
Abstract
The synthesis of carboxylated derivatives of poly(isobutylene-co-isoprene) (isobutylene–isoprene rubber, IIR) with substitution levels ranging from 1 to 4 mol% and different spacer lengths was accomplished through azide–alkyne Huisgen cycloaddition. Azido-functionalized IIR was first prepared by reacting brominated IIR with sodium azide to full conversion in a 90:10 tetrahydrofuran/N,N-dimethylacetamide mixture. The click reaction of azido-functionalized IIR with acetylenic acids, which was carried out using the copper(I) bromide/N,N,N′,N″,N″-pentamethyldiethylenetriamine catalyst system in tetrahydrofuran, yielded carboxylated IIRs. The products were characterized by 1H NMR and FT-IR spectroscopy, and their molecular weight was determined by size exclusion chromatography analysis. The conversion to carboxylated groups reached up to 100% as determined by NMR spectroscopy but was highly dependent on the type of solvent and the amounts of catalysts and reactants used in the procedures.
Similar content being viewed by others
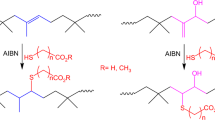

Introduction
Copolymers of isobutylene and isoprene, also known as isobutylene–isoprene rubber (IIR), have been used for multiple applications in different areas due to their unusual physical properties. The characteristics of IIR include excellent impermeability to air, good electrical resistivity, excellent resistance to moisture, oxidation and heat, and good flex properties resulting from its low unsaturation level [1,2,3,4]. The range of properties displayed by IIR was further extended by introducing a low concentration of ionic moieties, such as sulfonic acid groups bound along the polymer chains, to produce ionomers [5]. The synthesis of phosphonium and ammonium salts based on IIR has also been reported. Such derivatives are typically prepared from brominated IIR (BIIR) containing ca. 1 mol% of allylic bromide moieties [6]. The allylic bromide in BIIR can be displaced by a wide variety of nucleophiles since it is a good leaving group [7,8,9,10]. The postpolymerization chemical modification of BIIR by nucleophilic substitution is therefore useful to synthesize the derivatives (such as ionomers) that cannot be obtained by copolymerization.
Click chemistry is a powerful tool for polymer modification. Introduced by Sharpless in 2001 [11], the copper-catalyzed azide–alkyne Huisgen 1,3-dipolar cycloaddition reaction has attracted much interest because it is facile and selective, produces a high yield under mild conditions, and has an excellent tolerance for functional groups [12,13,14,15]. In contrast to the traditional 1,3-dipolar Huisgen cycloaddition, the copper-catalyzed azide–alkyne click reaction can be carried out at room temperature in a wide range of solvents including water, tetrahydrofuran (THF), and ethanol [16,17,18]. Additionally, the Cu(I)-catalyzed azide–alkyne cycloaddition reaction is usually much (≥107 times) faster than the uncatalyzed reaction [19] and is rather insensitive to the steric and electronic properties of the groups attached to the azide and alkyne centers [17]. In fact, primary, secondary, or even tertiary, electron-rich or electron-deficient azides usually react well with substituted terminal alkynes. Moreover, the triazole unit formed in the reaction has several interesting properties, including a high chemical stability (being inert to hydrolytic, oxidizing, and reducing conditions), good hydrogen bonding ability, and aromatic character [20]. The triazole group can also interact effectively with biological molecules, in analogy to amide bonds [21, 22]. Considering these factors, the azide–alkyne click reaction seems to be a good candidate for generating functional IIR derivatives.
Over the past few decades, different synthetic methods have been applied to modify IIR and improve its properties [6, 23,24,25,26]. Among these, the synthesis of carboxylated IIR is of particular interest because polar groups like carboxylate moieties enhance the adhesion of IIR, which is very hydrophobic in nature [27,28,29]. The introduction of carboxylate groups also increases the modulus and tensile strength of rubbery materials [30,31,32,33] and allows for further functionalization/modification of IIR. Gillies and coworkers recently reported the synthesis of carboxylated IIR via the ring opening of cyclic anhydrides or the introduction of poly(carboxylic acid)s [34]; however, to the best of our knowledge, there has been no report on the synthesis of carboxylated IIR by click chemistry. This study shows that under the appropriate conditions, azide–alkyne click chemistry can generate carboxylated IIR derivatives in high yields.
Experimental section
1H NMR spectra were recorded on a Bruker 300 MHz spectrometer (Milton, ON, Canada) in CDCl3 with tetramethylsilane (TMS) as the internal standard. FT-IR spectra were acquired on a Bruker Vector 22 FT-IR spectrophotometer from 400 to 4000 cm−1. The molecular weight of the IIR samples was determined on a Viscotek GPCmax size exclusion chromatography (SEC) instrument (Montreal, QC, Canada) with a VE 2001 GPC solvent/sample module. The unit was equipped with a TDA 305 triple detector array with refractive index (RI), light scattering, and viscometer detectors, as well as a UV detector (Viscotek 2600) and three PolyAnalytik organic mixed bed columns (London, ON, Canada), for an overall polystyrene molecular weight range of 103–107 g/mol. The samples were analyzed in THF at a flow rate of 1 mL/min.
Copper bromide (99.99%), sodium azide (99%), bromine (≥99.5%), sodium hydroxide (≥97%), N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA, 99%), cyclohexanone (≥99%), 10-undecenoic acid (95%), and PEG-200 were all purchased from Aldrich and used without further purification. Hydrogen peroxide (30%) was purchased from Fisher Scientific. Brominated poly(isobutylene-co-isoprene) (BIIR) samples containing 1, 1.9, or 4 mol% of brominated isoprene units (all of experimental grade with Mw = 280–380 kg/mol) were supplied by Lanxess Canada Inc. Solvents such as THF, N,N-dimethylacetamide (DMA), N,N-dimethylformamide (DMF), methanol, and chloroform were purchased from Caledon and were used as received.
Azidation of BIIR (4.0 mol% brominated isoprene units)
In a 250-mL round-bottomed flask (RBF), BIIR (1 g, 6.74 × 10−4 mol brominated isoprene units) was dissolved in 90 mL of THF before adding 10 mL of DMA. Sodium azide (0.88 g, 0.0135 mol, 20 eq with respect to the brominated isoprene units) was added and the mixture was stirred on a magnetic stirrer at room temperature for 4 days. The solution was then filtered to remove excess sodium azide, concentrated, and precipitated into methanol. The product was further purified twice by dissolution in 25 mL of THF followed by precipitation into methanol. Recovery yield = 0.87 g (90%), substitution level 4.0 mol% (100% conversion). 1H NMR (300 MHz, CDCl3) δ ppm: 5.45 (1H, b, C=CH), 5.16 and 5.03 (2H of isoprene, s), 3.82–3.61 (3H, bm, CH–N3 and CH2–N3), 2.4–0.75 (aliphatic protons from isobutylene and isoprene units). Note: Caution must be applied in designing reactions with sodium azide since it is dangerous, particularly in large quantities. Inorganic azides are highly toxic and are potentially explosive compounds. Metal spatulas should not be used to handle sodium azide to avoid the formation of shock-sensitive heavy metal azides. Solutions of sodium azide may be absorbed through the skin and can generate highly toxic hydrazoic acid when exposed to water. Therefore, the reactions should only be carried out in a well-ventilated fume hood and with appropriate personal protective equipment.
Click reaction of azidated IIR (4.0 mol% azidated isoprene units) with 4-phenyl-1-butyne
In a 100-mL RBF, PMDETA (0.3 g, 1.73 × 10−3 mol, 5 eq with respect to the azide) and 4-phenyl-1-butyne (0.23 g, 1.73 × 10−3 mol, 5 eq with respect to the azide) were added to a solution of azidated IIR (0.5 g in 50 mL of THF, 3.46 × 10−4 mol azide moieties), and the flask was purged with nitrogen for 30 min. CuBr (0.1 g, 7.01 × 10−4 mol, 2 eq with respect to the azide) was added to this mixture against the nitrogen flow. The reaction was allowed to proceed at room temperature for 3 days, the THF was evaporated, and the residue was redissolved in CHCl3 (100 mL) for extraction with water (5 × 50 mL) to remove the copper salts. The CHCl3 solution was then concentrated, precipitated into methanol or acetone, and further purified twice by dissolution in 15 mL of THF and precipitation into methanol or acetone. Recovery yield = 0.89 g (82%), substitution level 4.0 mol% (100% conversion). 1H NMR (300 MHz, CDCl3) δ ppm: 7.37–7.70 (5H, Ar), 6.98 (1H, s, N–CH=C), 5.43 (1H, b, C=CH), 5.18–5.03 (3H, b, CH–N3 and CH2–N3), 5.12 and 5.01 (2H of isoprene, s), 3.01 (4H, b, –CH2–CH2–Ar), 2.35–0.65 (aliphatic protons of isobutylene and isoprene units).
Synthesis of 5-hexenoic acid
Hydrogen peroxide (30% aqueous solution, 115 mL, 1 mol) was added to a stirred solution of cyclohexanone (49 g, 0.5 mol) in methanol (50 mL) over 30 min at 20–25 °C. This mixture was then added to a stirred solution of ferrous sulfate heptahydrate (138 g, 0.9 mol) and cupric sulfate pentahydrate (125 g, 0.8 mol) in water (900 mL) over 2 h while maintaining the mixture at 18–20 °C. The reaction was then stirred at the same temperature for 1 h before extraction with diethyl ether (6 × 100 mL), concentration to 150 mL, and washing with 20% aq. NaOH (4 × 50 mL). The alkaline extract was acidified to pH 2 with sulfuric acid and extracted with diethyl ether (3 × 100 mL). Removal of the solvent under reduced pressure yielded the crude 5-hexenoic acid as a clear oil, which was purified by distillation. Yield: 40 g (70%). 1H NMR (300 MHz, CDCl3) δ ppm: 11.1 (br s, 1H, CH2=CH–CH2–CH2–CH2–COOH), 5.74 (m, 1H, CH2=CH–CH2–CH2–CH2–COOH), 5.01 (dd, 2H, CH2=CH–CH2–CH2–CH2–COOH), 2.35 (t, 2H, CH2=CH–CH2–CH2–CH2–COOH), 2.08 (q, 2H, CH2=CH–CH2–CH2–CH2–COOH), 1.72 (quint, 2H, CH2=CH–CH2–CH2–CH2–COOH).
Synthesis of 5,6-dibromohexanoic acid
A solution of bromine (55 g, 0.34 mol) in dichloromethane (70 mL) was added at −40 °C with vigorous stirring over 1 h to a solution of 5-hexenoic acid (39.3 g, 0.34 mol) in CH2Cl2 (130 mL). The mixture was stirred at the same temperature for 1 h. Removal of the solvent yielded 5,6-dibromohexanoic acid as an orange-yellow oil. Yield: 93.5 g (99%). The product was used in the next step without additional purification. 1H NMR (300 MHz, CDCl3) δ ppm: 10.61 (br s, 1H, BrCH2–CHBr–CH2–CH2–CH2–COOH), 4.10 (m, 1H, BrCH2–CHBr–CH2–CH2–CH2–COOH), 3.83 (dd, 1H, BrCH2–CHBr–CH2–CH2–CH2–COOH), 3.61 (t, 1H, BrCH2–CHBr–CH2–CH2–CH2–COOH), 2.42 (t, 2H, BrCH2–CHBr–CH2–CH2–CH2–COOH), 2.28–2.10, 2.01–1.65 (both m, 1H and 3H, BrCH2–CHBr–CH2–CH2–CH2–COOH).
Synthesis of 5-hexynoic acid
A mixture of PEG-200 (180 mL) and NaOH pellets (36 g, 0.91 mol) was stirred in an oil bath at 80 °C until all the NaOH dissolved and 5,6-dibromohexanoic acid (50 g, 0.18 mol) was added dropwise to this solution over 30 min. The mixture was further heated at 80–85 °C for 3–4 h before adding water (150 mL), acidifying with HCl to pH 2 and cooling. The mixture was then extracted with diethyl ether and concentrated. The combined organic layers were washed with water and then a brine solution and dried. The crude product was distilled in a Kugelrohr apparatus under reduced pressure (80 °C, 5 Torr) to yield a colorless oil. Yield = 14 g (70%). 1H NMR (300 MHz, CDCl3) δ ppm: 11.56 (br s, 1H, CH≡C–CH2–CH2–CH2–COOH), 2.49 (t, 2H, CH≡C–CH2–CH2–CH2–COOH), 2.26 (m, 2H, CH≡C–CH2–CH2–CH2–COOH), 1.96 (t, 1H, CH≡C–CH2–CH2–CH2–COOH), 1.82 (quint, 2H, CH≡C–CH2–CH2–CH2–COOH). 13C NMR (75 MHz, CDCl3) δ ppm: 179.40 (C), 83.0 (C), 69.5 (CH), 32.6 (CH2), 23.1 (CH2), 17.7 (CH2). HRMS (ESI−) calculated for C6H7O2 [M − H]− 111.0446, found 111.0439.
Synthesis of 10,11-dibromoundecanoic acid
This acid was synthesized from 10-undecenoic acid (25 g) by the procedure described above for 5,6-dibromohexanoic acid. Removal of the solvent yielded 10,11-dibromoundecanoic acid as an orange-yellow crystalline compound. The product was used in the next step without additional purification. Yield = 46.2 g (99%). 1H NMR (300 MHz, CDCl3) δ ppm: 10.66 (br s, 1H, BrCH2–CHBr–CH2–(CH2)5–CH2–CH2–COOH), 4.15 (m, 1H, BrCH2–CHBr–CH2–(CH2)5–CH2–CH2–COOH), 3.82 (dd, 1H, BrCH2–CHBr–CH2–(CH2)5–CH2–CH2–COOH), 3.61 (t, 1H, BrCH2–CHBr–CH2–(CH2)5–CH2–CH2–COOH), 2.33 (t, 2H, BrCH2–CHBr–CH2–(CH2)5–CH2–CH2–COOH), 2.10, 1.76 (both m, 1H and 1H, BrCH2–CHBr–CH2–(CH2)5–CH2–CH2–COOH), 1.61 (m, 2H, BrCH2–CHBr–(CH2)5–CH2–CH2–CH2–COOH), 1.30 (m, 10H, BrCH2–CHBr–(CH2)5–CH2–CH2–CH2–COOH).
Synthesis of 10-undecynoic acid
The procedure used was as described for 5-hexynoic acid. The crude product was distilled in a Kugelrohr apparatus under reduced pressure (110 °C, 1 Torr) to yield a colorless oil, which solidified to a white powder at room temperature. Yield = 18.4 g (75%). 1H NMR (300 MHz, CDCl3) δ ppm: 11.37 (br s, 1H, CH≡C–CH2–(CH2)5–CH2–CH2–COOH), 2.32 (t, 2H, CH≡C–CH2–(CH2)5–CH2–CH2–COOH), 2.14 (m, 2H, CH≡C–CH2–(CH2)5–CH2–CH2–COOH), 1.91 (s, 1H, CH≡C–CH2–(CH2)5–CH2–CH2–COOH), 1.60, 1.49 (both m, 1H and 1H, CH≡C–CH2–(CH2)5–CH2–CH2–COOH), 1.28 (m, 10H, CH≡C–CH2–(CH2)5–CH2–CH2–COOH). 13C NMR (75 MHz, CDCl3) δ ppm: 180.4 (C), 84.7 (C), 68.1 (CH), 34.1 (CH2), 29.1 (CH2), 28.9 (CH2), 28.8 (CH2), 28.6 (CH2), 28.4 (CH2), 24.6 (CH2), 18.4 (CH2). HRMS (ESI−) calculated for C11H17O2 [M − H]− 181.1229, found 181.1226.
Synthesis of carboxylated IIR
The carboxylated IIR samples were prepared using BIIR containing 1.0–4.0 mol% of azidated isoprene units and the different acetylenic acids synthesized, following the procedure described above for 4-phenyl-1-butyne. Recovery yield for the reaction of IIR (1 mol% azidated isoprene units) with 10-undecynoic acid = 88%, substitution level 1 mol% (100% conversion). 1H NMR (300 MHz, CDCl3) δ ppm: 7.15 (1H, s, N–CH=C), 5.49 (1H, b, C=CH), 5.06 (3H, b, CH–N3, CH2–N3 and IIR), 4.93 and 4.84 (2H of isoprene, s), 2.78–2.36 (4H, C–CH2– and –CH2–C=O), 2.05–0.73 (aliphatic protons of isobutylene and isoprene units). Recovery yield for the reaction of IIR (1.9 mol% azidated isoprene units) with 10-undecynoic acid = 85%, substitution level 1.9 mol% (100% conversion). 1H NMR (300 MHz, CDCl3) δ ppm: 7.15 (1H, s, N–CH=C), 5.48 (1H, b, C=CH), 5.10 (3H, b, CH–N3 and CH2–N3), 4.93 and 4.85 (2H of isoprene, s), 2.68–2.40 (4H, C–CH2– and –CH2–C=O), 2.03–0.75 (aliphatic protons of isobutylene and isoprene units). Recovery yield for the reaction of IIR (4 mol% azidated isoprene units) with 10-undecynoic acid = 85%, substitution level 4 mol% (100% conversion). 1H NMR (300 MHz, CDCl3) δ ppm: 7.17 (1H, s, N–CH=C), 5.46 (1H, b, C=CH), 5.05 (3H, b, CH–N3 and CH2–N3), 4.91 and 4.81 (2H of isoprene, s), 2.65–2.34 (4H, C–CH2– and –CH2–C=O), 1.97–0.70 (aliphatic protons of isobutylene and isoprene units). The reactions of azidated IIR with 5-hexynoic acid produced yields similar to 10-undecynoic acid and 100% conversion.
Results and discussion
The copper-catalyzed azide–alkyne reaction based on the Huisgen 1,3-dipolar cycloaddition was employed to synthesize carboxylated derivatives of IIR. The synthesis process included two main steps, namely, the conversion of BIIR to the corresponding azide-functionalized polymer (IIR-N3), followed by clicking with the acetylenic acids. To optimize the reaction conditions, BIIR was selected to contain ~4 mol% of brominated isoprene units with an exomethylene structure. Azidated IIR was obtained by treating BIIR with a large excess of sodium azide (20 equiv. with respect to the brominated isoprene units) in THF/DMA 90:10 v/v under vigorous stirring for 3–4 days. The procedure used for the synthesis of the click-modified IIR and the corresponding 1H NMR spectra are provided in Scheme 1 and Fig. 1, respectively. The 1H NMR spectrum for BIIR (Fig. 1a) had two singlets at 5.39 and 5.02 ppm, which corresponded to the exomethylene structural unit, and a peak at 4.32 ppm for the allylic bromide proton. Upon azidation, the peak at 4.32 ppm vanished, and a new broad peak appeared at 3.82–3.61 ppm, indicating the complete conversion of bromide to azide. An azidation level of 4.0 mol% was calculated from the 1H NMR spectrum, corresponding to 100% conversion. The azidation reaction also led to the isomerization of IIR, as seen by the appearance of a peak at 5.45 ppm, corresponding mainly to an exomethylene microstructure (~55%) and approximately 45% of the endo isomer based on the peaks at 5.45 and 5.16 ppm [35].

Optimization of the azidation and azide–alkyne click reactions

1H NMR spectra for the optimization of the azidation and azide–alkyne click reactions: a BIIR; b IIR-N3; c IIR-Ph. The peak assignments are provided in Scheme 1
The azidation reaction was also attempted in THF/DMF mixtures, but this approach was found to produce gel particles, and the azidated polymer obtained was unstable. It should be considered that the BIIR samples used had very high molecular weights (M > 105 g/mol, corresponding to degrees of polymerization X > 1700), and the formation of a gel is expected when a mole fraction of only 1/1700 ≈ 0.06% of the structural units is involved in cross-linking reactions. This is well below the detection limit of any of the analytical techniques currently available for polymers; hence, any discussion of the nature of reactions leading to gel formation can only be speculative. Two hypotheses are suggested for the formation of gel particles in the THF/DMF mixtures. The photochemical degradation of DMF produces minute amounts of CN−/HCN as an impurity [36]. When DMF is used as a solvent for azidation, this can lead to the transformation of some alkyl halide groups to alkyl nitriles, which may react with the azide functionalities on other chains to form tetrazole cross-links (Scheme S1a). However, this possibility is somewhat unlikely since the formation of tetrazoles from azides and nitriles typically requires a temperature of approximately 100–150 °C [37], while the azidation reaction was performed at room temperature in the present case. Another (more likely) explanation for the gel formation is the presence of trace amounts of dimethylamine in DMF, arising from hydrolysis [38]. This compound reacts with two allylic bromide functionalities on different chains to yield quaternary amine cross-links (Scheme S1b). The reactions that were carried out with 5 or 10 equiv. of sodium azide in the THF/DMF mixtures did not lead to full azidation, as evidenced by the NMR analysis (Supporting Information, Figure S1). The slower rate of azidation achieved under these conditions made it more likely for competing side reactions involving nitrile or amine groups to induce gel formation. The choice of solvent and the amount of sodium azide used are therefore important parameters to be considered for azidation.
The click reaction of IIR-N3 with 4-phenyl-1-butyne served as the model reaction using the copper(I) bromide/N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA) catalyst system at room temperature. To assess the efficiency of the click reaction, a series of reactions were carried out by varying parameters such as the amount of catalyst and ligand and the solvent system used (Table 1). A THF/DMA 90:10 solvent mixture was investigated initially since DMA is useful for increasing the solubility of CuBr. Unfortunately, at high DMA concentrations, the IIR derivatives were insoluble. As shown for Entry 1 in Table 1, a reaction using only 1 equiv. of CuBr, PMDETA, and alkyne (1:1:1 with respect to the azide) in THF/DMA did not yield any product. A 1:2:2 ratio led to the same result, while increasing the amounts of the reagents to 2:2:2 led to gelation of the reaction in less than 10 min. It was therefore concluded that there was a strong correlation between the type of solvent used and the amount of catalyst required in the reaction [39, 40]. To confirm this, THF and toluene were also explored as solvents while varying the ratios of CuBr, PMDETA, and alkyne. The experimental details provided in Table 1 show that in THF, for a 2:2:2 ratio of CuBr:PMDETA:alkyne, the reaction proceeded without gelation but only reached 20% conversion in 2 days. Interestingly, the amount of 4-phenyl-1-butyne incorporated increased significantly in THF upon increasing the ratio to 2:5:5 (90% in 2 days). The progress of the reaction could also be monitored through a color change from pale green to orange, probably due to the formation of a complex of triazole with CuBr and PMDETA. Whenever a color change was not observed, the reaction failed. Further increase in substitution level was achieved when the reaction was allowed to proceed for 3 days. No gelation was observed under these conditions, and the product obtained had excellent solubility even in comparison to its precursor. While toluene is a good solvent for IIR, the click reactions in toluene yielded mixed and irreproducible results. This again demonstrates the strong influence of the solvent used on the success of the azide–alkyne click reactions [41]. Excess PMDETA also ensured the complete dissolution of CuBr, which led to homogeneous reactions, in contrast to lower ratios. A higher ligand concentration is expected to enhance the reaction rate by increasing the solubility of CuBr and protecting Cu(I) from oxidation, thereby maintaining a high concentration of the catalytically active complex throughout the reaction [42, 43].
The 1H NMR spectrum for the product of the reaction between the azidated butyl rubber and 4-phenyl-1-butyne (IIR-Ph) is provided in Fig. 1c. The click reaction resulted in the disappearance of the resonance at 3.82–3.61 ppm, corresponding to the protons close to the azide group, and the appearance of aromatic proton signals at 7.24–7.11 ppm and at 6.97 ppm [44] for the proton on the triazole ring. The broad singlet at 3.01 ppm is attributed to the aliphatic protons from 4-phenyl-1-butyne. On the basis of these results, it was concluded that a CuBr:PMDETA:alkyne ratio of 2:5:5 in THF worked best for the click reaction with azidated IIR. The FT-IR spectra obtained for the products (Figure S2) also show the appearance of a strong azide stretching vibration at 2092 cm−1 upon azidation, which disappeared after the click reaction.
Synthesis of acetylenic acids
The acetylenic acids were synthesized by modifying the procedure of Starostin et al. [45]. The synthesis of 5-hexynoic acid (6-COOH) is described in Scheme 2. Cyclohexanone was first treated with 30% H2O2 to form cyclohexanone peroxide, which upon reaction with FeSO4/CuSO4 decomposed to produce 5-hexenoic acid in a 70% yield. The addition of bromine to 5-hexenoic acid at −40 °C yielded 5,6-dibromohexanoic acid, which could be dehydrobrominated with NaNH2, KOH, or NaOH. While these bases are most commonly used for that purpose, NaNH2 and KOH have disadvantages: Impurities in NaNH2 can decrease its reactivity and cause explosions, and KOH promotes the migration of the acetylenic bond to the center of the chain [45, 46]. Consequently, we preferred using NaOH, with PEG-200 acting as a phase transfer catalyst. This method was found to favor the formation of 5-hexynoic acid compared with an aqueous NaOH solution (Figure S3). The dehydrobromination of 5,6-dibromohexanoic acid with NaOH yielded a mixture of 6-bromohex-5-enoic acid and 5-hexynoic acid, which were separated by distillation in a Kugelrohr apparatus under reduced pressure to yield 5-hexynoic acid as a colorless liquid in a 70% yield. It should be noted that the reaction temperature must be maintained between 80 and 85 °C, because higher temperatures lead to the formation of other isomeric products and 4-hexynoic acid. The 1H NMR spectra corresponding to the different steps of the 5-hexynoic acid synthesis are provided as Supporting Information (Figure S4), and the 13C NMR spectrum for the final product is shown in Figure S5. The synthesis of 10-undecynoic acid (11-COOH) started from 10-undecenoic acid by the same procedure described above. The 1H NMR and 13C NMR spectra for the products are shown in Figures S6 and S5, respectively. The spectral characteristics for these molecules were similar to previously reported data [47].

Synthesis of 5-hexynoic acid
Synthesis of carboxylated IIR
Samples of carboxylated IIR were prepared as depicted in Scheme 3, using the optimized reaction conditions described above. BIIR substrates with three different brominated isoprene unit contents, namely, 1, 1.9, and ~4 mol%, were used for this part of the investigation. The reaction of BIIR having 1.9 mol% brominated units with 5-hexynoic acid and 10-undecynoic acid is discussed below. The progress of these reactions was monitored by 1H NMR and FT-IR spectroscopy. The 1H NMR spectra for the reaction are provided in Fig. 2, starting with BIIR for comparison. The peak marked with an asterisk corresponds to nonbrominated isoprene units [48]. Similarly to the 4.0 mol% substrate, the azidation of BIIR in THF/DMA proceeded to full conversion. The success of the azidation reaction was further confirmed by FT-IR analysis, which showed the appearance of a peak at 2092 cm−1 (Fig. 3) [49]. Upon reaction with the acetylenic acids, a new signal appeared at 7.15 ppm for the proton on the 1,2,3-triazole ring [44], as well as a broad signal at 2.81–2.29 ppm for the aliphatic protons on the substituent closer to the triazole ring and the carboxylic acid group (Fig. 2c, d). The signal at 3.81–3.64 ppm in the azidated product disappeared, again confirming the purity of the product obtained. The disappearance of the strong azide stretching vibration at 2092 cm−1 in the FT-IR spectrum, as well as the appearance of a peak at 1720 cm−1 for the carboxylic acid C=O, further confirmed the success of the coupling reaction (Fig. 3c, d). Quantification of the substitution level by the 1H NMR analysis of the reaction products with both 5-hexynoic acid and 10-undecynoic acid confirmed the full conversion. The click reactions starting from BIIR with 1 and 4 mol% brominated isoprene units yielded results similar to the other reactions, except for their relative signal intensities. The 1H NMR and FT-IR spectra for these products are provided as Supporting Information (Figures S7–S9).

Synthesis of carboxylated IIR by the azide–alkyne click reaction

1H NMR spectra for the synthesis of carboxylated IIR (1.9 mol% brominated isoprene units) by the azide–alkyne click reaction. a BIIR, b IIR-N3, c IIR-6COOH, and d IIR-11COOH. The peak assignments are provided in Scheme 3

FT-IR spectra for the synthesis of carboxylated IIR (1.9 mol% brominated isoprene units) by the azide–alkyne click reaction. a BIIR, b IIR-N3, c IIR-6COOH, and d IIR-11COOH
SEC analysis was carried out to monitor potential changes in the molecular weight distribution of the products resulting from either cross-linking or chain cleavage. A series of SEC traces starting with BIIR containing 1.9 mol% brominated units and the different derivatives synthesized are provided in Fig. 4. The SEC analysis of IIR is known to be problematic [50, 51], and it was indeed difficult to prepare solutions of BIIR and the carboxylated IIR derivatives concentrated enough for the SEC measurements (0.5 mg/mL) while avoiding clogging of the columns. It was indeed reported that THF is a poor solvent as the mobile phase for both halogenated IIR and its derivatives [52]. Due to these considerations, the results are only provided to demonstrate the trends among the samples. The weight-average molar mass measured for BIIR (1.9 mol%) was Mw = 3.8 × 105 g/mol, compared to 2.3 × 105 and 2.7 × 105 g/mol for IIR-6COOH and IIR-11COOH, respectively. It is therefore clear from the SEC analysis that there were no significant changes in the molecular weight distribution of the carboxylated derivatives compared with BIIR, both on the order of 105 g/mol, which suggests that no significant degradation or cross-linking of the IIR derivatives took place in the azidation and click reaction steps. The analysis of the products derived from BIIR with 1 and ~4 mol% brominated isoprene units by SEC displayed similar trends (Supporting Information, Figures S10, S11).

SEC traces for the IIR derivatives (1.9 mol% brominated isoprene units)
Conclusions
Carboxylated BIIR derivatives with different substitution levels and alkyl spacer lengths were successfully synthesized by the azide–alkyne click coupling methodology. The success of the azidation and click reactions of IIR strongly depended on the type of solvent and the stoichiometry of the catalyst system used. This approach provides a new dimension to the modification of IIR. The high fidelity coupling of alkynes with azide-functionalized polymers offers a useful tool for the modification of IIR, potentially enabling the generation of a wide range of derivatives.
References
Thomas RM, Lightbown IE, Sparks WJ, Frolich PK, Murphree EV. Butyl rubber a new hydrocarbon product. Ind Eng Chem. 1940;32:1283–92.
Chu CY, Vukov R. Determination of the structure of butyl rubber by NMR spectroscopy. Macromolecules. 1985;18:1423–30.
Boyd RH, Pant PVK. Molecular packing and diffusion in polyisobutylene. Macromolecules. 1991;24:6325–31.
Dubey V, Pandey SK, Rao NBSN. Research trends in the degradation of butyl rubber. J Anal Appl Pyrolysis. 1995;34:111–25.
Brenner D, Oswald AA. Quaternary phosphonium ionomers. US4102876 (1978).
Parent JS, Penciu A, Guillén-Castellanos SA, Liskova A, Whitney RA. Synthesis and characterization of isobutylene-based ammonium and phosphonium bromide ionomers. Macromolecules. 2004;37:7477–83.
Parent JS, White GDF, Whitney RA. Synthesis of thioether derivatives of brominated poly(isobutylene-co-isoprene): direct coupling chemistry for silica reinforcement. J Polym Sci Part A Polym Chem. 2002;40:2937–44.
Parent JS, White GDF, Whitney RA, Hopkins W. Amine substitution reactions of brominated poly(isobutylene-co-isoprene): new chemical modification and cure chemistry. Macromolecules. 2002;35:3374–9.
Parent JS, White GDF, Thom DJ, Whitney RA, Hopkins W. Sulfuration and reversion reactions of brominated poly(isobutylene-co-isoprene). J Polym Sci Part A Polym Chem. 2003;41:1915–26.
Guillén-Castellanos SA, Parent JS, Whitney RA. Synthesis of ester derivatives of brominated poly(isobutylene-co-isoprene): solvent-free phase transfer catalysis. Macromolecules. 2006;39:2514–20.
Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed. 2001;40:2004–21.
Joralemon MJ, O’Reilly RK, Matson JB, Nugent AK, Hawker CJ, Wooley KL. Dendrimers clicked together divergently. Macromolecules. 2005;38:5436–43.
Hoyle CE, Bowman CN. Thiol–ene click chemistry. Angew Chem Int Ed. 2010;49:1540–73.
Sumerlin BS, Vogt AP. Macromolecular engineering through click chemistry and other efficient transformations. Macromolecules. 2010;43:1–13.
Becer CR, Hoogenboom R, Schubert US. Click chemistry beyond metal-catalyzed cycloaddition. Angew Chem Int Ed. 2009;48:4900–8.
Huisgen R. 1.3-Dipolare dycloadditionen rückschau und ausblick. Angew Chem. 1963;75:604–37.
Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective ‘ligation’ of azides and terminal alkynes. Angew Chem Int Ed. 2002;41:2596–9.
Tornøe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem. 2002;67:3057–64.
Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB. et al. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J Am Chem Soc. 2005;127:210–6.
Krivopalov VP, Shkurko OP. 1,2,3-Triazole and its derivatives. Development of methods for the formation of the triazole ring. Russ Chem Rev. 2005;74:339–79.
Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discov Today. 2003;8:1128–37.
Valverde IE, Bauman A, Kluba CA, Vomstein S, Walter MA, Mindt TL. 1,2,3-Triazoles as amide bond mimics: triazole scan yields protease-resistant peptidomimetics for tumor targeting. Angew Chem Int Ed. 2013;52:8957–60.
Parent JS, Porter AMJ, Kleczek MR, Whitney RA. Imidazolium bromide derivatives of poly(isobutylene-co-isoprene): a new class of elastomeric ionomers. Polym (Guildf). 2011;52:5410–8.
Ozvald A, Parent JS, Whitney RA. Hybrid ionic/covalent polymer networks derived from functional imidazolium ionomers. J Polym Sci Part A Polym Chem. 2013;51:2438–44.
Twigg C, Mueller E, Molloy BM, Parent JS. Peroxide-initiated chemical modification of poly(isobutylene-co-isoprene): H-atom transfer yields and regioselectivity. J Polym Sci Part A Polym Chem. 2016;54:3102–9.
Kleczek MR, Whitney RA, Daugulis AJ, Parent JS. Synthesis and characterization of thermoset imidazolium bromide ionomers. React Funct Polym. 2016;106:69–75.
Sahoo RR, Biswas SK. Frictional response of fatty acids on steel. J Colloid Interface Sci. 2009;333:707–18.
Bellamine A, Degrandi E, Gerst M, Stark R, Beyers C, Creton C. Design of nanostructured waterborne adhesives with improved shear resistance. Macromol Mater Eng. 2011;296:31–41.
Vendamme R, Olaerts K, Gomes M, Degens M, Shigematsu T, Eevers W. Interplay between viscoelastic and chemical tunings in fatty-acid-based polyester adhesives: engineering biomass toward functionalized step-growth polymers and soft networks. Biomacromolecules. 2012;13:1933–44.
Visser SA, Cooper SL. Comparison of the physical properties of carboxylated and sulfonated model polyurethane ionomers. Macromolecules. 1991;24:2576–83.
Santerre JP, Brash JL. Physical properties of nonionomeric and ionomeric segmented polyurethanes: effect of sulfonate, carboxylate, and quaternary ammonium ions in the hard segment. Ind Eng Chem Res. 1997;36:1352–9.
Mashita R, Kishimoto H, Inoue R, Kanaya T. Small-angle X-ray and neutron scattering analyses of highly crosslinked rubber with unsaturated carboxylic acid. Polym J. 2013;45:57–63.
Mashita R, Kishimoto H, Inoue R, Kanaya T. Structure analyses of polybutadiene rubber crosslinked with unsaturated carboxylate using contrast variation small-angle neutron scattering. Polym J. 2016;48:239–45.
McEachran MJ, Trant JF, Sran I, de Bruyn JR, Gillies ER. Carboxylic acid-functionalized butyl rubber: synthesis, characterization, and physical properties. Ind Eng Chem Res. 2015;54:4763–72.
McNeish JR, Parent JS, Whitney RA. Halogenated poly(isobutylene-co-isoprene): influence of halogen leaving-group and polymer microstructure on chemical reactivity. Can J Chem. 2013;91:420–7.
Trisler JC, Freasier BF, Wu S-M. The detection of hydrogen cyanide present as an impurity in N,N-dimethyformamide. Tetrahedron Lett. 1974;15:687–90.
Himo F, Demko ZP, Noodleman L, Sharpless KB. Mechanisms of tetrazole formation by addition of azide to nitriles. J Am Chem Soc. 2002;124:12210–6.
Juillard J. Dimethylformamide: purification, tests for purity and physical properties. Pure Appl Chem. 1977;49:885–92.
Lonsdale DE, Bell CA, Monteiro MJ. Strategy for rapid and high-purity monocyclic polymers by CuAAC ‘click’ reactions. Macromolecules. 2010;43:3331–9.
Geng J, Lindqvist J, Mantovani G, Haddleton DM. Simultaneous copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) and living radical polymerization. Angew Chem Int Ed. 2008;47:4180–3.
Bell CA, Jia Z, Perrier S, Monteiro MJ. Modulating catalytic activity of polymer-based cuAAC ‘click’ reactions. J Polym Sci Part A Polym Chem. 2011;49:4539–48.
Faucher S, Okrutny P, Zhu S. Catalyst solubility and experimental determination of equilibrium constants for heterogeneous atom transfer radical polymerization. Ind Eng Chem Res. 2007;46:2726–34.
Meldal M, Tornøe CW. Cu-catalyzed azide–alkyne cycloaddition. Chem Rev. 2008;108:2952–3015.
Carvalho I, Andrade P, Campo VL, Guedes PMM, Sesti-Costa R, Silva JS. et al. ‘Click chemistry’ synthesis of a library of 1,2,3-triazole-substituted galactose derivatives and their evaluation against Trypanosoma cruzi and its cell surface trans-sialidase. Bioorg Med Chem. 2010;18:2412–27.
Starostin EK, Lapitskaya MA, Ignatenko AV, Pivnitsky KK, Nikishin GI. Practical synthesis of hex-5-ynoic acid from cyclohexanone. Russ Chem Bull. 1990;49:81–4.
Lespieau R, Bourguel M. 2,3-Dibromopropene. Org Synth. 1925;5:49.
Starostin EK, Ignatenko AV, Lapitskaya MA, Pivnitsky KK, Nikishin GI. Synthesis of ω- and (ω−1)-acetylenic acids from five-, six-, or seven-membered cycloalkanones. Russ Chem Bull. 2001;50:833–7.
McLean JK, Guillén-Castellanos SA, Parent JS, Whitney RA, Kulbaba K, Osman A. Phase-transfer catalyzed esterification of brominated poly(isobutylene-co-isoprene). Ind Eng Chem Res. 2009;48:10759–64.
Wei X, Chen W, Chen X, Russell TP. Disorder-to-order transition of diblock copolymers induced by alkyne/azide click chemistry. Macromolecules. 2010;43:6234–36.
Bonduelle CV, Karamdoust S, Gillies ER. Synthesis and assembly of butyl rubber–poly(ethylene oxide) graft copolymers: from surface patterning to resistance to protein adsorption. Macromolecules. 2011;44:6405–15.
Karamdoust S, Bonduelle CV, Amos RC, Turowec BA, Guo S, Ferrari L. et al. Synthesis and properties of butyl rubber–poly(ethylene oxide) graft copolymers with high PEO content. J Polym Sci Part A Polym Chem. 2013;51:3383–94.
Yamashita S, Kodama K, Ikeda Y, Kohjiya S. Chemical modification of butyl rubber. I. Synthesis and properties of poly(ethylene oxide)-grafted butyl rubber. J Polym Sci Part A Polym Chem. 1993;31:2437–44.
Acknowledgements
The authors thank ARLANXEO Canada Inc. and the Natural Sciences and Engineering Research Council (NSERC) of Canada for their generous support of this work. We are also grateful to the late Dr. Lorenzo Ferrari and to Dr. Dana Adkinson for useful discussions in relation to this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Deepak, V.D., Mahmud, I. & Gauthier, M. Synthesis of carboxylated derivatives of poly(isobutylene-co-isoprene) by azide–alkyne “click” chemistry. Polym J 51, 327–335 (2019). https://doi.org/10.1038/s41428-018-0130-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-018-0130-y
