different C-terminal ends (α, β, γ, δ, and ε) [3,4,5]. All isoforms contain a DNA-binding domain (DBD) and an oligomerization domain (OD), but differ in the TA domain, sterile-alpha-motif domain (SAM), auto-regulatory TA inhibiting domain (TI domain), and a second TA domain, TA2 (Fig. 1) [3, 4, 6].

Schematic representation of the deletions and point variants in TP63 identified in affected individuals and controls. The upper part of the figure depicts the complete deletions and the genes covered by deletions in the three families, W11–4934, W12–0831, and W16–065, respectively. The middle part of the figure presents a zoom-in image of the TP63 gene depicting the deletions in the families at the exon level and the splicing routes for various isoforms. The lower part of the figure shows the protein structure highlighting the various domains between different isoforms, with the variants in affected individuals and controls indicated
p63 is engaged in dimeric and tetrameric complexes regulating a network of genes important for development of ectodermal structures. Variants in TP63 lead to three major phenotypes: ectodermal dysplasia, orofacial clefting (OFC) and split-hand/split-foot malformations (SHFMs) [7]. Heterozygous TP63 variants have been associated with five different syndromes with overlapping phenotypic features. These syndromes are: ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome 3 (EEC3 [MIM: 604292] [8]), ankyloblepharon–ectodermal defects–cleft lip/palate syndrome (AEC [MIM: 106260]), Rapp–Hodgkin syndrome (RHS [MIM: 129400]), acro-dermato-ungual-lacrimal-tooth syndrome (ADULT [MIM: 103285]), and limb mammary syndrome (LMS [MIM: 603543]). Rare TP63 variants are causative for nonsyndromic OFC (OFC8 [MIM: 129400]) [9], and for nonsyndromic SHFM4 (MIM: 605289) [10].
So far, a broad spectrum of different heterozygous variants have been reported in TP63, leading to the developmental disorders stated above. The vast majority of variants give rise to amino-acid substitutions. A striking genotype–phenotype correlation for TP63 missense variants is apparent in EEC and AEC syndromes [10,11,12]. Variants causing EEC syndrome are almost exclusively found in the DBD, whereas the variants causing AEC syndrome are either found in the SAM and TI domains or in the TAΔN domain of the ΔN isoforms. This distinctive pattern of variants highlights specific pathogenic mechanisms for each syndrome. The EEC variants, which are heterozygous and found in DBD abolish the DNA binding fully or partially in all p63 isoforms, and affect the TA capacity of the protein in a dominant-negative fashion [7]. This is in agreement with the fact that p63 acts as a transcriptional regulator in homomeric complexes. AEC variants in the SAM domain also affect the TA activity in a dominant-negative way [11]. ADULT syndrome variants uniquely affect amino-acid Arginine 337 (298 according to former nomenclature). Arginine 337 also maps to the DBD like in EEC syndrome variants, but exerts gain-of-function effects by conferring novel TA properties to the ΔNp63γ isoform [13].
Rare variants have been reported that lead to truncations at the C-terminal end of the α isoform [12] and at the N-terminal end of the TA isoform in individuals affected with AEC [14]. All of these variants are shown or predicted to give rise to mutant proteins carrying N-terminal or C-terminal truncations but fully maintain their DBDs and ODs. Thus, these variants do not lead to haploinsufficient alleles. Few individuals with a constitutive terminal deletion of the long arm of chromosome 3 encompassing TP63 along with other genes have been reported, who did not have the typical p63-related phenotype [15]. Also, mice carrying heterozygous deletion of Trp63 present no p63-related phenotype, whereas homozygous Trp63 knockout mice present features similar to EEC, AEC, and LMS syndrome [1, 3]. Taken together, the spectrum of variants affecting TP63 has led to the rejection of a haploinsufficiency model [7], and supported the notion that TP63 variants exhibit dominant-negative and gain-of-function effects [7, 9,10,11,12].
In the current study, we identified deletions of TP63 in individuals from three families affected with OFC and/or hypodontia and occasional other ectodermal features. We also identified two variants that are predicted to lead to a premature stop codon in individuals affected with nonsyndromic orofacial clefting (nsOFC). Finally, we identified the first missense variants in the OD, one of which was shown to abrogate oligomerization of mutant p63 protein into oligomeric complexes. Together, our data provide evidence for loss-of-function variants in TP63, giving rise to a phenotype dominated by OFC.
Results
Using single nucleotide polymorphism (SNP) microarray analysis at our diagnostics services, deletions encompassing parts of the TP63 gene were identified in three families (Fig. 1, Table S1, Figure S1A). Family W11–4934 contained a deletion of ~528 kbp at 3q28, affecting TP63, CLDN1, and CLDN16. The proximal endpoint of the deletion in TP63 was identified between exon 13 and 14 using quantitative PCR (qPCR) (Figure S1B). The segregation of the deletion was confirmed in other affected individuals in the family. Family W12–0831 contained a deletion of ~810 kbp at 3q28, affecting TP63, CLDN1, CLDN16, and TMEM207. The deletion was confirmed to segregate in another affected member of the family. Family W16–065 contained a deletion of ~880 kbp at 3q28, affecting TP63 and TPRG1. This deletion was confirmed to be de novo.
TP63 variants identified in individuals affected with OFC
In order to find novel rare variants in TP63 responsible for nonsyndromic OFC, we performed targeted multiplex sequencing using molecular inversion probes (MIPs), a reliable and economical approach for deep sequencing of individual genes in large patient cohorts [16]. All of the samples (1072 OFC and 706 population-matched controls) had an average coverage > 1000-fold over all TP63 targeting MIPs. The average coverage for the samples from affected individuals (1010-fold) and controls (1023-fold) was comparable. In total, seven variants were identified in the individuals affected with OFC and four in the controls. All of the latter variants were missense substitutions, of which three were present in the DBD and one in the TA domain. Each of the seven variants identified in OFC individuals were present in a single person and were not found in controls (Table 2). These variants were located in different domains of p63, i.e., the DBD, OD, and TA2 domains. Among these variants, we found two variants, which represent loss-of-function alleles, including one frameshift and one stop variant in DBD. Moreover, the missense variants affecting the OD might impair the capacity of p63 oligomerization (see below).
The frameshift and stop variants in DBD
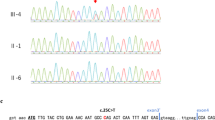
Among the confirmed TP63 variants are two variants that predict the generation of premature stop codons. A deletion of eight nucleotides (NM_003722.4:c.567_574delCCACCTGG) (p.(Ala190Aspfs*5)) was identified in an individual with a unilateral cleft lip and alveolus on the left, and in his/her unaffected parents. This deletion was paternally inherited and creates a frameshift starting at codon Ala190 and a new reading frame that ends in a stop codon five positions downstream. Different from previously reported premature stop variants, the transcript carrying the c.567_574del variant is likely to be targeted for nonsense-mediated decay (NMD) (Table 2). The other premature stop variant is a c.997C>T nonsense variant predicting a premature stop codon p.(Gln333*) in the DBD. This variant was identified in an individual with bilateral cleft lip and cleft palate (CLP). Due to unavailability of parental DNA, it could not be confirmed whether the variant was inherited or occurred de novo. This premature stop codon is also predicted to lead to NMD (Table 2).
Missense variants in the OD of p63
The two variants identified in the OD of the p63 protein are c.1213G>A (p.(Val405Met)) and c.1223G>A (p.(Arg408His)). Variant c.1213G>A (p.(Val405Met)) was identified in an individual with a cleft lip and alveolus on the left side. The variant inherited from an unaffected parent occurs at a moderately conserved nucleotide (phyloP: 3.76) and affects an amino-acid conserved up to frog. A small physicochemical difference exists between the reference amino-acid valine and the variant amino-acid methionine (Table 2). The other variant in the OD, c.1223G>A (p.(Arg408His)), was identified in an individual with a cleft lip and alveolus (left), and a split velum and uvula (hard palate intact), with unaffected parents. This known rare variant, rs751698974 (minor allele frequency (MAF) = 0.000008; ExAC database), was inherited from an unaffected parent. It occurs at a highly conserved nucleotide (phyloP: 4.40) and affects an amino-acid conserved up to zebrafish. The physicochemical difference between the reference amino-acid arginine and the variant amino-acid histidine is small (Table 2).
Other missense variants in affected individuals
Besides the above-mentioned variants, three other missense variants were identified in affected individuals. The c.670G>A (p.(Val224Ile)) in DBD was identified in an individual with bilateral CLP with unaffected parents. This is a known rare variant, rs757669482 (MAF = 0.000008; ExAC database). This paternally inherited variant occurs at a highly conserved nucleotide (phyloP: 4.32) and affects an amino-acid conserved up to zebrafish. The physicochemical difference between the reference amino-acid valine and the variant amino-acid isoleucine is small (Table 2). The variant c.1459C > T (p.(Arg487Cys)) in the TA2 domain of p63 was identified in a CLP individual with unaffected parents. This is a known rare variant, rs777306829 (MAF = 0.0001; ExAC database). This maternally inherited variant occurs at a moderately conserved nucleotide (phyloP: 2.87) and affects an amino-acid conserved up to zebrafish. The physicochemical difference between the reference amino-acid arginine and the variant amino-acid cysteine is large (Table 2). Finally, one γ form-specific variant, c.1390C>T (p.(Arg464Trp)), was identified in an individual with cleft lip and alveolus of the left side, with unaffected parents. The variant was maternally inherited and present in exon 10ʹ. This is a known rare variant, rs369826042 (MAF = 0.0001), with ExAC frequency 0.00004. It occurs at a weakly conserved nucleotide (phyloP: 1.66) and affects a weakly conserved amino acid. There is a moderate physicochemical difference between the reference amino-acid arginine and the variant amino-acid tryptophan (Table 2).
Isomerization domain variant c.1213G>A (p.(Val405Met)) affects p63 oligomerization
A thermal shift assay (TSA) was performed to determine whether the stability of the mutant tetramerization domains was affected compared with the wild-type protein (Fig. 3). The c.1213G>A (p.(Val405Met)) variant indeed leads to a significant reduction of the unfolding equilibrium temperature by ~24 °C, whereas the c.1223G>A (p.(Arg408His)) variant causes only a slight reduction in melting temperature. Investigation of the oligomeric state of the mutant domains by size exclusion chromatography revealed that the predicted p.(Val405Met) variant tetramerization domain is less stable than the wild-type domain showing a significant dimer population. In contrast, no reduction in the population of the tetrameric state could be detected for the p.(Arg408His) substitution.
Isomerization domain variant c.1213G>A (p.(Val405Met)) decreases the transactivation capacity of p63
A transactivation assay was performed to analyze the effects of the variants identified in this study. The fold induction of a luciferase reporter gene under control of a minimal promoter and p63 binding site was compared between the empty pcDNA vector and the p63WT vector [17]. One of the identified variants, c.1390C>T (NM_001114979.1) (p.(Arg464Trp)), was specific to p63γ isoforms. In this case, the variant was created in the ΔNp63γ isoform, which contains a smaller but TA-competent domain, called TAΔN. The measurements of all constructs was normalized to the respective p63WT construct. A clear luciferase induction was measured for all p63WT constructs compared with the empty pcDNA vector (Fig. 4). This means that the WT p63 protein binds to the TP63 enhancer element and induces the expression of downstream luciferase. For the positive control, a missense variant p.(Arg343Trp) in p63 known to cause EEC syndrome was used, which has been previously shown to reduce TA activity in a dominant-negative pattern (Fig. 4) [7].
The EEC-associated variant p.(Arg343Trp) reduced the induction of the luciferase reporter by 82% (P-value = 0.011) (Fig. 4). The previously published variant p.(Arg352Gly) identified in an individual affected with nsOFC was also tested [9]. This variant, which resides in the DBD, reduced TA activity by 63% (P-value = 0.025) (Fig. 4). The two variants in OD, p.(Val405Met) and p.(Arg408His), reduced the activation of luciferase reporter, but only by 32% (P-value = 0.048) and 10% (P-value = 0.094), respectively (Fig. 4). These data are consistent with our results from structural studies of these variants/functional characterization (Fig. 3).
The remaining variants identified in affected individuals were also tested and were found not to affect the activation drastically (Figure S2).
Discussion
So far, the variants identified in TP63, which lead to various developmental disorders, were found to have dominant-negative or gain-of-function effects only. Also, individuals identified earlier with deletions including TP63 did not present the typical features seen for TP63 variants [11, 13]. In this study, we report three families with deletions encompassing part of the TP63 with affected individuals presenting OFC, hypodontia, nail abnormalities, and other phenotypic features (Fig. 2, Table 1). Previously, a systematic analysis of copy number variants of a large cohort 312 OFC patients did not identify any CNVs affecting TP63 [18]. SNP microarray analysis in a cohort of 2366 healthy individuals reported a 174.5-kb deletion encompassing exons 2–4 and exon 3ʹ in 161 individuals [19]. However, this is likely to be a technical artifact, as we have never observed this deletion nor any other one in our in-house exome sequencing and SNP array pipelines conducted in over 30,000 individuals (Dr. R. Pfundt, personal communication).

Pedigree structures of the three orofacial clefting (OFC) families carrying a deletion affecting the TP63 gene. Black symbols indicate those individuals who are clinically affected with OFC and/or other features of ectodermal dysplasia. A complete overview of clinical features for each individual is provided in Table 1. Individuals used for SNP microarray analysis are indicated by the arrow. Individuals with underlined identifiers were tested by quantitative PCR (qPCR) (Table S2) for the presence of the deletion, which was confirmed in all cases
According to the ExAC browser, there is a strong constraint for loss-of-function variants (pLI = 0.98) and a moderate constraint for TP63 missense variants (z = 2.65) [20]. The identification of partial deletions in three OFC families in this study is inconsistent with the earlier notion that TP63 does not function in a dosage-dependent manner. We also report seven variants in individuals affected with nsOFC identified using MIPs analysis. These seven variants identified in the affected individuals were not seen in the control cohort. These variants include two variants c.569_576del (p.(Ala190Aspfs*5)) and c.997C>T (p.(Gln333*)) in the DBD predicting a premature stop, which can lead to complete loss-of-function by NMD or otherwise the ablation of most of the critical p63 protein domains, including DBD, OD, SAM, and TI domains. Two variants that were identified in the OD c.1213G>A (p.(Val405Met) and c.1223G>A (p.(Arg408His)) were shown to cause partial loss-of-function in the TA assay (Figs. 3, 4).

Melting points of different tetramerization domain mutants and wild type. The melting point of the wild-type p63 tetramerization domain (blue line) as well as the p.Val405Met (orange line) and the p.Arg408His (gray line) variants was determined by a thermal shift assay. Due to the extraordinary stability of the tetramerization domains, the pH had to be shifted to 4.5 to ensure melting in a temperature range, which is accessible to this assay. Under these conditions, the measured melting temperatures were 85.6 ± 0.1 °C for the wild-type tetramerization domain, 51.6 ± 2.5 °C for the p.Val405Met variant, and 82.1 ± 0.1 °C for the p.Arg407His variant. The reduction in stability of the p.Val405Met variant is also reflected in a shift of ΔNp63α from a clear tetramer in wild-type protein towards a dimer-tetramer mix in the p.Val405Met variant on analytical SEC

Transient transfection assay (luciferase induction) for the oligomerization domain (OD) variants. The fold inductions for the different mutant constructs are calculated relative to the WT isoform. p.Arg343Trp, an EEC causing mutant was used as control. p.Val405Met and p.Arg408His are the two OD variants tested. p.Arg352Gly, the only variant previously found responsible for nsOFC was also tested. The data are represented as the mean ± SEM
The variants identified so far in the individuals affected with p63 syndromes lie outside the OD. Interestingly, the two OD variants reported in this study were identified in individuals affected with nsOFC who presented no additional TP63-related phenotype. This difference in distribution of variants between nsOFC and p63 syndromes revealed different pathogenic mechanisms in syndromic versus nonsyndromic conditions. The two variants in the OD reduced the activation of luciferase reporter by 32 and 10%, respectively, whereas the other variants identified in the study lead to no or only a slight increase compared with the respective wild-type isoforms (Fig. 4). We propose a model to explain the functional effects of different kinds of variants in p63. The normal tetrameric complex with all wild-type p63 molecules leads to a normal level of TA. A heterozygous dominant-negative variant in TP63, as seen in individuals affected with p63 syndromes, produces only half the total wild-type copies of p63 molecules [21]. The remaining half are mutant copies, which retain the ability to oligomerize. Theoretically, 6.25% of the total tetrameric complexes will be formed by all wild-type monomers, which will have normal TA activity. The rest of the complexes will have one or more mutant monomers, which will affect TA. The loss-of-function variants, which can be deletions or nonsense variants as reported in this study, also produce only half the total wild-type copies of p63 molecules. With only half the protein molecules, complexes formed will be normal but 50% less abundant. The heterozygous OD variants, as reported in this study, will produce only half the normal copy of the protein, leaving the other half with an impaired ability to oligomerize. As a result, there will be normal protein complexes but they will be almost half in number. These OD variants will therefore lead to partial loss-of-function (Fig. 5). This model is also supported by analysis of the biophysical properties of the respective mutant proteins, which showed reduced thermo-stability in p.Val405Met mutant proteins compared with wild type. Where the normal protein denatures at 85.6 ± 0.1 °C, the p.Val405Met mutant protein unfolds at 51.6 ± 2.5 °C (Fig. 3). In contrast to the p.Val405Met mutant protein, no obvious explanation based on the biophysical properties of the p.Arg408His is possible. Nevertheless, our data suggest that dominant-negative or gain-of-function variants in TP63 cause developmental disorders including more than one phenotype, whereas the loss-of-function variants lead to nsOFC.

Model explaining the functional effects of different kind of variants in p63 on the transactivation (TA) activity. The normal complex with all wild-type p63 molecules leads to regular level of TA. A heterozygous dominant-negative variant in TP63 will lead to half mutant copies of p63 molecules. Theoretically, 6.25% of the total tetrameric complexes will be formed by all wild-type monomer, which will have normal TA activity. The rest of the complexes will have at least one (or more) mutant monomer, which will block TA. The loss-of-function variants will lead to production of only half the total wild-type copies of p63 molecules. With only half the protein molecules, complexes formed will be normal but 50% less abundant. The heterozygous oligomerization domain (OD) variants will produce only half normal copy of the protein, leaving the other half incapable to oligomerize. As a result, there will be normal protein complexes but they will be almost half in number. These OD variants will therefore lead to partial loss-of-function
All seven variants in affected individuals except for c.997C>T (p.(Gln333*)) were inherited from an unaffected parent. Due to unavailability of parental DNA, we could not confirm whether the c.997C>T variant was de novo or inherited. This suggests that the variants are not always completely penetrant. Incomplete penetrance has also been observed for other TP63 variants, including common variants such as those affecting arginine 319 (280 according to former nomenclature). Arg319 is considered a hotspot for EEC syndrome variants [10, 22], but there are also reports on families in which Arg319Cys is inherited as isolated SHFM. These families have cases of non-penetrance as well [23]. Thus, clinical variability and reduced penetrance is typical for TP63 variants. Four missense were identified in the control cohort. Two of these, c.85T>G (p.(Phe29Val)) and c.406A>G (p.(Thr136Ala)), were predicted to be tolerated (Table 2). Two others, c.725A>G (p.(Lys242Arg)) and c.1121C>T (p.(Thr374Met)), were predicted to be deleterious, similar as the missense variants identified in the CLP cohort. We cannot exclude that these variants represent CLP risk alleles.
Recent genome-wide association studies (GWAS) have identified intronic variants in TP63, which impart the risk of nsOFC [24]. The other variants c.670G>A (p.(Val224Ile), NM_001114979.1:c.1390C>T (p.(Arg464Trp)), and c.1459C>T (p.(Arg487Cys)) identified in our study are located in various domains of p63 and did not show any dramatic decreases in TA activity in our assay. These variants could represent risk factors, which would cause only nsOFC in conjunction with other genetic and/or environmental factors with strong effect sizes. Importantly, all seven TP63 variants identified were present in individuals affected with CL/P (n = 878) and none in individuals with CPO (n = 194). This has also been seen previously in a genome-wide meta-analysis, which identified a statistically significant association between TP63 and CL/P but not CPO [24].
In conclusion, we identified three deletions and seven probable disease-causing variants in TP63, which uncover a dosage-dependent function of p63 and a loss-of-function mechanism behind nsOFC. Our findings also change the previous assumption that loss-of-function alleles in TP63 do not give rise to any clinical phenotype.
Materials and methods
Subjects
Probands from three families were referred to a Clinical Genetics Center because of the presence of cleft lip palate. Standard diagnostic testing was offered, including microarray-based CNV analysis. A CNV affecting TP63 was identified in three families.
In family W11–4934, a 53-year-old male (II4) and his 55-year-old brother (II3), third and fourth child from Dutch non-consanguineous parents, were born with a CLP, and showed hypodontia and nail hypoplasia (Fig. 2, Table 1). Their parents are of Caucasian origin. Interestingly, seven family members (I1, II2, II5, III1, III2, III4, and III5) demonstrated a variety of clinical features that might be related to the mutant TP63 phenotypic spectrum, ranging from prominent to very subtle ectodermal symptoms. Most striking were the hypodontia and tooth anomalies in most affected family members and the choanal atresia in a daughter of the eldest male with CLP (III2), and in one affected sister (II5) and her offspring (III4 and III5). Both children (III4 and III5) had coarse fair hair. The mildest features in affected family members (I1 and II2) were hypoplastic nails and dry skin. To the best of our knowledge, Nabothian cysts, epithelial inclusion cysts of the cervix, were not previously reported as p63-related features. No congenital limb anomalies, lacrimal duct abnormalities, ankyloblepharon nor intellectual disability were present in affected members.
In family W12–0831, a 37-year-old male (IV3), the only child of non-consanguineous Dutch parents with a Caucasian origin, was born with an unilateral cleft lip and cleft palate and showed a mild hypodontia, with agenesis of three teeth (Fig. 2, Table 1). The mother (III4) was born with a cleft palate. Family history revealed two additional family members (III3 and V1) with a cleft lip and cleft palate. In addition, two family members (II2 and IV1) were born with a duplicated thumb. In one family member (II4), a duplication of the thumb was suspected. SHFM or other limb anomalies were not reported. There was no history of ankyloblepharon, lacrimal duct anomalies, or ectodermal involvement in affected family members, except for dry skin in the mother (III4). No intellectual disability was present.
Finally, in family W16–065 the affected individual is a now 33-year-old woman, born as the second of four children of non-consanguineous Danish parents. She was born with a unilateral cleft lip and cleft palate. She had several teeth extracted and had mandibular surgery for correction of asymmetry and overbite. She was also diagnosed with lichen sclerosus.
A cohort of 1072 individuals affected with OFC and 706 population-based controls was analyzed for variants in TP63 using MIPs sequencing. All of these individuals were of European ancestry. The OFC cohort included 647 CLP, 194 cleft palate only (CPO), 127 cleft lip alveolus (CLA), and 104 cleft lip only (CLO) affected individuals. The MIPs analysis involved subjects from Bonn, Leuven, and Nijmegen as part of a larger project in which other genes were also analyzed [25]. Approval was obtained from the Institutional Review Boards of the respective University Hospitals. Informed consent was obtained from all individuals who participated in the study.
DNA variant data have been submitted to the gene variant database at www.LOVD.nl/TP63 (patient IDs: 00208593, 00208594, 00208598, 00208628, 00208629, 00208633, 00208754, and 00208769–00208775).
CNV microarray analysis
We assessed copy number variations in the three families by genome-wide high-resolution cytogenetic array analysis (CytoScan HD, Affymetrix, Santa Clara, CA, USA). Data analysis was performed with the Chromosome Analysis Suite software (ChAS, Affymetrix; V3.0.0.42) using default settings (Table S1).
Confirmation of the identified deletions and their segregation within families was carried out by qPCR. SYBR green-based genomic real-time qPCR analysis was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) by using Power SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer’s instructions. Primers were developed by the Primer3 program (Table S2) and validated as previously described [26].
MIPs sequencing
All 17 coding exons of TP63 were sequenced using a total of 40 MIPs (Supplemental spreadsheet), which represented a subset of MIPs that were part of a previously performed experiment [16]. The final pooled MIPs libraries were run on a NextSeq500 sequencer (Illumina, San Diego, CA, USA) using 2 × 79 bp reads. Data were analyzed with an in-house analysis pipeline [16]. The identified variants from affected individuals and control cohorts were filtered for quality-by-depth higher than 500, rare (dbSNP freq ≤0.1 and <5 samples in MIPs screen), nonsynonymous variants with CADD PHRED-like score >15 (Table 1, Fig. 2) [27]. Alamut® Visual was used to evaluate the effect of the variants using genetic and genomic information from different sources (phyloP, Grantham, SIFT) as described. The variants in affected individuals were validated using Sanger sequencing and segregation analysis was performed in all available family members.
Analysis of functional effects of missense variants in the OD
The TSA was performed using an iCycler IQ Real Time PCR Detection System (Bio-Rad, Hercules, California, USA). Highly concentrated protein samples were diluted at least 100-fold into the measurement buffer (10 mM Na-Phosphate, 140 mM NaCl, pH 4.5) to a final concentration of 20 µM. In all, 36 µl of protein solution was combined with 4 µl of a 1:200 dilution of SYPRO Orange (ThermoFisher, Waltham, Massachusetts, USA). Samples were measured in MicroAmp Optical 96-well plates (ThermoFisher, Waltham, Massachusetts, USA). Temperature was increased at a rate of 1 °C/min, while fluorescence measurements were taken at 0.2 °C increments. The resulting data were smoothed by a rolling average over 5 data points. The first derivative of the result was plotted to obtain the melting point. The experiment was performed in duplicates. Analytical SEC of rabbit reticulocyte lysate expressed ΔNp63α variants was done as described previously [28].
Constructs and site-directed mutagenesis
Modified pcDNA3 vectors, containing various forms of human and mouse TP63 were used to create the variants identified by MIPs analysis (Table 2). Variants were created through polymerase chain reaction, using Phusion® High-Fidelity DNA Polymerase (New England Biolabs, Thermo Scientific) and polyacrylamide gel electrophoresis purified primers containing the variants. The PCR program consisted of an initial denaturation at 95 °C for 5 min followed by 18 cycles of 30-s denaturation at 95 °C, 1-min annealing at 55 °C and 7-min extension at 68 °C. This was followed by final extension at 68 °C for 5 min.
Cell culture and transfection
The Saos-2 cell line, which lacks endogenous p63, was maintained in Gibco™ Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, Waltham, Massachusetts, USA) with 10% fetal bovine serum, 1% sodium pyruvate at 37 °C, 5% CO2. For transfection, approximately 2 × 105 cells were seeded into one well of a six-well tissue culture plate. Cells were transfected with the pcDNA, a mammalian expression vector containing mutant TP63, Renilla pRL-SV40 reporter vector and modified pGL3 enhancer vector (Promega, Fitchburg, Wisconsin, United States) containing a TP63 binding site that has been shown before to work as an enhancer [17]. Effectene™ Transfection Reagent (Qiagen, The Netherlands) was used as per the protocol described previously [14]. pcDNA vector containing wild-type inserts of TP63 isoforms were used as negative control and pcDNA vector with mutated TP63 encoding the EEC-associated missense variant p.(Arg343Trp) was used as positive control [14].
Transactivation assays
Twenty-four hours after the transfection, subconfluent cells were collected for luciferase transactivation assay. Cells were lysed and the luciferase assay was performed using the dual luciferase assay system (Promega, Fitchburg, Wisconsin, United States) according to the manufacturer’s instructions. For the measurements, a Lumat LB 9507 (Berthold Technologies, Germany) was used. The level of activation was calculated in comparison with transfection with an empty pcDNA3 vector. Data were normalized to the Renilla reporter signal [14].
References
Mills AA, Zheng B, Wang X-J, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–13.
Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–8.
Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–16.
Mangiulli M, Valletti A, Caratozzolo MF, Tullo A, Sbisà E, Pesole G, et al. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res. 2009;37:6092–104.
Vanbokhoven H, Melino G, Candi E, Declercq W. p63, a story of mice and men. J Invest Dermatol. 2011;131:1196–207.
Serber Z, Lai HC, Yang A, Ou HD, Sigal MS, Kelly AE, et al. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol Cell Biol. 2002;22:8601–11.
Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999;99:143–53.
Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), 1996. https://omim.org/.
Leoyklang P, Siriwan P, Shotelersuk V. A mutation of the p63 gene in non-syndromic cleft lip. J Med Genet. 2006;43:e28.
van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, et al. p63 gene mutations in EEC syndrome, limb-mammary syndrome, and isolated split hand–split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet. 2001;69:481–92.
McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 2001;10:221–9.
Rinne T, Hamel B, Bokhoven H, van, Brunner HG. Pattern of p63 mutations and their phenotypes—update. Am J Med Genet Part A. 2006;140A:1396–406.
Duijf PH, Vanmolkot KR, Propping P, Friedl W, Krieger E, McKeon F, et al. Gain-of-function mutation in ADULT syndrome reveals the presence of a second transactivation domain in p63. Hum Mol Genet. 2002;11:799–804.
Rinne T, Clements SE, Lamme E, Duijf PH, Bolat E, Meijer R, et al. A novel translation re-initiation mechanism for the p63 gene revealed by amino-terminal truncating mutations in Rapp-Hodgkin/Hay-Wells-like syndromes. Hum Mol Genet. 2008;17:1968–77.
Chitayat D, Babul R, Silver MM, Jay V, Teshima IE, Babyn P, et al. Terminal deletion of the long arm of chromosome 3 [46,XX,del(3)(q27→qter)]. Am J Med Genet. 1996;61:45–8.
O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science (80-). 2012;338:1619–22.
Thomason HA, Zhou H, Kouwenhoven EN, Dotto GP, Restivo G, Nguyen BC, et al. Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J Clin Invest. 2010;120:1561–9.
Conte F, Oti M, Dixon J, Carels CE, Rubini M, Zhou H. Systematic analysis of copy number variants of a large cohort of orofacial cleft patients identifies candidate genes for orofacial clefts. Hum Genet. 2016;135:41–59.
Campbell CD, Sampas N, Tsalenko A, Sudmant PH, Kidd JM, Malig M, et al. Population-genetic properties of differentiated human copy-number polymorphisms. Am J Hum Genet. 2011;88:317–32.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
van Bokhoven H, Brunner HG. Splitting p63. Am J Hum Genet. 2002;71:1–13.
Brunner HG, Hamel BCJ, Van Bokhoven H. The p63 gene in EEC and other syndromes. J Med Genet. 2002;39:377–81.
Ianakiev P, Kilpatrick MW, Toudjarska I, Basel D, Beighton P, Tsipouras P. Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am J Hum Genet. 2000;67:59–66.
Leslie EJ, Carlson JC, Shaffer JR, Butali A, Buxó CJ, Castilla EE, et al. Genome-wide meta-analyses of nonsyndromic orofacial clefts identify novel associations between FOXE1 and all orofacial clefts, and TP63 and cleft lip with or without cleft palate. Hum Genet. 2017;136:275–86.
Ockeloen CW, Khandelwal KD, Dreesen K, Ludwig KU, Sullivan R, van Rooij IALM. et al. Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet Med. 2016;18:1158–62.
Marcelis CL, Hol FA, Graham GE, Rieu PN, Kellermayer R, Meijer RP, et al. Genotype-phenotype correlations in MYCN-related Feingold syndrome. Hum Mutat. 2008;29:1125–32.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Deutsch GB, Zielonka EM, Coutandin D, Weber TA, Schäfer B, Hannewald J, et al. DNA damage in oocytes induces a switch of the quality control factor TAp63α from dimer to tetramer. Cell. 2011;144:566–76.
Acknowledgements
All the affected individuals, their parents, families, and control individuals from Bonn, Nijmegen, and Leuven who participated in this study. Members of the Nijmegen Cleft Lip and Palate Team: Dr R. Admiraal, Dr W. Borstlap, Dr T. Wagner, Dr M. Nienhuis, Mrs V. Engels, Drs M. Kuijpers, Mrs J. Vanderstappen, Mrs E. Kerkhofs, Prof AM Kuijpers-Jagtman, Mrs M. Kloen, Mrs J. Buchner, and Mrs M. van der Looy and Drs M. Thonissen for their help in the recruitment of the affected individuals; Margrieta Alblas for preparing the DNAs for MIPs. Members of the Cleft Lip and Palate Team of the University Hospitals KU Leuven: Prof Dr V. Vander Poorten, Prof Dr A. Verdonck, Dr T. Dormaar, Drs C. Vergalle, Dr J. Roosenboom for their help in recruiting the affected individuals; Mr B. Vankeirsbilck for preparing the DNAs, and Dr. L. Veken and Dr R. Pfundt for conducting microarray analyses. The research was funded by the DFG (DO 545/8–1, LU 1944–2/1 to KUL)) and the Cluster of Excellence Frankfurt (Macromolecular Complexes).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Khandelwal, K.D., van den Boogaard, MJ.H., Mehrem, S.L. et al. Deletions and loss-of-function variants in TP63 associated with orofacial clefting. Eur J Hum Genet 27, 1101–1112 (2019). https://doi.org/10.1038/s41431-019-0370-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0370-0
This article is cited by
-
Heterozygous TP63 pathogenic variants in isolated primary ovarian insufficiency
Journal of Assisted Reproduction and Genetics (2023)
-
Identification of a novel heterozygous missense TP63 variant in a Chinese pedigree with split-hand/foot malformation
BMC Medical Genomics (2022)
-
Familial cleft tongue caused by a unique translation initiation codon variant in TP63
European Journal of Human Genetics (2022)
-
Identification of a novel TP63 mutation causing nonsyndromic cleft lip with or without cleft palate
BMC Medical Genomics (2021)
