
Abstract
A new compound, fusaramin (1), along with three known compounds, sambutoxin (2), N-demethylsambutoxin (3) and (−)-6-deoxyoxysporidinone (4), was isolated from a culture broth of Fusarium sp. FKI-7550 by bioassay-guided fractionation using multidrug-sensitive Saccharomyces cerevisiae 12geneΔ0HSR-iERG6. The chemical structure of 1 was elucidated by NMR studies and electronic circular dichroism spectrum. Compound 1 showed antibacterial activity against some Gram-positive and Gram-negative bacteria and inhibited the growth of S. cerevisiae 12geneΔ0HSR-iERG6 grown on glycerol-containing medium. The MICs of 1 against wild-type and multidrug-sensitive yeasts grown on glycerol-containing medium were >128 μg ml−1 and 0.64 μg ml−1, respectively. However, MICs of 1 against both yeast strains grown on glucose-containing medium were >128 μg ml−1. All compounds showed inhibition of ATP synthesis via oxidative phosphorylation using isolated S. cerevisiae mitochondria.
Similar content being viewed by others

Introduction
Secondary metabolites produced by microorganisms display chemical diversity and have been used for a wide variety of purposes, including drugs and pesticides. Such important drugs derived from natural products as streptomycin, adriamycin, pravastatin, FK506 and amphotericin B, have been clinically used following the discovery of penicillin by Fleming in 1928. Compounds derived from natural products also play an important role in pesticides, and some inhibitors of mitochondrial electron transport chain have been commercially available [1, 2]. In addition, a research group has reported a clinical-grade small-molecule inhibitor of complex I of the mitochondrial electron transport chain for a tumour inhibitor [3]. Therefore, inhibitors of mitochondrial electron transport chains may help increase yields in agriculture and be useful in human cancer chemotherapy. Therefore we undertook a programme to search for new mitochondrial inhibitors from microbial secondary metabolites, using a yeast screening system.
The budding yeast Saccharomyces cerevisiae is a useful tool for identification and evaluation of the targets of bioactive compounds. However, the high-level drug resistance of S. cerevisiae often makes the discovery of useful bioactive compounds difficult. An important factor in some resistance mechanisms is the ATP-binding cassette (ABC) transporter family that exports compounds from cells. Therefore, using the budding yeast system and examining expression of the transporter, we expected to find bioactive compounds that were undetectable using previous methods [4]. We exploited the multidrug-sensitive budding yeast 12geneΔ0HSR-iERG6 strain [5] where 12 genes related to ABC transporters had been destroyed. We selected microbial culture broths that showed growth inhibition against the drug-sensitive 12geneΔ0HSR-iERG6 only on glycerol-containing medium (YPG agar medium; 1% yeast extract, 2% peptone, 3% glycerol and 1.5% agar) but which did not cause growth inhibition on glucose-containing medium (YPD agar medium; 1% yeast extract, 2% peptone, 3% glucose and 1.5% agar). Some compounds active against mitochondrial functions have been isolated by this method using glycerol- or glucose-containing medium [4, 6].
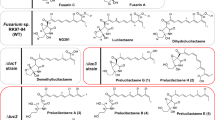
The screening system led to the discovery that Fusarium sp. FKI-7550 produced a new tetramic acid derivative (fusaramin, 1) and three known compounds, sambutoxin [7] (2), N-demethylsambutoxin [8] (3) and (−)-6-deoxyoxysporidinone [9] (4) (Fig. 1). Here, we report the fermentation, isolation, structure elucidation and biological profiles of 1–4.

Structures of fusaramin (1), sambutoxin (2), N-demethylsambutoxin (3) and (−)-6-deoxyoxysporidinone (4)
Results
Structure elucidation of fusaramin (1)
The molecular formula of 1 was elucidated as C27H39NO4 by HR-ESI-MS data ([M + H]+, m/z 442.2946: calculated for C27H40NO4, 442.2957), requiring nine degrees of unsaturation. All connections for 1H and 13C in 1 were elucidated by HSQC study. The NMR data measured in DMSO-d6 and the molecular formula indicated the presence of five methyls, five methylenes, five sp3 methines including one oxygenated and one nitrogenated methine, six sp2 methines and six fully-substituted sp2 carbons (Table 1). 1H–1H COSY of 1 indicated alignments from H2-7 (δH 2.54 and 2.63) to H2-10 (δH 1.65 and 1.90) and from H-12 (δH 4.83) to H3-17 (δH 0.79), plus connections between H-9 (δH 1.50) and H3-18 (δH 0.73), H-13 (δH 2.39) and H3-20 (δH 0.83), H-15 (δH 1.29) and H3-21 (δH 0.79) and H-5 (δH 4.17) and H-22 (δH 4.94) as shown in Fig. 2. The HMBC correlations from H2-10 to C-11 (δC 131.1), C-12 (δC 133.1) and C-19 (δC 15.7), from H-12 to C-10 (δC 47.0) and C-19 and from H3-19 (δH 1.49) to C-10, C-11 and C-12 suggested the alignment of C-10, C-11, C-12 and C-19. 1H, 13C and 2D NMR data suggested that 1 has a phenyl group, and the HMBC correlations from phenyl protons (δH 7.16–7.22) to C-22 (δC 72.8) and from H-22 to phenyl carbons (δC 139.5 and 127.0) deduced the connection between C-22 and the phenyl group. Moreover, six remaining 13C broad signals of 1 at δC 175.1 (C-2), 101.3 (C-3), 192.9 (C-4), 67.4 (C-5) and 188.7 (C-6) (Fig. S2) were assigned as a tetramic acid moiety after comparing with other tetramic acids [10,11,12,13]. It was supported by four remaining degrees of unsaturation. Due to the tautomerism of tetramic acid, the 13C signals of C-2 to C-6 were suggested to be broad. The HMBC correlations from H2-7 to C-3 and C-6 and from H2-8 (δH 1.15 and 1.39) to C-6 indicated the alignment of C-3, C-6 and C-7. Finally, the planar structure of 1 was elucidated by the HMBC correlations from H-5 to C-2, C-4 and C-23 and from H-22 to C-4. Furthermore, the geometry of a double bond between C-11 and C-12 was elucidated as being (E)-configuration by the NOESY cross peak between H2-10 and H-12 (Fig. 2). The relative configuration of C-5 and C-22 was elucidated as 5S* and 22S* by large 3J coupling constant of 7.2 Hz at H-5/H-22 in 1 (in CDCl3, Table S1) compared with the previous study [12] of tetramic acids, F-14329 and chaunolidines A–C. The absolute configuration of C-5 was elucidated as 5S by negative cotton effects at 226 and 285 nm in ECD spectra of 1 (in MeOH, Fig. S18) compared with the previous study [12]. Thus, the absolute configurations of 1 were elucidated to be 5S and 22S.

Structure elucidation of fusaramin (1). 1H–1H COSY (bold lines), key HMBC (arrows) and key NOESY correlation (dashed arrow)
Evaluation of antimicrobial activity of fusaramin (1) by the agar diffusion method
The antimicrobial activities of 1 were evaluated by the agar diffusion method using some Gram-positive and Gram-negative bacteria, yeasts (including Candida albicans and wild-type and multidrug-sensitive budding yeast S. cerevisiae) and a filamentous fungus (Mucor racemosus). The result of antimicrobial evaluation is shown in Table S2. Compound 1 showed growth inhibition against some Gram-positive bacteria (Staphylococcus aureus, Bacillus subtilis and Kocuria rhizophila) and one Gram-negative bacterium (Xanthomonas oryzae pv. oryzae). Although 1 did not exhibit growth inhibition against wild-type S. cerevisiae KF237 and BY4741, S. cerevisiae BY25929, C. albicans FK1 and multidrug-sensitive S. cerevisiae 12geneΔ0HSR-iERG6 grown on YPD agar medium, it inhibited the growth of S. cerevisiae 12geneΔ0HSR-iERG6 on YPG agar medium. As 1 inhibited yeast growth only in glycerol medium, which is similar to mitochondrial respiration inhibitors, such as ascosteroside C [6], trichopolyn VI [14] and decatamariic acid [4], we hypothesized that 1 might inhibit mitochondrial functions.
Evaluation of MIC values of 1–4 against yeasts
We measured the minimum inhibition concentrations (MICs) of 1–4 against wild-type and multidrug-sensitive S. cerevisiae (Table 2). All compounds did not demonstrate any inhibition against the wild-type yeast grown on YPD medium, and 4 slightly inhibited the multidrug-sensitive yeast grown on YPD medium. It is interesting that all compounds showed strong inhibition against the multidrug-sensitive yeast grown on YPG medium. Compound 2 was reported to be an inhibitor of electron transfer of quinol-cytochrome c oxidoreductase (complex III) [15] and showed >2000-fold more potent growth inhibition against multidrug-sensitive S. cerevisiae growth on YPG medium than on YPD medium. The selectivity of 1, 3 and 4 was >200, >400 and 16 times, respectively. Therefore, we assumed that the target of 1–4 is any one of the mitochondrial functions, such as the electron transfer system. As most of the compounds did not show growth inhibition against the wild-type yeast, it is strongly suggested that the multidrug-sensitive budding yeast is a useful tool to find new growth inhibition compounds.
Evaluation of inhibition of mitochondrial functions
With the assay monitoring overall ADP-uptake/ATP-release reactions [16], using mitochondria (respiring with α-ketoglutarate) isolated from wild-type S. cerevisiae, we evaluated the inhibitory effects of 1–4 on mitochondrial functions. This assay method principally enabled us to determine potential inhibitors of any of the mitochondrial mechanisms presiding over ATP synthesis via oxidative phosphorylation. Compounds 1–4 inhibited overall ADP-uptake/ATP-release reactions; the IC50 (the molar concentration required for 50% inhibition) are listed in Table 3. Of the four compounds, 2 turned out to be the most potent.
To know whether the compounds inhibit ATP synthesis by acting on the electron transfer system, we examined their effects on the NADH-cytochrome c oxidoreductase activity [17] (covering NADH dehydrogenase to complex III). The concentrations of 1–4 were set to 72, 2.6, 28 and 370nmol/mg of proteins, respectively, equivalent to 3 × IC50 obtained in the above ADP-uptake/ATP-release reactions (Table 3). Compounds 2–4, but not 1, significantly inhibited NADH-cytochrome c oxidoreductase activity (Fig. 3), indicating that 2–4 are inhibitors of complex III, as reported previously [15]. The potential target of 1 may be one of the transporters of substrates that are essential for ATP synthesis, such as an ADP/ATP carrier (though we have not yet identified the target).

Effects of 1–4 on the electron transfer system in yeast mitochondria. Isolated yeast mitochondria were permeabilized by repeated freeze-thawing under N2 atmosphere and the NADH-cytochrome c oxidoreductase activity was measured in the presence of 1–4, as described in the experimental procedures. The concentrations of 1–4 were set to 72, 2.6, 28 and 370 nmol/mg of proteins, respectively, which are equivalent to 3 × IC50 that was obtained in the ADP-uptake/ATP-release reactions (Table 3). Values show means ± S.E.M. (n = 3)
Cytotoxic evaluation
We tested the cytotoxic activity of compounds 1–4 against various cancer cell lines (floating cell lines, HL-60, Jurkat and THP-1; adherent cell lines, HeLa S3, HT29, A549, H1299 and Panc1). Compound 4 showed cytotoxic against both floating and adherent cell lines. Compounds 1, 2 and 3 exhibited potent cytotoxicity against floating cell lines (Table 4).
Discussion
We isolated 1–4 using an S. cerevisiae 12geneΔ0HSR-iERG6 obtained from the culture broth of Fusarium sp. FKI-7550.
Compound 1 is a tetramic acid derivative composed of an aromatic amino acid and a polyketide. These types of compounds are produced by fungi, such as epicoccarines A and B, F-14329, chaunolidines A and B, militarinone B, tolypoalbin and pretenellin A [10, 12, 13, 18]. Among them, militarinone B and pretenellin A are co-produced with militarinone A and tenellin, respectively, which have a pyridone-type rearranged ring. Biosynthesis study of tenellin revealed that pretenellin A was a precursor of tenellin [18]. Compound 1 may be a precursor of 2–4 based on their structural resemblance. Several tetramic acid derivatives composed of an aromatic amino acid and a polyketide have been reported, and their aromatic amino acid parts were tyrosine, β-hydroxytyrosine or phenylalanine. Compound 1 is the first compound having β-hydroxyphenylalanine as the aromatic amino acid part.
Evaluation of the growth inhibition of the yeast by 1 suggested that it may be an inhibitor of mitochondrial functions because this compound inhibited the growth of the multidrug-sensitive budding yeast S. cerevisiae 12geneΔ0HSR-iERG6 grown on YPG agar medium but did not inhibit the yeast grown on YPD agar medium. The biochemical evaluation using isolated yeast mitochondria verified this notion. As for the cytotoxicity, 1 preferentially inhibited growth inhibition against floating cell lines, HL-60, Jurkat and THP-1 but did not show growth inhibition against adherent cell lines, HeLa S3, HT29, A549, H1299 and Panc1. Tetramic acid analogues have been reported to display antimicrobial, antitumor, antiviral and antimitochondrial activities [19, 20]. The inhibitions of tetramic acids on mitochondrial functions have previously only been reported for equisetin and equisetin-like compounds [20, 21], but the structure of 1 does not resemble equisetin. Further chemical and biological studies of 1 may lead to the development of a new type of mitochondrial inhibitor.
Experimental section
General experiments
Reverse- and normal-phase column chromatography was conducted on YMC-gel ODS-A (150 μm) and Silica gel 60 (0.063–0.200 mm), respectively. A Chromatorex C8 SPS100-5HE column was purchased from Fuji Silysia Chemical Co. (Aichi, Japan). High- and low-resolution mass data were measured on a JEOL JMS-T100LP (JEOL, Tokyo, Japan). NMR spectra were measured on a Bruker Avance III HD600 (Bruker Corp, Germany) with 1H NMR at 600 MHz and 13C NMR at 125 MHz and on a Varian XL-400 spectrometer (Agilent Technologies, CA, USA) with 1H NMR at 400 MHz and 13C NMR at 100 MHz in DMSO-d6 and CDCl3. The chemical shifts were expressed in ppm and were referenced to DMSO-d6 (2.48 ppm) and CDCl3 (7.26 ppm) in the 1H NMR spectra and DMSO-d6 (39.5 ppm) and CDCl3 (77.0 ppm) in the 13C NMR spectra.
IR spectra (ATR) were measured on an FT-210 Fourier transform IR spectrometer (Horiba Ltd, Kyoto, Japan). UV spectra were recorded on a Hitachi U-2801 spectrophotometer (Hitachi Ltd, Tokyo, Japan). Optical rotation was measured on a JASCO P-2200 polarimeter (JASCO Corporation, Tokyo, Japan). OD600 was measured on a Corona Grating Microplate Reader SH-9000 (Corona Electric, Ibaraki, Japan). ECD spectra were measured on a J-720 (JASCO Corporation, Tokyo, Japan).
Taxonomic studies
Fungal strain FKI-7550 was isolated from soil around the root of Cerasus × yedoensis collected in Tokushima, Japan. This strain was identified with members of the genus Fusarium by its producing lunate to falcate macroconidia and ellipsoidal microconidia. The ITS sequence of strain FKI-7550 was compared to sequences in the GenBank database by BLASTN 2.7.1 analysis [22]. The sequence of FKI-7550 was 99.6% similar to that of NRRL 25181 (ex-type of Fusarium concentricum, GenBank accession number NR_111886). The producing strain FKI-7550 was assigned to the genus and was designated as Fusarium sp. based on its morphology and sequence analysis.
Fermentation
One loopful of strain FKI-7550 grown on an LcA slant (0.1% glycerol, 0.08% KH2PO4, 0.02% K2HPO4, 0.02% MgSO4·7 H2O, 0.02% KCl, 0.2% NaNO3, and 1.5% agar, pH 6.0) was inoculated into a 500-ml Erlenmeyer flask containing 100 ml of a seed culture medium [2% glucose, 0.2% yeast extract, 0.05% MgSO4·7 H2O, 0.5% Polypepton (Wako Pure Chemical Industries, Ltd), 0.1% KH2PO4, and 0.1% agar, pH 6.0 and incubated on a rotary shaker at 27 °C for 4 days. Twenty-five millilitres of the seed culture was inoculated into each of 200 Ulpack 47 culture bags (Hokken Co. Ltd, Tochigi, Japan) containing a production medium (500 g of water-sodden rice). Static fermentation was continued at 27 °C for 12 days.
Isolation and identification of 1–4
The stationary culture was extracted with acetone (120 l) and the extract was filtrated. The filtrate was concentrated in vacuo to remove acetone. The remaining aqueous solution (35 l) was extracted three times with an equal volume of EtOAc (total 105 l). The organic layer was concentrated to dryness to afford a crude extract (628 g). The extract was chromatographed on a Diaion HP20 column and eluted stepwise with a mixture of MeOH–H2O (20:80, 50:50 and 100:0). Compounds were eluted with the 100:0 eluate, and the solution was concentrated in vacuo to remove MeOH. The 100:0 fraction (129 g) was applied to an ODS gel column (55 i.d. × 215 mm) and eluted stepwise with a mixture of MeOH–H2O (20:80, 50:50, 70:30, 80:20, 90:10 and 100:0). The 80:20 fraction was concentrated to dryness to afford a crude extract (33 g). The extract was chromatographed on a silica gel column and eluted stepwise with a mixture of CHCl3–MeOH (100:0, 100:1, 100:5, 90:10, 1:1 and 0:100). The active fraction (90:10) was concentrated in vacuo to remove organic solvent. Finally, the concentrated material (1.6 g) was applied to an HPLC (Chromatorex C8 SPS100-5HE, 20 i.d. × 250 mm) with an isocratic solvent system of 70% acetonitrile–water with 0.1% trifluoroacetic acid solution at a flow rate of 10 ml min−1 to give the new compound (34.4 mg), fusaramin (1).
The active fraction (100:5) from silica gel column chromatography (0.6 g) was applied to an HPLC (Chromatorex C8 SPS100-5HE, 20 i.d. × 250 mm) with an isocratic solvent system of 65% acetonitrile–water with 0.1% trifluoroacetic acid solution at a flow rate of 10 ml min−1 to give active peaks at 33–35 and 45–48 min. The peak of 45–48 min was identified as sambutoxin (33.9 mg, 2) using ESI-MS data and NMR data. The peak of 33–35 min (35.5 mg) was further applied to an HPLC (Chromatorex C8 SPS100-5HE, 20 i.d. × 250 mm) with an isocratic solvent system of 55% acetonitrile–water with 0.1% trifluoroacetic acid solution at a flow rate of 10 ml min−1 to give (−)-6-deoxyoxysporidinone (12.6 mg; retention time 72–79 min; 4) and N-demethylsambutoxin (13.6 mg; retention time 81–91 min; 3).
Fusaramin (1): yellow oil; soluble in DMSO, MeOH, acetone and CHCl3; insoluble in H2O; \([{\rm{\alpha}}]^{25}_{\mathrm{D}}\) –127.2 (c = 0.1, MeOH); IR νmax (ATR) cm−1 3332, 2954, 2919, 1650, 1596, 1450, 1342, 1106 and 1041; UV (MeOH) λmax (ε) 203 (20 100), 242 (8500) and 281 (13 100) nm; CD (MeOH) λmax (Δε) 226 (–18 412), 242 (–10 585), 262 (–9472) and 285 (–9759) nm. 1H and 13C NMR data are shown in Table 1.
Sambutoxin (2): yellow oil; 1H NMR (400 MHz, CDCl3) δ 10.08 (–OH, br s), 7.24 (2H, d, J = 8.4 Hz), 7.12 (1H, s), 6.29 (2H, d, J = 8.8 Hz), 5.18 (1H, d, J = 9.2 Hz), 5.04 (1H, dd, J = 11.0, 1.8 Hz), 3.51 (1H, m), 3.51 (3H, s), 2.46 (1H, m), 2.07 (1H, d, J = 13.6 Hz), 1.89 (1 H, m), 1.71–1.58 (2H, overlapped), 1.61 (3H, s), 1.45–1.16 (4H, overlapped), 1.03 (2H, m), 0.89 (3H, d, J = 6.8 Hz), 0.82 (3H, t, J = 7.2 Hz), 0.82 (3H, d, J = 6.4 Hz) and 0.73 (3H, d, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 162.7, 161.3, 156.5, 138.0, 135.9, 130.2, 130.2, 130.0, 124.8, 116.0, 115.4, 115.4, 110.4, 92.5, 77.6, 44.6, 37.4, 32.2, 32.0, 31.9, 30.6, 29.5, 28.8, 20.6, 19.5, 17.5, 11.5 and 11.1; ESI-MS (+) mode m/z 454 [M + H]+ and ESI-MS (−) mode m/z 452 [M − H]−. The NMR and MS data showed good correlation with those of sambutoxin [7].
N-Demethylsambutoxin (3): pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 10.25 (–OH, s), 7.16 (2H, d, J = 8.4 Hz), 6.95 (2H, d, J = 8.8 Hz), 6.64 (1H, s), 5.21 (1H, d, J = 13.2 Hz), 5.05 (1H, d, J = 12.0 Hz), 3.55 (1H, d, J = 9.6 Hz), 2.46 (1H, m), 2.07 (1H, m), 1.91 (1H, d, J = 10.8 Hz), 1.68 (1H, m), 1.62 (3H, s), 1.62 (1 H, m), 1.46–1.17 (4 H, overlapped), 1.04 (2H, m), 0.90 (3H, d, J = 6.4 Hz), 0.83 (3H, t, J = 7.2 Hz), 0.82 (3H, d, J = 6.0 Hz) and 0.74 (3H, d, J = 6.4 Hz); 13C NMR (100 MHz, CDCl3) δ 164.4, 163.4, 156.6, 138.2, 132.8, 130.5, 130.5, 130.0, 125.2, 116.6, 115.6, 115.6, 110.1, 92.7, 77.2 44.7, 32.3, 31.9, 31.9, 31.0, 29.6, 28.8, 20.6, 19.6, 17.5, 11.6 and 11.2; ESI-MS (+) mode m/z 440 [M + H]+ and ESI-MS (−) mode m/z 438 [M − H]−. These data showed good correlation with those of N-demethylsambutoxin [8].
(−)-6-Deoxyoxysporidinone (4): pale yellow oil; \({[{\rm{\alpha}}]^{23}_{\mathrm{D}}}\) –100.5 (c = 0.1, MeOH); 1H NMR (400 MHz, CDCl3) δ 10.49 (–OH, br s), 7.14 (1H, s), 5.20 (1H, d, J = 9.2 Hz), 4.94 (1H, d, J = 10.8 Hz), 3.50 (1H, d, J = 10.0 Hz), 3.44 (3H, s), 2.89 (2H, m), 2.48 (1H, m), 2.36–2.03 (8H, overlapped), 1.89 (1H, m), 1.68 (1H, m), 1.65 (3H, s), 1.44–1.17 (4H, overlapped), 1.04 (2H, m), 0.91 (3H, d, J = 6.8 Hz), 0.83 (6H, overlapped), 0.74 (3H, d, J = 6.4 Hz); 13C NMR (100 MHz, CDCl3) δ 211.7, 162.1, 161.1, 138.1, 133.2, 129.9, 117.5, 111.1, 92.4, 78.0, 70.2, 44.6, 37.2, 36.8, 36.8, 36.1, 36.0, 32.4, 31.9, 31.9, 30.6, 29.6, 28.8, 20.6, 19.5, 17.5, 11.8 and 11.1; ESI-MS (+) mode m/z 496 [M + Na]+ and ESI-MS (−) mode m/z 472 [M − H]−. These data showed good correlation with those of (−)-6-deoxyoxysporidinone [9].
Evaluation of antimicrobial activity using the agar diffusion method
All of evaluation was performed by the paper disc method [4] using 6 mm discs.
Evaluation of MIC values of 1–4
We evaluated the MIC of each compound against yeasts using a broth microdilution method [23]. The test compounds were dissolved in MeOH. The yeasts (1.0 × 104 cells/100 μl/well in a 96-well plate) were cultured in either YPD or YPG liquid medium with 1 × 10−4% TWEEN 20 (Sigma-Aldrich, Co., St. Louis, USA) in the presence of various concentrations of 1–4 for 2 and 3 days, respectively. MICs were evaluated by measuring OD600 on a Corona Grating Microplate Reader SH-9000 (Corona Electric, Ibaraki, Japan).
Evaluation of the inhibition of ADP-uptake/ATP-release in isolated mitochondria
The measurement of ADP-uptake/ATP-release in isolated mitochondria was performed according to the method reported by Lousa et al. [16]. Isolated mitochondria (50 µg of proteins/ml) were suspended in 2.5 ml of reaction buffer (0.60 M mannitol, 0.10 mM EGTA, 2.0 mM MgCl2, 10 mM KPi, 5.0 mM α-ketoglutarate, and 10 mM Tris-HCl, pH 7.4) at 30 °C in the presence of an ATP detecting system (2.5 mM glucose, hexokinase (1.7 E.U.), glucose-6-phosphate dehydrogenase (0.85 E.U.), 0.20 mM NADP+ and 10 µM Ap5A). Externally added ADP (25 mM) initiated exchange reaction with ATP synthesized in mitochondrial matrix. The formation of NADPH, which is proportional to ATP efflux, was measured continuously for 10 min spectrophotometrically on a Shimadzu UV3000 (340 nm; ɛ = 6.2 mM−1 cm−1).
Evaluation of NADH-cytochrome c oxidoreductase activity
Isolated yeast mitochondria were permeabilized by repeated freeze-thawing in 50 mM KPi buffer (pH 7.4) to improve the accessibility of substrates (NADH and cytochrome c). The NADH-cytochrome c oxidoreductase activity was followed by the reduction of cytochrome c (550–540 nm; ε = 21 mM−1 cm−1) in buffer (2.5 ml) containing 50 mM KPi (pH 7.4), 4.0 mM KCN and 50 µM cytochrome c (from horse heart, Sigma-Aldrich) at 30 °C. Protein concentration was set to 12 μg ml−1, and the reaction was initiated by the addition of NADH (the final concentration of 50 µM).
Cytotoxic evaluation
Cytotoxicity of 1–4 was evaluated using three floating cell lines, HL-60 (human promyelocytic leukaemia cell line), Jurkat (human acute lymphocytic leukaemia cell line) and THP-1 (human acute monocytic leukaemia cell line), along with five adherent cell lines, HeLa S3 (human cervical cancer cell line), HT29 (human colorectal adenocarcinoma cell line), A549 (human adenocarcinoma cell line derived from lung cancer), H1299 (human non-small cell lung carcinoma cell line derived from lymph nodes) and Panc1 (human cell line derived from pancreatic cancer).
Culture conditions were as follows: floating cell lines [RPMI medium (Nacarai Tesque, Inc, Kyoto, Japan) added to 10% FBS, 1% sodium pyruvate and 1% penicillin streptomycin, 37 °C, under 5% CO2] and adherent cell lines [DMEM medium (Wako Pure Chemical Industries, Osaka, Japan) added to 10% FBS and 1% penicillin streptomycin, 37 °C, under 5% CO2].
The cultivated floating (3 × 105 cells per well) and adherent (5 × 103 cells per well) cell lines were seeded in 96-well plates. After culturing overnight, MeOH solutions of 1–4 were added into each well. After 2 days of incubation at 37 °C, WST-8 solution was added to each well and kept for 4 h at 37 °C. The absorbance of each well was measured on a Corona Grating Microplate Reader SH-9000 at 460 nm.
References
Balba H. Review of strobilurin fungicide chemicals. J Environ Sci Health B. 2007;42:441–51.
Eisinger M, Almog Y. Primidifen intoxication. Ann Emerg Med. 2003;42:289–91.
Molina JR, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24:1036–46.
Watanabe Y, et al. Decatamariic acid, a new mitochondrial respiration inhibitor discovered by pesticidal screening using drug-sensitive Saccharomyces cerevisiae. J Antibiot. 2017;70:395–9.
Chinen T, Nagumo Y, Usui T. Construction of a genetic analysis-available multidrug sensitive yeast strain by disruption of the drug efflux system and conditional repression of the membrane barrier system. J Gen Appl Microbiol. 2014;60:160–2.
Suga T, et al. Ascosteroside C, a new mitochondrial respiration inhibitor discovered by pesticidal screening using recombinant Saccharomyces cerevisiae. J Antibiot. 2015;68:649–52.
Kim JC, Lee YW, Tamura H, Yoshizawa T. Sambutoxin: a new mycotoxin isolated from Fusarium sambucinum. Tetrahedron Lett. 1995;36:1047–50.
Jayasinghe L, Abbas HK, Jacob MR, Herath WH, Nanayakkara NP. N-Methyl-4-hydroxy-2-pyridinone analogues from Fusarium oxysporum. J Nat Prod. 2006;69:439–42.
Zhan J, Burns AM, Liu MX, Faeth SH, Gunatilaka AA. Search for cell motility and angiogenesis inhibitors with potential anticancer activity: beauvericin and other constituents of two endophytic strains of Fusarium oxysporum. J Nat Prod. 2007;70:227–32.
Kemami Wangun HV, Hertweck C. Epicoccarines A, B and epipyridone: tetramic acids and pyridone alkaloids from an Epicoccum sp. associated with the tree fungus Pholiota squarrosa. Org Biomol Chem. 2007;5:1702–5.
Xu W, Cai X, Jung ME, Tang Y. Analysis of intact and dissected fungal polyketide synthase-nonribosomal peptide synthetase in vitro and in Saccharomyces cerevisiae. J Am Chem Soc. 2010;132:13604–7.
Shang Z, et al. New PKS-NRPS tetramic acids and pyridinone from an Australian marine-derived fungus, Chaunopycnis sp. Org Biomol Chem. 2015;13:7795–802.
Fukuda T, et al. Tolypoalbin, a new tetramic acid from Tolypocladium album TAMA 479. J Antibiot. 2015;68:399–402.
Suga T, et al. Trichopolyn VI: a new peptaibol insecticidal compound discovered using a recombinant Saccharomyces cerevisiae screening system. J Gen Appl Microbiol. 2015;61:82–7.
Kawai K, Suzuki T, Kitagawa A, Kim JC, Lee YW. A novel respiratory chain inhibitor, sambutoxin from Fusarium sambucinum. Cereal Res Commun. 1997;25:325–6.
De Marcos Lousa C, Trézéguet V, Dianoux AC, Brandolin G, Lauquin GJ. The human mitochondrial ADP/ATP carriers: kinetic properties and biogenesis of wild-type and mutant proteins in the yeast S. cerevisiae. Biochemistry. 2002;41:14412–20.
Unten Y, et al. Pentendiol-type compounds specifically bind to voltage-dependent anion channel 1 in Saccharomyces cerevisiae mitochondria. Biochemistry. 2019;58:1141–54.
Halo L, et al. Late stage oxidations during the biosynthesis of the 2-pyridone tenellin in the entomopathogenic fungus Beauveria bassiana. J Am Chem Soc. 2008;130:17988–96.
Mo X, Li Q, Ju J. Naturally occurring tetramic acid products: isolation, structure elucidation and biological activity. RSC Adv. 2014;4:50566–93.
Quek NC, et al. The novel equisetin-like compound, TA-289, causes aberrant mitochondrial morphology which is independent of the production of reactive oxygen species in Saccharomyces cerevisiae. Mol Biosyst. 2013;9:2125–33.
Köning T, Kapus A, Sarkadi B. Effects of equisetin on rat liver mitochondria: evidence for inhibition of substrate anion carriers of the inner membrane. J Bioenerg Biomembr. 1993;25:537–45.
Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Sakai K, et al. Pastiocandin, a new papulacandin class antibiotic isolated from Pestalotiopsis humus. J Antibiot. 2018;71:1031–5.
Acknowledgements
This research was supported financially by The Public Foundation of Elizabeth Arnold–Fuji, Japan. We are grateful to Dr Kenichiro Nagai and Ms Noriko Sato, School of Pharmacy, Kitasato University, for help in obtaining NMR and MS data.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sakai, K., Unten, Y., Iwatsuki, M. et al. Fusaramin, an antimitochondrial compound produced by Fusarium sp., discovered using multidrug-sensitive Saccharomyces cerevisiae. J Antibiot 72, 645–652 (2019). https://doi.org/10.1038/s41429-019-0197-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0197-5
