
Abstract
We report the ion-conductive and electrochemical properties of solid polymer electrolytes (SPEs) based on poly(ethylene carbonate) (PEC) and magnesium bis(trifluoromethanesulfonyl)imide, Mg(TFSI)2, with the inclusion of small amounts of lithium bis(fluorosulfonyl)imide, LiFSI. The addition of LiFSI improved the ionic conductivity of PEC-Mg(TFSI)2 from ~2.3 × 10−6 S cm−1 to ~1.0 × 10−5 S cm−1 at 80 °C. Cyclic voltammetry revealed that the plating/stripping reaction of Mg is enhanced by the addition of LiFSI. Linear sweep voltammetry found that the addition of LiFSI has no negative influence on the electrochemical oxidation stability of the electrolyte, which reached 2 V vs. Mg/Mg2+. The addition of a small amount of LiFSI proves to be an effective method for enhancing the ion-conductive and electrochemical performance of PEC-based Mg electrolytes.
Similar content being viewed by others
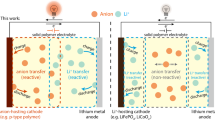


Introduction
Studies of multivalent electrolytes for use in batteries are increasing rapidly because of their high energy density, safety, and low material cost [1,2,3]. Mg metal is an excellent candidate because of its high volumetric capacity, high theoretical specific capacity (3832 mAh/cm3 and 2230 mAh/g) and low reduction potential (−2.356 V vs. standard hydrogen electrode) [4]. The earliest study on a reversible Mg battery was by Gregory et al. in 1990 and focused on plating Mg metal via the electrolysis of Grignard reagents, RMgX, where R is an alkyl or aryl group and X is Cl or Br for oxidative stability [5]. In 2000, Aurbach et al. reported a breakthrough for preparing an electrolyte with higher oxidative stability by combining Grignard reagents with an Al-based Lewis acid [6]. The higher boiling points of ionic liquids and glymes were then exploited as solvents for Mg electrolytes [7,8,9]. Unfortunately, the electrochemical performance of the resulting Mg-ion-conductive electrolytes requires improvement because of the difficulty of the Mg plating/stripping process [10, 11]. The flammable and leaky character of the liquid solution is also problematic.
The use of solid polymer electrolytes (SPEs) offers greater safety and prevents leakage in battery systems [12,13,14]. Mg-ion-conductive SPEs based on poly(ethylene oxide) (PEO), poly(vinyl alcohol) (PVA), and poly(methyl methacrylate) (PMMA) have already been reported [15,16,17,18,19], and detailed discussions have been given of their electrochemical properties, particularly the Mg plating/stripping behavior of the SPEs. Polymeric gel systems have also been proposed as Mg-ion conductors having high ionic mobility [20,21,22,23], but only a few studies have attained high coulombic efficiency for the Mg redox reaction and high cycling stability [24]. The deployment of SPE-based Mg electrolytes is hindered by the difficulty of enabling the Mg plating/stripping process between electrode and electrolyte because Mg ions form clusters with solvent molecules and then make up a passivation layer [25, 26].
Recently, an innovative electrolyte system that combines the advantages of Mg and Li has been explored. The first such study examined a mixed solution of Mg(BH4)2 and LiBH4. This system can achieve a coulombic efficiency of 94% for the Mg plating/stripping process in dimethoxyethane (DME) when LiBH4 is used as an additive [27]. Further research has been done by Ichitsubo et al., who found many advantages, including high energy density and rate capability for two kinds of salts (LiBF4 and phenylmagnesium chloride) as electrolytes [28]. Additionally, the hybrid electrolyte of phenylmagnesium bromide (PhMgBr) with LiBr gave an improvement in the kinetics of the Mg plating/dissolution process [29]. Despite these features, most previous hybrid systems that use liquid electrolytes still face safety and environmental concerns.
The present study examines the effect of the addition of Li salt on the electrochemical properties of a poly(ethylene carbonate) (PEC)-magnesium bis(trifluoromethanesulfonyl)imide (Mg(TFSI)2) solid electrolyte. In our recent work, we found that PEC is a good polymer candidate for SPEs because of its specific ion-conductive properties. Our previous studies investigated PEC-based Li electrolytes and found excellent electrochemical performance with high conductivities and Li+ transference numbers [30,31,32]. We have also examined Mg-ion-conductive PEC electrolytes, comparing different types of Mg salts, such as Mg(TFSI)2 and MgClO4, and found that the PEC-Mg(TFSI)2 system at 40 mol% has reasonable ionic conductivity and adequate electrochemical stability [33]. The conductivity value still requires improvement and an adequate Mg redox reaction. To our knowledge, no studies of hybrid SPE systems of Li and Mg salts have previously been conducted. We report here that the addition of a small amount of Li salt, lithium bis(fluorosulfonyl)imide (LiFSI), into a PEC-Mg salt electrolyte improved its ion-conductive and electrochemical properties.
Experimental
Materials and sample preparation
Poly(ethylene carbonate) (PEC, QPAC®25, Empower Materials, Mn = 1.0 × 105, Mw/Mn = 2.7) was dissolved into acetonitrile. The solution was added to excess methanol to generate a precipitate, which was then rinsed and dried under vacuum at 60 °C for 24 h. Magnesium bis(trifluoromethanesulfonyl)imide (Mg(TFSI)2, battery grade) and lithium bis(fluorosulfonyl)imide (LiFSI, battery grade) were purchased from Kishida Chemical Co. and were used without purification. The salt concentration of PEC-Mg(TFSI)2 was fixed at 40 mol%, which was noted as P2.5Mg, by the ratio of Mg2+ to the EC monomer unit of PEC ([Mg]/[EC unit] = 1/2.5) because this electrolyte gave the highest conductivity and had good electrochemical properties [33]. A small amount of LiFSI was incorporated into the P2.5Mg, and the mixed electrolyte was noted as P2.5Mg-xLi (x is the wt.% of LiFSI; ((wt. of LiFSI)/(wt. of P2.5Mg)) × 100 = 0.5, 2.0, 5.0, and 10.0 wt.% (in mol%, ((LiFSI)/(P2.5Mg)) × 100 = 0.003, 0.010, 0.030, and 0.050, respectively)). A PEC-LiFSI electrolyte at 3.5 mol%, (x mol% = [Li+]/[EC unit] × 100) was also prepared using the same method, for comparison. These predetermined quantities of the purified PEC, Mg(TFSI)2 and LiFSI were dissolved into acetonitrile until the solutions were homogenous. The resulting solutions were dried under vacuum at 60 °C for 24 h.
Quantification
The ionic conductivity of all electrolytes was measured by the electrochemical impedance method, using a potentiostat/galvanostat SP-150 (Biologic Co. Ltd.) in the frequency range of 100 Hz to 1 MHz in an Ar-filled glovebox. A two-electrode cell using stainless steel (SS|SPE|SS) was tested in order to determine the conductivity value. The glass transition temperature (Tg) was determined using a DSC7020 (Hitachi High Tech Co.). The sample was sealed in an aluminum pan in the Ar-filled glovebox. The sample pans were heated from 25 °C to 80 °C and then cooled to −90 °C, followed by a second heating scan to 90 °C under a dry N2 gas flow at a scan rate of 10 °C min−1. Thermogravimetric analysis (TGA) was conducted using a TG/DTA7200 (Hitachi High Tech Co.) under dry N2 gas flow over a temperature range from 30 to 500 °C at a heating scan rate of 10 °C min−1. FT-IR spectra for pure PEC and all electrolytes were determined by an FT/IR-4100 spectrometer (JASCO Co.), using an ATR unit (ZnSe lens) from 400 to 4000 cm−1 with a resolution of 4 cm−1, under N2 gas. For electrochemical studies, we performed cyclic voltammetry (CV) and linear sweep voltammetry (LSV), using a potentiostat/galvanostat SP-150 (Biologic) at 80 °C. The CV was carried out using a symmetric AZ31|SPE|AZ31 cell in the potential range of −3 V to 3 V at a scan rate of 0.01 V s−1. To confirm Mg deposition, CV measurements were made using an asymmetric SS|SPE|AZ31 cell. The surface morphology of the SS working electrode after the CV measurement was observed by a scanning electron microscope (SEM) equipped with an energy dispersive X-ray spectroscopy (EDX) unit (JEOL JCM6000 Plus). The surface of the electrode was washed with THF to remove residual electrolyte.
Results and discussion
The thermal properties of pure PEC and P2.5Mg electrolytes with and without LiFSI were measured by DSC and TGA. DSC thermograms of PEC and electrolytes are shown in Figure S1. From the thermograms, we determined the Tg for the PEC matrix in the electrolytes; the results are presented in Table 1. The Tg value of pure PEC decreased upon adding Mg(TFSI)2, from 17 °C to −16 to −21 °C. This fall in Tg upon adding salt is a typical behavior in PEC and is also observed in PEC-Li salt electrolytes [34]. Upon addition of LiFSI, the Tg value of P2.5Mg decreased slightly. The Li salt may weaken the strong cohesion among the polymer chains with Mg2+ ions in the P2.5Mg electrolyte by penetrating into the polymer matrix, altering the physical properties and causing Tg to decrease [35]. This decrease in Tg reflects the faster segmental motion of the polymer chains and promotes fast ionic conduction [36].
Thermal stability is vital for the fabrication of electrochemical cells, for which it is important to understand the thermal degradation, especially when two different cations are involved. Figure S2 shows the TGA results, and the thermal decompositions of PEC and salts can clearly be seen. For pure PEC, there is only one weight loss step at ~190 °C; the addition of Mg(TFSI)2 and LiFSI gives rise to a multistep pattern. The first step, at low temperatures, corresponds to the degradation of PEC, and the second step at 280–370 °C is due to the degradation of mixed Li and Mg. The thermal stability of the P2.5Mg-Li electrolytes is maintained over 143 °C, and the addition of LiFSI slightly promotes the thermal decomposition of PEC. The addition of LiFSI causes the polymer membranes to be more flexible; therefore, less energy is needed, and the electrolyte may decompose at lower temperatures [37].
The P2.5Mg electrolytes with added LiFSI underwent impedance spectroscopy. Figure 1a displays Arrhenius plots of the conductivity with Nyquist plots at 80 °C for the P2.5Mg with differing LiFSI concentrations. In the Nyquist plots, the circular portion is due to the bulk resistance for all electrolytes. The bulk conductivities were calculated from the bulk resistances acquired from the impedance spectra and the area and thickness of the electrolyte in the measurement cell. The relation between the ionic conductivity and Tg was determined, as shown in Fig. 1b, c. The conductivity of the P2.5Mg at 80 °C increased from ~2.3 × 10−6 S cm−1 to ~9.4 × 10−6 S cm−1 upon adding 0.5 wt.% LiFSI. The conductivity increases with increasing LiFSI concentration up to ~1.0 × 10−5 S cm−1 for 2.0 wt.% LiFSI. This result indicates that the addition of extra cations into the P2.5Mg ensures adequate transport of Mg ions inside the electrolytes [38], where the LiFSI increased the amorphous phase. As the value of Tg decreases with increasing LiFSI concentration, the polymer chains become more flexible in the amorphous state and enhance the conductivity [39]. We suggest that the dissociation of the Mg salt is enhanced by adding LiFSI, increasing the plasticizing effect on polymer chains of PEC and consequently influencing Tg. Similar results have been reported previously on the addition of LiBH4 to Mg(BH4)2/THF electrolyte; the addition gives rise to the dissociation of Mg(BH4)2, which induces the improvement in the conductivity [40]. However, in this study, the conductivity decreased slightly with P2.5Mg at 5 and 10 wt.% of LiFSI. This decrease may be due to excess Li salt blocking aggregates, hindering the migration of mobile ions and restricting the segmental relaxation [41, 42].

a Temperature dependence of the ionic conductivity (inset: Nyquist plots at 80 °C), b dependence of the ionic conductivity on the LiFSI concentration at 80 °C and c the glass transition temperature (Tg) for pure PEC and the P2.5Mg electrolytes with different LiFSI concentrations
For further analysis of the effect on salt dissociation by adding LiFSI, FT-IR spectra were observed. These can detect the stretching vibrational mode of C = O groups in the carbonate units of PEC chains. The Mg(TFSI)2 and LiFSI salts incorporated into PEC drastically affect the stretching vibration mode of C = O, as shown in Fig. 2(ii). The peak shifted to lower wavenumbers with increasing LiFSI concentration as a result of the increase in interacting ions. At higher LiFSI concentrations (e.g., P2.5Mg-10.0Li), the peak shifted to higher wavenumbers as a result of the increase in the free ions. As seen in Fig. 2(ii), there are two peaks, which correspond to free C = O at 1740 cm−1 and C = O interacting with cations at 1710 cm−1 [43]. According to the calculation of peak fractions, a slight increase is visible in the fraction of the interacting C = O, from 60% for P2.5Mg to 64% for P2.5Mg-2.0Li. The increase in the interacting ions and decrease in the free ions therefore indicates that the LiFSI salt is essential in enhancing the dissociation of Mg(TFSI)2. In case of P2.5Mg-5.0Li, the peak fraction of interacting ions continues to increase up to 70%, but the conductivity is low compared to the P2.5Mg-2.0Li system, due to the aggregation of ions. For P2.5Mg-10.0Li, the peak fraction of interacting C = O decreased drastically to 15%. The higher content of Li salt reduces the number of interacting ions, leading to a large decrease in the conductivity.

FT-IR spectra of C = O stretching vibrations in carbonate units for pure PEC and P2.5Mg with differing LiFSI concentrations a 0.5Li b 2.0Li, c 5.0Li d 10.0Li ((i) original spectra, (ii) peak separation analysis of (i))
Figure 3 shows cyclic voltammograms of AZ31 symmetric cells for P2.5Mg electrolytes with different LiFSI concentrations and PEC-LiFSI at 3.5 mol% for comparison. For P2.5Mg, the CV displays significantly lower cathodic and anodic currents, suggesting a limited extent of the Mg plating/stripping process. Surprisingly, the addition of Li salt of just 0.5 wt.% (P2.5Mg-0.5Li) resulted in a drastic increase in the anodic current density, from 3 μA cm−2 (of P2.5Mg) to 16 μA cm−2. This result shows that the addition of LiFSI enhances the reactivity of Mg ions on the AZ31 surface. In the P2.5Mg and P2.5Mg-0.5Li electrolytes, the anodic peak appeared at approximately 2.4 V. In the P2.5Mg-2.0Li electrolyte, a new broad peak appeared at approximately 1 V. The CV of P2.5Mg-2.0Li was determined and compared with the PEC-LiFSI electrolyte (3.5 mol%), which has the same amount of Li salt added. The conductivity of the PEC-LiFSI electrolyte (3.5 mol%) is ~8.0 × 10−7 S cm−1 at 80 °C; this conductivity influenced the electrochemical response. In comparing the anodic peaks in P2.5Mg-2.0Li at ~1 V and in PEC-LiFSI electrolyte at ~0 V, it is likely that a substantial amount of Li ions reacted during the CV measurement. Moreover, the anodic voltage shifts from 1 V to ~0.4 V with increasing LiFSI, a value very close to the potential for the PEC-LiFSI electrolyte. For P2.5Mg-0.5Li electrolytes, there is no anodic peak; this suggest that the addition of a small amount of LiFSI facilitated the Mg plating/stripping kinetics without any significant deposition of Li metal. This behavior is consistent with the CV data reported previously, for which the potential-dependent Mg deposition kinetics are enhanced by the addition of Li salt, with the reduction kinetics involving Mg ions favoring the formation of the Mg-Li alloy [44, 45]. In fact, the current density of P2.5Mg-5.0Li and P2.5Mg-10.0Li electrolytes decreased slightly compared to P2.5Mg-2.0Li, indicating that the higher content of LiFSI attenuated the Mg plating/stripping kinetics.

Cyclic voltammograms of P2.5Mg with differing LiFSI concentrations and of PEC-LiFSI (3.5 mol%) for comparison (scan rate 0.01 Vs−1 at 80 °C)
The oxidation stability of the P2.5Mg-2.0Li electrolyte having the greatest conductivity was studied by linear sweep voltammetry (LSV), using SS as the working electrode. As shown in Figure S3, the oxidation stability was similar to that of the P2.5Mg electrolyte without LiFSI and extended over 2 V vs. Mg/Mg2+. This stability is comparable to a previously studied system in which SS was used as the working electrode [46]. This indicates that LiFSI does not extend any negative influence on the oxidation stability of the PEC-Mg(TFSI)2 electrolyte and is a suitable additive for improving the conductivity and the redox activity of the Mg metal.
The composition of the electrochemical deposit on the SS working electrode was analyzed by SEM-EDX for the P2.5Mg-2.0Li electrolyte before and after the CV measurement at 4 cycles. To assist in determining the composition, the SEM and EDX analyses were compared before and after the CV measurement. The cell before the CV measurement was heated in the same way as that after the CV measurement. In Figure S4, the composition of the SS surface for before the CV measurement is almost identical to that of pure SS. On the other hand, Mg was clearly deposited on the surface of the SS after the CV measurement, according to analysis of the particulate deposit on the surface of the stainless steel, as shown in the SEM image in Figure S5b). Other deposits, such as sulfur, were also confirmed as the decomposition products of anions as a passivation layer. These results clearly indicate that Mg was deposited during the CV process, although the unclear composition and structure of the surface, including sulfur, remain to be interpreted.
Conclusion
The ion-conductive and electrochemical properties of hybrid solid polymer electrolytes based on PEC with Mg(TFSI)2 and LiFSI have been studied. The ionic conductivity increased upon adding a small amount of LiFSI (0.5~2.0 wt.%) to the P2.5Mg electrolyte. Higher current densities were was also achieved for the Mg plating/stripping process in the solid Mg electrolyte, even with the addition of a small amount of LiFSI. The P2.5Mg-2.0Li electrolyte had oxidation stability up to approximately 2 V vs. Mg/Mg2+, which is almost identical to that of the original P2.5Mg electrolyte. These results show that the Mg-Li hybrid system has clear potential to overcome the sluggish diffusion of Mg ions and to achieve a reversible Mg plating/stripping reaction without deterioration of the oxidation stability. For practical deployment of the Mg-Li hybrid SPE system, further studies are needed of the electrochemical properties to elucidate the mechanism of enhancement of Mg plating/stripping reactivity upon the addition of Li salt.
References
Gu S, Wang H, Wu C, Bai Y, Li H, Wu F. Confirming reversible Al3+ storage mechanism through intercalation of Al3+ into V2O5 nanowires in a rechargeable aluminum battery. Energy Storage Mater. 2017;6:9–17.
Ponrouch A, Frontera C, Bardé F, Palacín MR. Towards a calcium-based rechargeable battery. Nat Mater. 2016;15:1–5.
Mandai T, Akita Y, Yagi S, Egashira M, Munakata H, Kanamura K. A key concept of utilization of both non-Grignard magnesium chloride and imide salts for rechargeable Mg battery electrolyte. J Mater Chem A. 2017;5:3152–6.
Song S, Kotobuki M, Zheng F, Li Q, Xu C, Wang Y. et al. A composite polymer electrolyte for safer Mg batteries. J Electrochem Soc. 2017;164:A741–3.
Gregory TD, Hoffman RJ, Winterton RC. Nonaqueous electrochemistry of magnesium applications to energy storage. J Electrochem Soc. 1990;137:775–80.
Aurbach D, Lu Z, Schechter A, Gofer Y, Gizbar H, Turgeman R. Prototype systems for rechargeable magnesium batteries. Nature. 2000;407:724–7.
Cho J, Hoon J, Lee S, Won B, Yoon K, Na B. et al. Effect of 1-allyl-1-methylpyrrolidinium chloride addition to ethylmagnesium bromide electrolyte on a rechargeable magnesium battery. Electrochim Acta. 2017;231:379–85.
Watkins T, Buttry DA. Determination of Mg2+ speciation in a TFSI—-based ionic liquid with and without chelating ethers using raman spectroscopy. J Phys Chem B. 2015;119:7003–14.
Kitada A, Kang Y, Uchimoto Y, Murase K. Room-temperature electrodeposition of Mg metal from amide salts dissolved in glyme-ionic liquid mixture. J Electrochem Soc. 2014;161:D102–6.
Zhao Q, Nuli Y, Nasiman T, Yang J, Wang. J. Reversible deposition and dissolution of magnesium from BMIMBF4 ionic liquid. Int J Electrochem. 2005;7:1105–10.
Cheek GT, Grady WEO, El Abedin SZ, Moustafa EM, Endres F. Studies on the electrodeposition of magnesium in ionic liquids. J Electrochem Soc. 2008;155:91–5.
Bandara TMWJ, Dissanayake MAKL, Albinsson I, Mellander BE. Mobile charge carrier concentration and mobility of a polymer electrolyte containing PEO and Pr4N+I− using electrical and dielectric measurements. Solid State Ion. 2011;189:63–8.
Abreha M, Subrahmanyam AR, Kumar JS. Ionic conductivity and transport properties of poly (vinylidene fluoride-co-hexafluoropropylene)-based solid polymer electrolytes. Chem Phys Lett. 2016;658:240–7.
Yoshimoto N, Tomonaga Y, Ishikawa M, Morita M. Ionic conductance of polymeric electrolytes consisting of magnesium salts dissolved in cross-linked polymer matrix with linear polyether. Electrochim Acta. 2001;46:1195–200.
Polu AR, Kumar R, Rhee HW. Magnesium ion conducting solid polymer blend electrolyte based on biodegradable polymers and application in solid-state batteries. Ionics. 2015;21:125–32.
Polu AR, Rhee H. Ionic liquid doped PEO-based solid polymer electrolytes for lithium-ion polymer batteries. Int J Hydrog Energy. 2016;42:7212–9.
Zainol NH, Samin SM, Othman L, Isa KB, Chong WG, Osman Z. Magnesium ion-based gel polymer electrolytes: Ionic conduction and infrared spectroscopy studies. Int J Electrochem Sci. 2013;8:3602–14.
Anilkumar KM, Jinisha B, Manoj M, Jayalekshmi S. Poly(ethylene oxide) (PEO) - poly(vinyl pyrrolidone) (PVP) blend polymer based solid electrolyte membranes for developing solid state magnesium ion cells. Eur Polym J. 2017;89:249–62.
Sharma J, Hashmi SA. Magnesium ion transport in poly(ethylene oxide)-based polymer electrolyte containing plastic-crystalline succinonitrile. J Solid State Electrochem. 2013;17:2283–91.
Morita M, Shirai T, Yoshimoto N, Ishikawa M. Ionic conductance behavior of polymeric gel electrolyte containing ionic liquid mixed with magnesium salt. J Power Sources. 2005;139:351–5.
Wang J, Song S, Gao S, Muchakayala R, Liu R. Mg-ion conducting gel polymer electrolyte membranes containing biodegradable chitosan: Preparation, structural, electrical and electrochemical properties. Polym Test. 2017;62:278–86.
Pandey GP, Agrawal RC, Hashmi SA. Magnesium ion-conducting gel polymer electrolytes dispersed with fumed silica for rechargeable magnesium battery application. J Solid State Electrochem. 2010;15:2253–64.
Pandey GP, Agrawal RC, Hashmi SA. Performance studies on composite gel polymer electrolytes for rechargeable magnesium battery application. J Phys Chem Solids. 2011;72:1408–13.
Shao Y, Rajput NN, Hu J, Hu M, Liu T, Wei Z. et al. Nanocomposite polymer electrolyte for rechargeable magnesium batteries. Nano Energy. 2015;12:750–9.
Aurbach D, Weissman I, Gofer Y, Levi E. Nonaqueous magnesium electrochemistry. Chem Rec. 2003;3:61–74.
Aurbach D, Schechter A, Moshkovich M, Cohen Y. On the mechanisms of reversible magnesium deposition processes. J Electrochem Soc. 2001;148:1004–14.
Mohtadi R, Matsui M, Arthur TS, Hwang S. Magnesium borohydride: from hydrogen storage to magnesium battery. Angew Chem. 2012;51:1–5.
Yagi S, Ichitsubo T, Shirai Y, Yanai S, Doi T, Murase K. et al. A concept of dual-salt polyvalent-metal storage battery. J Mater Chem A. 2014;2:1144–9.
Chang Z, Yang Y, Wang X, Li M, Fu Z, Wu Y. et al. Hybrid system for rechargeable magnesium battery with high energy density. Sci Rep. 2015;5:11931
Kimura K, Yajima M, Tominaga Y. A highly-concentrated poly(ethylene carbonate)-based electrolyte for all-solid-state Li battery working at room temperature. Electrochem Commun. 2016;66:46–48.
Kimura K, Motomatsu J, Tominaga Y. Highly concentrated polycarbonate-based solid polymer electrolytes having extraordinary electrochemical stability. Electrochim Acta. 2016;56:2442–7.
Tominaga Y. Ion-conductive polymer electrolytes based on poly(ethylene carbonate) and its derivatives. Polym J. 2016;49:291–9.
Aziz, AA & Tominaga, Y Magnesium ion-conductive poly(ethylene carbonate) electrolytes. Ionics. https://doi.org/10.1007/s11581-018-2482-x. Accessed on 3 August 2018.
Kimura K, Motomatsu J, Tominaga Y. Correlation between solvation structure and ion-conductive behavior of concentrated poly(ethylene carbonate)-based electrolytes. J Phys Chem C. 2016;120:12385–91.
Nadimicherla R, Kalla R, Muchakayala R, Guo X. Effects of potassium iodide (KI) on crystallinity, thermal stability, and electrical properties of polymer blend electrolytes (PVC / PEO: KI). Solid State Ion. 2015;278:260–7.
Wu HY, Chen YH, Saikia D, Pan YC, Fang J, Tsai LD. et al. Synthesis, structure and electrochemical characterization, and dynamic properties of double core branched organic–inorganic hybrid electrolyte membranes. J Memb Sci. 2013;447:274–86.
Shen Z, Simon GP. Saturation ratio of poly (ethylene oxide) to silicate in melt intercalated nanocomposites. Eur Polym J. 2003;39:1917–24.
Cristina P, Daza C, Arbey R, Meneses M, Luiz J, Ferreira DA. et al. Influence of microstructural characteristics on ionic conductivity of ceria based ceramic solid electrolytes. Ceram Int. 2018;44:2138–45.
Correa E, Moncada ME, Zapata VH. Electrical characterization of an ionic conductivity polymer electrolyte based on polycaprolactone and silver nitrate for medical applications. Mater Lett. 2017;205:155–7.
Su S, NuLi Y, Wang N, Yusipu D, Yang J, Wang J. Magnesium borohydride-based electrolytes containing 1-butyl-1-methylpiperidinium bis(trifluoromethyl sulfonyl)imide ionic liquid for rechargeable magnesium batteries. J Electrochem Soc. 2016;163:D682–8.
Reddy MJ, Chu PP. Ion pair formation and its effect in PEO: Mg solid polymer electrolyte system. J Power Sources. 2002;109:340–6.
Gamal R, Sheha E, Shash N, El-Shaarawy MG. Preparation and characterization of Mg-ion conducting composite based on poly(vinyl alcohol) with various concentrations of Li2O. Mater Express. 2014;4:293–300.
Wang J, Wu Y, Xuan X, Wang H. Ion-molecule interactions in solutions of lithium perchlorate in propylene carbonate+diethyl carbonate mixtures: an IR and molecular orbital study. Spectrochim Acta Part A. 2002;58:2097–104.
Chang J, Haasch RT, Kim J, Spila T, Braun PV, Gewirth AA. et al. Synergetic role of Li+ during Mg electrodeposition/dissolution in borohydride diglyme electrolyte solution: voltammetric stripping behaviors on a Pt microelectrode indicative of Mg-Li alloying and facilitated dissolution. ACS Appl Mater Interfaces. 2015;7:2494–502.
Shimamura O, Yoshimoto N, Matsumoto M, Egashia M, Morita M. Electrochemical co-deposition of magnesium with lithium from quaternary ammonium-based ionic liquid. J Power Sources. 2011;196:1586–8.
Tang X, Muchakayala R, Song S, Zhang Z, Polu AR. A study of structural, electrical and electrochemical properties of PVdF-HFP gel polymer electrolyte films for magnesium ion battery applications. J Ind Eng Chem. 2016;37:67–74.
Acknowledgements
This work was supported financially by the Advanced Low Carbon Technology Research and Development Program, Specially Promoted Research for Innovative Next Generation Batteries (ALCA-SPRING) from the Japan Science and Technology Agency (JST) and a Grant-in-Aid for Challenging Exploratory Research of JSPS KAKENHI (16K14098), Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Aziz, A.A., Tominaga, Y. Effect of Li salt addition on electrochemical properties of poly(ethylene carbonate)-Mg salt electrolytes. Polym J 51, 61–67 (2019). https://doi.org/10.1038/s41428-018-0113-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-018-0113-z
