
Abstract
Organic materials with unpaired electrons along with the discovery of their unique properties have fascinated scientists for over a century. The development of open-shell molecules has recently been rekindled as the result of improved synthetic strategies and spectroscopic techniques. In this focus review, we provide an overview of open-shell polymers and small-molecules organic semiconductors. We review strategies toward molecular magnets or spin-polarized magnetic organic semiconductors that encompasses incorporation of stable radical groups directly into the backbone of organic semiconductors or preparing highly conjugated ladder-type molecules based on open–shell Kekulé–type structures to enable efficient spin delocalization along the conjugation length. These novel materials have the potential to make significant societal impacts in the areas of information, energy and human health technologies.
Similar content being viewed by others
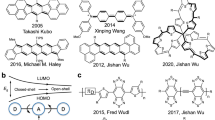

Introduction
Open-shell organic-free radicals with one or more unpaired electrons are often regarded as highly reactive intermediates and historically associated with poorly controlled chaotic reactions [1]. The discovery of the triphenylmethyl radical by Gomberg has intrigued scientists and inspired developments of radicals [2]. Modern synthetic organic chemistry takes great advantage of utilizing radicals in a variety of novel strategies [3, 4], and multiple types of controlled radical polymerizations have enabled preparation of functional polymers with well-defined architectures and uniform molecular weight [5,6,7,8]. In the meantime, the spin of an unpaired electron in radicals gives rise to magnetism, where the bulk magnetic property depends on the interaction of the unpaired electron spin moments [9]. Conventional organic electronic devices often ignore the spin property and rely strictly on the transport of the electrical charge of electrons upon an external stimulus. In principle, adding the spin degree of freedom can result in new phenomena, functionalities and technologies [10]. Indeed, the emerging field of spintronics or magneto-electronics/optoelectronics explores the phenomena that interlink the spin degree of freedom of electrons and charge degree of freedom [11,12,13,14,15,16,17]. Because spin can be manipulated at a faster speed and with less energy than charges, spin-based devices are expected to exhibit faster switching times and decreased electric power consumption than conventional semiconductor devices [15, 18]. Metal-based spintronics have undeniably transformed magnetic data storage and memory applications through giant and tunnel magnetoresistance in layered magnetic structures [19]. Numerous applications have been explored in semiconductor-based spintronics and are envisioned for multifunctional devices including non-volatile memories [20], spin-valves [21], spin-transistors [22], spin-photovoltaics [23], spin-biosensors [24, 25], and spin-light emitting diodes [26]. While organic semiconductor-based spintronics have scarcely been investigated, they have the potential to transform the field of spin-based electronics, optoelectronics, and diagnostics. Molecular-scale organic spintronics offer the potential for miniaturization of spintronic devices in the semiconductor and microelectronics industries, ease of fabrication such as roll-to-roll printing, and the ability to simultaneously engineer the molecular, magnetic, and electronic structures.
Our group and others have been interested in the design and synthesis of novel open-shell polymers semiconductors for electronics and optoelectronics. A central theme is the fundamental understanding of the structural and physical factors that control photo-induced spin-alignment in oligomer/polymer semiconductors, to elucidate the role of electron spin on optoelectronic devices such as organic light-emitting diodes (OLEDs) and organic photovoltaics (OPVs), and to explore open-shell organic semiconductors in magneto-optoelectronic devices.
In this focus review, we survey two strategies in the design of molecular magnets or spin-polarized magnetic organic semiconductors. First, we review the strategy of incorporating stable pendant radicals directly into the backbone of organic semiconductors to enable spin-delocalization and as an approach to engineer excited-states in organic semiconductors. Second, we take a brief overview of the highly conjugated ladder–type molecules based on Kekulé–type structures, “biradicaloid” molecules, with closed-shell and open-shell dual structures as a result of the resonance π-electrons, which leads to biradical characteristics in the ground state. The unique structures of biradicaloid have attracted significant research efforts to elucidate optical, and magnetic properties. Significant efforts have been dedicated to diradicaloids and it would not be possible to cover all research in this focus review. We thus direct readers to the cited reviews on particular topics alongside our discussions. Finally, we conclude with future perspectives and potential applications of open-shell organic semiconductors.
Incorporation of pendant radicals to modulate excited-states in organic semiconductors
Controlling the magnetic properties or electron spin using light radiation can have significant implications in OPVs and OLEDs [23, 26,27,28,29,30,31]. Magnetism or spin-coupling in organic molecules can be achieved by incorporation of radical groups and exploiting the “through-bond” approach [9]. Theoretical studies have long suggested that polymers containing radical groups can lead to magnetically coupled or high-spin alignment between the unpaired electrons of the pendant radicals and the backbone of conjugated polymers [32, 33]. Unfortunately, early reports of ferromagnetic conjugated polymers were plagued by problems [34]. Careful and rigorous examinations eventually led to the realization that the observed ferromagnetism was due impurities of iron oxide rather than due to the intrinsic properties of the conjugated polymers [35, 36]. Nonetheless, several groups thereafter have demonstrated ferromagnetism or high-spin order in conjugated polymers as a result of intramolecular spin-exchange interactions through the conjugated backbone (Fig. 1a) [37,38,39,40,41,42,43]. For example, polythiophene with pendant phenoxyl radicals exhibited a high-spin ground state and an intramolecular ferromagnetic spin coupling through the conjugated polymer backbone [44]. In 2001, experimental studies validated the theoretical prediction and the influence of dimensionality on spin alignment numbers with dendritic structures that exhibits the highest spin-alignment for purely organic molecules (S ~ 5000) [10].

Examples of conjugated polymers (a) and small molecules (b) functionalized with pendant radicals (highlighted in red)
Elegant examples of photo-responsive small molecules bearing stable radicals have illustrated intramolecular or intermolecular spin-based properties (Fig. 1b) [13, 45]. Exchange interactions between two radicals were studied with EPR and SQUID measurements to determine the strength and the sign (ferro- or antiferromagnetic) of the two unpaired electrons connected by a series of thiophene and bithiophene rings [46], and phenylene-ethynylene spacers [47]. Results have shown that short inter-spin distance leads to strong exchange, and the substituent position of the radical has a significant effect on the spin interaction. For example, the 4,5-IB between the iminyl nitroxide radicals caused a weak ferromagnetic coupling, while 5,5-IB resulted in an antiferromagnetic coupling (Fig. 1b). Investigation of intermolecular radical-chromophore interactions was enabled by time-resolved EPR (TREPR). The absorptive/emissive signal patterns give a clear indication of multiplet precursors. Intermolecular interactions between chromophores and radicals have been extensively studied and explained by the radical-triplet pair mechanism (RTPM) [48,49,50]. Thus, we will focus on intramolecular-radical interactions upon photoexcitation. The concept for controlling magnetic properties by photoexcitation was examined using SiPc-TEMPO, a silicon phthalocyanine bearing two TEMPO radicals, and zinc porphyrins coordinated by pyridyl nitronyl nitroxide radicals [51, 52]. In SiPc-TEMPO, modulating the population of singlet and triplet ground state was difficult to achieve. However, photoexcitation of the phthalocyanines directly resulted in a selective population from the excited multiplet states to the triplet ground state [52]. Further studies of SiPc-TEMPO using TREPR and transient absorption revealed that the spin polarization originates from the spin-selective relaxation of the excited states [53]. TREPR of intramolecular radical-excited state pair was accurately simulated on zinc porphyrins, which interpreted the quartet state as a result of selective ISC [51]. Incorporation of nitroxide radicals in fullerene [54, 55] and anthracene [56, 57] derivatives were discovered to result in a photo-induced spin alignment that yields photoexcited quintet state generated by interactions of the chromophore excited triplet states and the radicals. The unpaired spin in the nitroxide radical DPA-IN published in 2001 shown in Fig. 1, was found to be coupled to the diphenyl anthracene moiety through π-conjugation, and the photo-induced spin alignment was apparently dependent on the topology of the organic semiconductor relative to the organic radical [56, 58].
Early interests in combining chromophores and nitroxide radicals emerged because of the unique photo-magnetic switching that can lead to excited states with multi-spin systems and for potential applications as molecular fluorescent probes [59]. Examples of intramolecular chromophore-nitroxide radical interactions have been reviewed by Likhtenstein and coworkers [59]. Figure 2 highlights the common decay pathways of quenching as a result of appending radicals to a fluorescent chromophore. In the absence of a pendant radical, photoexcitation of molecule A leads to the singlet excited state (1A*). For a highly fluorescent molecule, fluorescence is the major decay pathway from 1A* to the ground state 1A, and the spin-forbidden ISC from 1A* to the triplet excited state (3A*) is usually very slow [60]. However, a pendant radical (R●) would complicate the situation by introducing a variety of other possible decay pathways (Fig. 2). In general, if a radical can cause any of these pathways to operate efficiently with a large rate constant, depletion of 1A* would happen more rapidly than without a radical. Depending on redox potentials, electron transfer (ET) may happen between 1A* and R●, which leads to the formation of radical cation or anion (A●+ or A●−). For instance, conjugation of phenoxyl radical to highly fluorescent perylene diimide (PDI) results in fluorescence quenching of PDI [61]. The fluorescence quenching in PDI is attributed to ET, that is visible from steady-state spectroscopy due to the intramolecular charge-transfer complex observed at near-IR wavelengths [61]. Although the two radicals connected to the 1,7-positions of PDI could possibly lead to a quinoidal closed-shell resonance structure, experimental and theoretical evaluations concluded a large singlet diradical character of y = 0.72, and a small singlet-triplet energy gap of 0.041 eV for the PDI diradicals, due to the gain in aromaticity in the diradical form [61]. Förster or Dexter types of energy transfer (EnT) from 1A* to R● that forms excited radical (R●*) are also possible, given considerable spectral and wavefunction overlap [62, 63]. Enhanced intersystem crossing (EISC) is an important and widely recognized pathway that the radical triggers the conversion of 1A* to 3A* in rigid chromophores such as PDI [64], pentacene [65], boron dipyrromethene (BODIPY) [66], due to the strong spin-orbit coupling caused by the paramagnetic species. Spin-flip of the radical in EISC could keep the spin multiplicity of the radical-chromophore ensemble constant (doublet), thus accelerating the formerly spin-forbidden process. Studies of the PDI-BPNO radicals present a great example that systematically investigated each possible photophysical pathway using various spectroscopic methods, including steady-state and ultrafast transient absorption (TA) spectra, as well as TREPR and other characterizations [64]. On the basis of the energy levels of PDI with respect to BPNO radicals and lack of solvent polarity dependence, the authors were able to immediately exclude ET and EnT as major contributing pathways. On the other hand, observation of triplet PDI in TA spectroscopy and its associated TREPR pattern confirmed the significance of EISC following the excitation of PDI [64]. The environment can also play a significant role in whether promoting ISC or other competing pathways. For example, EISC of a photoexcited PDI-nitroxide radical in toluene was very efficient, but ET was the major process in more polar solvents such as tetrahydrofuran (THF) [67]. Pentacene is well-known to be sensitive to ambient environments. Conjugation of pentacene with nitronyl nitroxide (Pn-NN) or oxoverdazyl radicals at the central position resulted in a dramatically robust photostability and prevented photo-oxidation and photobleaching [68, 69]. Ultrafast transient-absorption spectroscopy and simulation studies suggested that the improved photostability is due to rapid depletion of the 1A* to 3A*. This process was attributed to both EISC and singlet fission, which is a relatively rare circumstance in solution [65]. Interestingly, varying the radical structure and the radical-chromophore linkage on the same pentacene chromophore gave rise to largely different photophysical properties. In contrast to the Pn-NN, the appended TEMPO radicals did not quench the fluorescence of pentacene in Pn-TEMPO derivatives [70]. Ultrafast TA suggests that rather than de-population of 1A*, the radicals selectively engaged 3A* through the electron spin polarization transfer (ESPT) mechanism [70, 71].

Common decay pathways of singlet excited molecule A with a pendant radical R●
In addition to the above-highlighted examples of EISC, incorporation of radical may also lead to enhanced internal conversion (EIC). The EIC is a competing process to EISC that leads to the recovery of ground state 1A. Although EIC is a common process, it is generally very difficult to measure directly because typical spectroscopic methods do not provide direct confirmation. Nevertheless, EIC can still be quantified indirectly after carefully determination of the kinetics of other competitive pathways [64].
In complex situations where the chromophore component is a donor-acceptor assembly, electron-/spin- transfer was elucidated in a series of covalent donor-acceptor-radical (D-A-R●) triads by Wasielewski and coworkers [72,73,74,75]. Briefly, selective excitation of D initiates electron transfer that generates D+●-A–●-R●, a triradical where the spin of only D+●-A–● is correlated but uncorrelated with R● [75]. Subsequent electron transfer and the reduction R● within this triradical became spin-selective. The formation of monoradical species D+●-A-R– only originated from D+●-1(A–●-R●) but not D+●-3(A–●-R●) [75]. These spin-selective redox reactions in D-A-R● systems may help to explore control of spin interactions and new approaches to quantum information processing applications [75].
In addition, excitation of photo-responsive radicals can lead to subsequent structural changes which may affect radical exchange interactions. The cis-diarylethylenes with two terminal NN radicals can undergo reversible photochromic reactions upon alternate UV/visible irradiation [76]. EPR spectra suggested the radical interactions also reversibly changed with the photochromism, and a 150-fold difference was observed between open- and closed-ring form isomers [76]. Stronger magnetic interactions were detected through thiophene rather than through phenylene spacers, and the photochromic reactivity with thiophene spacers was reduced.
Recently, there has been a surge in exploring chromophores bearing radicals in applications ranging from biomedical to electronics and optoelectronics. For example, in 2012, a rhodamine-TEMPO dye was utilized to probe intracellular ROS (reactive oxygen species) detection and has been shown to be a promising candidate with high sensitivity, selectivity, and photostability [77]. Fluorescence emission of rhodamine is deactivated by incorporation of TEMPO radical as a pendant group. The rhodamine-TEMPO reacts with ROS which results in a turn-on strong fluorescence of rhodamine [77]. Studies in 2017 of BODIPY-anthroxyl radical as a ROS probe emphasized the significance of kinetic blocking to achieve reasonably stable radical product, while unblocked radical easily decomposed in ambient environment [78]. In 2017, EISC was observed in the BOD-TEMPO by appending TEMPO radicals to the BODIPY core using click chemistry [66]. The triplet BODIPY formation was highly effective and was further employed as a triplet sensitizer for photon upconversion, where triplet energy transfer occurred from BODIPY sensitizer to the perylene acceptor, followed by triplet–triplet annihilation (TTA) of perylene [66]. Recently, chlorinated-triphenylmethyl radicals were shown to be stable radicals that exhibit high quantum yield of photoluminescence [79]. A carbazole derivative of the same class of radicals was examined as an emissive material in OLEDs and showed comparable performance to non-radical deep-red organic semiconductors. This approach exploits the potential of open-shell molecules as the emissive layer in OLEDs devices [80]. This is a strategy that provides an alternative route to harvesting triplet excitons and spin-forbidden triplet decay because the emission directly comes from the radical and involves the singly-occupied molecular orbitals of the radical [80].
Ground state open-shell radical characters
While the work mentioned above mostly focused on the influence of radicals on photoexcited states of organic chromophores, efforts have also focused on exploring the intrinsic radical characters of some unconventional structures, seemingly closed-shell molecules without a persistent radical. These structures usually contain conjugated double bonds that are highly delocalized along the π-conjugated backbone. The p-quinodimethane (p-QDM) building block is essential for the two important resonance forms: closed-shell “quinoidal” and open-shell “aromatic” diradical dual structures (Fig. 3a). On one hand, the diradicals benefit from gains in aromaticity, which stabilizes the structure. On the other hand, according to topology rules widely acknowledged previously [81], the two radicals in a p-QDM unit would incur strong antiferromagnetic coupling, for which the bond formation would be an extreme case. Unsubstituted p-QDM or its longer analogs with extended conjugation were never isolated due to its high reactivity [82], thus strategies to stabilize the p-QDM analog radicals have been continuously pursued. Thiele’s [83] and Tschitschibabin’s [84] hydrocarbons were the earliest examples in 1900’s of using bulky phenyl groups to stabilize the radicals [85, 86]. Tetrabenzo-annulation further increased the stability of Tschitschibabin’s hydrocarbons by incorporating more fused benzene rings, therefore, more aromatic sextets [87]. The chemically stable tetrabenzo-Tschitschibabin’s diradical showed an intriguing slow transformation from open-shell to closed-shell ground state, likely due to the huge energy barrier produced by the adjacent anthracene rings [87]. Phenoxyl radicals bridged by two anthracenes formed bisanthryl and bisanthenequinone, which exhibited closed-shell ground state character, intense near-IR absorption and reversible ambipolar redox behavior [88]. Electron-withdrawing moieties such as cyanoquinoid structures, as two terminal dicyanomethylene groups, were also efficient to accommodate quinoidal diradicals and led to primitive highly conducting donor-acceptor charge-transfer complex [89]. These aromatic and quinoidal structures are also of great significance and give rise to interesting electronic and optical properties in conjugated polymers [90, 91]. To fully understand the structures, properties, and behaviors of open-shell polymer semiconductors in optoelectronics, studies of simple oligomeric analogs serve as a good model. Using the “oligomer approach” [92] allows for the synthesis of well-defined chain-length for probing the structure-property relationship along with using theoretical computations to guide design and predict properties. Review on aromaticity and its influence on oligomeric ground state radical characters in various types of small molecules was written by Wu and coworkers in 2015 [82].

a Resonance structures of Thiele’s and Tschitschibabin’s hydrocarbons with p-quinodimethane (p-QDM) highlighted in red. b Cyanoquinoid oligothiophene in open-shell aromatic diradical and closed-shell quinoidal resonance structures
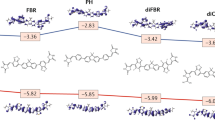
The broader scope of these diradicaloids includes variations of the p-QDM unit, such as replacing the backbone benzene rings with thiophenes for oligothiophene diradicals (Fig. 3b). It was known from extensive polythiophene research that twisting around the C–C single bond between thiophene units in its aromatic form may reduce the effective conjugation length [93], while the formation of quinoid segments is known during the oxidative p-doping of polythiophenes [94], which increases the planarity of the polymer and conductivity. Combination of cyanoquinoid and oligothiophenes offered a great platform for systematic linear extension of conjugation length or fused-ring varieties, for establishing ground state character as a function of oligomer structures. A detailed review has been published in 2012 by Navarrete and coworkers on the unconventional electronic structure, optical and magnetic properties, and material research of quinoidal oligothiophenes [94]. Briefly, significant red-shift of absorption wavelengths was observed for each quinoidal oligothiophene compared to their non-substituted counterparts [95]. For example, the maximum absorption of Q5 reached 913 nm and an extra thiophene unit in the series had an absorption peak beyond 1000 nm, both attributed to intramolecular charge-transfer transition [95]. Although shorter quinoidal oligothiophene tends to exhibit closed-shell properties, it gradually transformed into an open-shell with prominent diradical characters upon an increase in the conjugation length, as a result of the restoration of aromaticity [95]. In both experimental and theoretical studies, quinoidal oligothiophenes resulted in a very small singlet–triplet (ΔEST) energy gap, only of about 2.6 kcal/mol for Q5, thus allow thermal population of triplet states [96]. Among quinoidal oligothiophene, Q5 shows sufficient stability due to a considerable number of aromatic rings that compensate for the open-shell diradical [94]. Further computational work summarized the length-dependent preference of open-shell vs. closed-shell singlet ground states (Fig. 4) [97]. From structure manipulation aspect, an increase in the number of thiophene rings of the quinoid oligothiophene extends the conjugation length and in turn also increases the distance between terminal radicals. This results in a decrease in the ΔEST, the energy gap between singlet and triplet (Fig. 4). The continuous decrease in the ΔEST eventually reaches a negative value, which is indicative of preferred lower energy triplet state diradical.

Calculated relative energies between the singlet closed-shell (SCS), singlet open-shell (SOS) and triplet (T) states for increasing number of thiophene rings in the quinoid oligothiophenes. Reprinted with permission from reference [97]. Copyright 2011 American Chemical Society
In addition, polyradicals were demonstrated with fluorene and carbazole units in linear conjugated [98] and macrocyclic fused [99] structures. In both cases, the backbone of the oligomer was repeating Tschitschibabin’s hydrocarbon analog and the entire molecule behaves as a singlet ground state as a result of the antiferromagnetic coupling of polyradicals [98, 99]. In other diradicaloids that can be regarded as variations of conjugated p-QDM blocks, this length-dependent ground state preference was also a major contributing factor that determines radical characteristics in the ground state. Compared to the same number of repeating thiophene units, connection through the 3,10-positions of perylene effectively doubled the conjugation length [100], thus enabled studies of long-range diradical behavior with the span of oligo(p-QDM) up to 12 p-phenylene linkers [101]. As the number of N-annulated perylene spacer increased, the ground state of the oligomers gradually evolved across three characters: (a) closed-shell quinoid with only one perylene spacer unit; to (b) open-shell singlet diradical with 2~4 units, and eventually (c) triplet diradical in the presence of 5 units [101].
The highly-fused open-shell polycyclic aromatic hydrocarbon (PAH) is another class of biradicaloid due to its aromatic/quinoid resonance forms [82]. PAHs are excellent candidates for organic optoelectronic materials, and relevant reviews are available on topics such as functionalized (hetero)acenes [102, 103], low-band-gap PAHs [104], and advances in nano-graphene chemistry [105]. Visualized by Clar’s sextet rule [106], many large PAHs yield ground state radical characters as a result of restored aromaticity. PAHs can be considered as structural combinations of p-QDM and other non-Kekulé aromatics such as the phenalenyl radical (Fig. 5). The bisphenalenyls prototype that connects two phenalenyl with one p-QDM was reported to possess moderate singlet biradical character with a small HOMO-LUMO energy gap [107]. The two delocalized radicals in the bisphenalenyls have strong intramolecular interactions, and because of the flat geometry that facilitates π-stacking, strong intermolecular interactions between the unpaired electrons were also characterized. In contrast to quinoidal oligothiophene and analogs, the spacer between the two phenalenyls was typically extended by fusing p-QDM units that resemble acenes and keep all 5- and 6-member rings fused together. Reports in 2012 demonstrated that extension of the conjugated length provide small optical band gap, large absorption cross sections, and increased semiconducting properties [108, 109]. Increase in the fused p-QDM ladder bridge length can also lead to the larger planar conjugated core that facilitates intermolecular π-stacking, as well as further spatial separation of intramolecular phenalenyl terminals. The anthracene-linked bisphenalenyl exhibits highly ordered slip packing in crystals, with an intermolecular distance of only 3.1 Å, even shorter than the van der Waals contact of carbon atoms [109]. Such close contact and covalent bonding interaction between molecules painted a picture of strong intermolecular and weak intramolecular interactions in the bisphenalenyls crystals [108, 109].

Bisphenalenyls and zethrenes as structural combinations of phenalenyl and p-QDM blocks
Similarly, the zethrene biradicaloid can be seen as two phenalenyl radicals sharing carbon atoms with the p-QDM bridge (Fig. 5). The basic zethrene favors a closed-shell quinoid ground state[82]. Extension of the central fused p-QDM ladder bridge affords the longer derivatives of zethrene, namely heptazethrene and octazethrene [110]. Steric-group substituted zethrenes are decently stable and radical characters becomes more prominent at extended zethrenes, especially the singlet diradical ground state of octazethrene [110]. Interestingly, a series of substituted zethrene diradicaloids were found to result in a formation of the triplet-pair state upon photoexcitation that leads to efficient singlet fission [111]. It could be envisioned that should the fused p-QDM bridge be sufficiently long, the ground state of bisphenalenyls and zethrenes can also lean toward triplet diradicals, similar to finding in the oligo(N-annulated perylene) discussed above.
Structures with 4n π-electrons classified as anti-aromatic by Hückel's rule emerged as a new approach for ground state diradicals. An example among the latest development is the quinoidal methano[10]annulene, which exhibits small singlet-triplet energy gap and a significant geometry change from contorted S0 to planar T1 [112]. Its stability was attributed to Baird’s rule in which excited state aromaticity consists of 4n π-electrons [112]. The p-QDM bridged bisphenalenyls can be regarded as the core-extension analog of indenofluorene [113], known as a series of 20-electron PAH structures (Fig. 6a). Indenofluorenes have been studied by Haley and others as a prototype for ladder polyradicals, and several fusion modes that provide stable indenofluorene derivatives were identified [114,115,116]. While most of the basic indenofluorene structures proved to be closed-shell quinoid at ground state [82], radical characters emerge as the result of an increase in π-conjugation length. Examples include diindeno-fused naphthalene and anthracene which exhibit radical characters (Fig. 6b). Fusion of QDM rings provides a strategy to enable facile synthesis of DIAn as a stable singlet diradical with thermally accessible triplet excited state (~4 kcal/mol) [117, 118]. Further studies revealed that DIAn can also form stable charged species with its rich and reversible redox activities, and could be potentially excellent materials for ambipolar organic field-effect transistors [119]. Expansion of the indenofluorene structure can be carried out in two directions: along with or perpendicular to the ring alignment. Both were demonstrated in new stable PAHs of indenotetraphene, indenopicene, and phenanthrotetraphene in 2017 [120]. These large anti-aromatic PAHs consist of 4n π-electron systems and slightly curved molecular structures [120]. They exhibit low band gapp and reversible redox activities, and preliminary results show promising potential as charge transport materials in OFETs.

a Quinoid and diradical structures of indenofluorene in different fusion modes, and examples of (b) diindenoanthracene, (c) bis(benzothia)quinodimethanes and (d) dibenzopentalene
Generally, in the case of open-shell polycyclic aromatics containing heteroatoms, an increase in the conjugation length leads to an increase in the diradical character (Fig. 6c) [121], similar to what has been reported with open-shell PAH. This effect of length dependence was found to be more pronounced in zethrene derivatives than in (hetero)acenes [122]. Meanwhile, fusion mode is another factor that plays a role in influencing the radical character in the ground state. For example, the recently reported dibenzo[a,f]pentalene exhibits strong anti-aromatic character and unusual singlet diradical features in a rather short conjugation length (Fig. 6d) [123], contrary to the dibenzo[a,e]pentalene known as a closed-shell molecule.
The m-QDM is known as a ferromagnetic spin coupling unit which leads to persistent diradical properties, contrary to the antiferromagnetic spin coupling unit p-QDM bridge [81]. While the core structure of Dibenzopentacene (DP) has the desirable m-QDM at the central part of its structure (Fig. 7a), isolation of the core ring without side groups remained elusive due to the high reactivity DP [124]. Wu and coworkers were able to synthesize and isolate DP-Mes by kinetically blocking the reactive 5,7-positions with mesityl groups. DP-Mes is stable at −78 °C and exhibits rare persistent triplet diradical characteristics (Fig. 7a) [124]. The triplet character was attributed mainly to the spin delocalization in the DP framework. In some cases, p-QDM or m-QDM architecture may not play a significant role in influencing the diradical character. This was evidence in the case of BODIPY bridged by p-QDM or m-QDM coupling units (Fig. 7b) [125]. More recent studies on benzothia-fused thienoacene derivatives (Fig. 7c) revealed that diradical character (y) not only depends with extended ring fusion but is also related to the anti-aromatic or pro-aromatic nature of its structure, with the latter giving higher y values [126, 127]. Open-shell ground state diradical can be also obtained by chemical reduction of neutral species, as demonstrated by phosphorus-centered diradical dianion synthesized from diphosphaalkene and isolated as crystals [128].

Recent examples of polycyclic diradicals: (a) dibenzopentacene, (b) BODIPY, and (c) thienoacenequinodimethanes derivatives
Future prospects
There has been tremendous interests and developments in the field of open-shell organic semiconductors. Singlet diradicaloid with moderate diradical character exhibits very interesting properties that range from small energy gap, unique magnetic and thermo/photo-magnetic behavior, to enhanced third non-linear responses. Certainly, these properties are poised to enable new functions and applications ranging from field-effect transistors, solar cells, RFIDs, to organic spintronics. While there are diverse and impressive structures of diradicaloid reported to date, most studies have focused on small molecules and oligomers. This is because the synthesis of highly conjugated stable structures and polymers remains to be elusive and a significant challenge. Current synthetic strategies of open-shell PAHs often require multiple steps in series, and the final open-shell PAH products are not always shelf-stable. PAHs with even longer π-conjugated systems are expected to show intriguing properties including quartet radical characters or even polyradical characters that may lead to ferromagnetism and superconductivity. Thus, the development of synthetic strategies to produce polyradicals is a crucial aspect toward propelling this field to the forefront. Open-shell PAH polymers could be the conduit to the realization of anticipated novel properties and enable broader application in magneto-electronic devices [129]. In addition, from the observed EPR signals, diradical characters were proposed in a variety of low-band-gap small molecules and polymers with conjugated donor-acceptor moieties as the molecular backbone [130, 131]. The push-pull effect and electron delocalization in these examples is crucial in stabilizing the intrinsic diradicals, providing another strategy to design new polymer semiconductor structures and study its relationship with radical property [131, 132].
Incorporations of radical pendants to π-conjugated molecules can be a powerful strategy toward engineering excited-states. There is great synthetic flexibility that allows for systematic structure-property correlation studies. Although handful studies have investigated the influence of the distance between pendant radicals and the π-system on the ISC process, possible structural factors such as π-conjugation length, number of radical units per conjugation length, electronic coupling, and geometrical influence are yet to be determined. For example, it is not clear yet what are the important structural and electronic parameters that are required to mediate EISC and that determine excited state energy levels upon photoexcitation in open-shell organic semiconductors. Recently, there has been significant interest in harvesting triplet exciton through EISC for potential in optoelectronic devices. Thus, this area of research would benefit tremendously from combinations of materials design, spectroscopic studies, and computational theory.
References
Gomberg M. Organic Radicals. Chem Rev. 1924;1:91–141.
Gomberg M. An instance of trivalent carbon: triphenylmethyl. J Am Chem Soc. 1900;22:757–71.
Yan M, Lo JC, Edwards JT, Baran PS. Radicals: reactive intermediates with translational potential. J Am Chem Soc. 2016;138:12692–714.
Hawker CJ. Molecular weight control by a ‘living’ free-radical polymerization process. J Am Chem Soc. 1994;116:11185–6.
Wang J-S, Matyjaszewski K. Controlled/‘living’ radical polymerization. atom transfer radical polymerization in the presence of transition-metal complexes. J Am Chem Soc. 1995;117:5614–5.
Matyjaszewski K, Xia J. Atom transfer radical polymerization. Chem Rev. 2001;101:2921–90.
Meijs GF, Rizzardo E, Thang SH. Preparation of controlled-molecular-weight, olefin-terminated polymers by free radical methods. Chain transfer using allylic sulfides. Macromolecules. 1988;21:3122–4.
Moad G, Rizzardo E. Alkoxyamine-initiated living radical polymerization: factors affecting alkoxyamine homolysis rates. Macromolecules. 1995;28:8722–8.
Itoh K, Kinoshita M. Molecular magnetism—new magnetic materials. MRS Bull. 2002;27,1.
Rajca A, Wongsriratanakul J, Rajca S. Magnetic ordering in an organic polymer. Science. 2001;294:1503–5.
Affronte M. Molecular nanomagnets for information technologies. J Mater Chem. 2009;19:1731–7.
Camarero J, Coronado E. Molecular vs. inorganic spintronics: the role of molecular materials and single molecules. J Mater Chem. 2009;19:1678.
Ratera I, Veciana J. Playing with organic radicals as building blocks for functional molecular materials. Chem Soc Rev. 2012;41:303–49.
Sanvito S. Molecular spintronics. Chem Soc Rev. 2011;40:3336.
Stamp PCE, Gaita-Ariño A. Spin-based quantum computers made by chemistry: hows and whys. J Mater Chem. 2009;19:1718–30.
Sugawara T, Matsushita MM. Spintronics in organic π-electronic systems. J Mater Chem. 2009;19:1738–53.
Wolf SA, Awschalom DD, Buhrman RA, Daughton JM, von Molnár S, Roukes ML, Chtchelkanova AY, Treger DM. Spintronics: a spin-based electronics vision for the future. Science. 2001;294:1488–95.
Troiani F, Affronte M. Molecular spins for quantum information technologies. Chem Soc Rev. 2011;40:3119.
Baibich MN, Broto JM, Fert A, Van Dau FN, Petroff F, Etienne P, Creuzet G, Friederich A, Chazelas J. Giant magnetoresistance of (001)Fe/(001)Cr magnetic superlattices. Phys Rev Lett. 1988;61:2472–5.
Yonekuta Y, Susuki K, Oyaizu K, Honda K. Battery-inspired, nonvolatile, and rewritable memory architecture: a radical polymer-based organic device. J Am Chem Soc. 2007;129:14128–9.
Dediu VA, Hueso LE, Bergenti I, Taliani C. Spin routes in organic semiconductors. Nat Mater. 2009;8:707–16.
Urdampilleta M, Klyatskaya S, Cleuziou J-P, Ruben M, Wernsdorfer W. Supramolecular spin valves. Nat Mater. 2011;10:502–6.
Zhang Y, Basel TP, Gautam BR, Yang X, Mascaro DJ, Liu F, Vardeny ZV. Spin-enhanced organic bulk heterojunction photovoltaic solar cells. Nat Commun. 2012;3:1043.
Gaster RS, Xu L, Han S-J, Wilson RJ, Hall DA, Osterfeld SJ, Yu H, Wang SX. Quantification of protein interactions and solution transport using high-density GMR sensor arrays. Nat Nanotechnol. 2011;6:314–20.
González JW, Delgado F, Fernández-Rossier J. Graphene single-electron transistor as a spin sensor for magnetic adsorbates. Phys Rev B. 2013;87:85433.
Wang F, Vardeny ZV. Organic spin valves: the first organic spintronics devices. J Mater Chem. 2009;19:1685–90.
Wang J, Chepelianskii A, Gao F, Greenham NC. Control of exciton spin statistics through spin polarization in organic optoelectronic devices. Nat Commun. 2012;3:1191.
Nguyen TD, Ehrenfreund E, Vardeny ZV. Spin-polarized light-emitting diode based on an organic bipolar spin valve. Science. 2012;337:204–9.
Joo Y, Agarkar V, Sung SH, Savoie BM, Boudouris BW. A nonconjugated radical polymer glass with high electrical conductivity. Science. 2018;359:1391–5.
Xue Q, Liu M, Li Z, Yan L, Hu Z, Zhou J, Li W, Jiang X-F, Xu B, Huang F, Li Y, Yip H-L, Cao Y. Efficient and stable perovskite solar cells via dual functionalization of dopamine semiquinone radical with improved trap passivation capabilities. Adv. Funct. Mater. 2018. https://doi.org/10.1002/adfm.201707444
Xue Y, Guo P, Yip H-L, Li Y, Cao Y. General design of self-doped small molecules as efficient hole extraction materials for polymer solar cells. J Mater Chem A. 2017;5:3780–5.
Klein DJ, Nelin CJ, Alexander S, Matsen FA. High‐spin hydrocarbons. J Chem Phys. 1982;77:3101–8.
Ovchinnikov AA. Multiplicity of the ground state of large alternant organic molecules with conjugated bonds. Theor Chim Acta. 1978;47:297–304.
Korshak YV, Medvedeva TV, Ovchinnikov AA, Spector VN. Organic polymer ferromagnet. Nature. 1987;326:370–2.
Miller JS. The quest for magnetic polymers-caveat emptor. Adv Mater. 1992;4:298–300.
Nishide H. High-spin alignment in π-conjugated Polyradicals: A Magnetic polymer. Adv Mater. 1995;7:937–41.
Miura Y, Ushitani Y, Inui K, Teki Y, Takui T, Itoh K. Syntheses and magnetic characterization of poly(1,3-phenylene-ethynylene) with pendant nitronyl nitroxide radicals. Macromolecules. 1993;26:3698–701.
Nishide H, Nambo M, Miyasaka M. Hyperbranched poly(phenylenevinylene) bearing pendant phenoxys for a high-spin alignment. J Mater Chem. 2002;12:3578–84.
Selby TD, Blackstock SC. Naphthyldiamine diradical dications. triplet dications of 2,7-bis(amino)naphthalene and 2,7-bis(phenylenediamino)naphthalene. J Am Chem Soc. 1999;121:7152–3.
Nishide H, Miyasaka M, Doi R, Araki T. Poly(1,2-phenylenevinylene) ferromagnetically 3,5-bearing phenoxyl radicals. Macromolecules. 2002;35:690–8.
Nishide H, Kaneko T, Nii T, Katoh K, Tsuchida E, Yamaguchi K. Through-bond and long-range ferromagnetic spin alignment in a .pi.-conjugated polyradical with a poly(phenylenevinylene) skeleton. J Am Chem Soc. 1995;117:548–9.
Hayashi H, Yamamoto T. Synthesis of regioregular π-conjugated poly(thienyleneethynylene) with a hindered phenolic substituent. Macromolecules. 1997;30:330–2.
Anderson KK, Dougherty DA. An improved model for one-dimensional polaronic ferromagnetism: electrochemically doped poly(m-phenylenefuchsone). Adv Mater. 1998;10:688–92.
Miyasaka M, Yamazaki T, Tsuchida E, Nishide H. Regioregular polythiophene with pendant phenoxyl radicals: a new high-spin organic polymer. Macromolecules. 2000;33:8211–7.
Nakatsuji S. Recent progress toward the exploitation of organic radical compounds with photo-responsive magnetic properties. Chem Soc Rev. 2004;33:348.
Mitsumori T, Inoue K, Koga N, Iwamura H. Exchange interactions between two nitronyl nitroxide or iminyl nitroxide radicals attached to thiophene and 2,2’-bithienyl rings. J Am Chem Soc. 1995;117:2467–78.
Wautelet P, Le Moigne J, Videva V, Turek P. Spin exchange interaction through phenylene-ethynylene bridge in diradicals based on iminonitroxide and nitronylnitroxide radical derivatives. 1. experimental investigation of the through-bond spin exchange coupling. J Org Chem. 2003;68:8025–36.
Blättler C, Jent F, Paul H. A novel radical-triplet pair mechanism for chemically induced electron polarization (CIDEP) of free radicals in solution. Chem Phys Lett. 1990;166:375–80.
Kawai A, Okutsu T, Obi K. Spin polarization generated in the triplet-doublet interaction: hyperfine-dependent chemically induced dynamic electron polarization. J Phys Chem. 1991;95:9130–4.
Kawai A, Obi K. First observation of a radical-triplet pair mechanism (RTPM) with doublet precursor. J Phys Chem. 1992;96:52–6.
Ishii K, Fujisawa J, Adachi A, Yamauchi S, Kobayashi N. General simulations of excited quartet spectra with electron-spin polarizations: the excited multiplet states of (tetraphenylporphinato)zinc(ii) coordinated by p- or m-pyridyl nitronyl nitroxides. J Am Chem Soc. 1998;120:3152–8.
Ishii K, Hirose Y, Kobayashi N. Selective population from the excited multiplet states to the triplet ground state in a phthalocyanine: a new concept for controlling magnetic properties by photoexcitation. J Am Chem Soc. 1998;120:10551–2.
Takeuchi S, Ishii K, Kobayashi N. Time-resolved EPR and transient absorption studies on phthalocyaninatosilicon covalently linked to two PROXYL radicals. J Phys Chem A. 2004;108:3276–80.
Mizuochi N, Ohba Y, Yamauchi S. First observation of the photoexcited quintet state in fullerene linked with two nitroxide radicals. J Phys Chem A. 1999;103:7749–52.
Conti F, Corvaja C, Toffoletti A, Mizuochi N, Ohba Y, Yamauchi S, Maggini M. EPR Studies on a binitroxide fullerene derivative in the ground triplet and first photoexcited quintet state. J Phys Chem A. 2000;104:4962–7.
Teki Y, Miyamoto S, Nakatsuji M, Miura Y. π-topology and spin alignment utilizing the excited molecular field: observation of the excited high-spin quartet (s=3/2) and quintet (s=2) states on purely organic π-conjugated spin systems. J Am Chem Soc. 2001;123:294–305.
Matsumoto I, Ciofini I, Lainé PP, Teki Y. Intramolecular spin alignment within mono-oxidized and photoexcited anthracene-based π radicals as prototypical photomagnetic molecular devices: relationships between electrochemical, photophysical, and photochemical controlpathways. Chem-A Eur J. 2009;15:11210–20.
Ciofini I, Adamo C, Teki Y, Tuyèras F, Lainé PP. Reaching optimal light-induced intramolecular spin alignment within photomagnetic molecular device prototypes. Chemistry. 2008;14:11385–405.
Likhtenstein GI, Ishii K, Nakatsuji S. Dual chromophore-nitroxides: novel molecular probes, photochemical and photophysical models and magnetic materials. Photochem Photobiol. 2007;83:871–81.
Levanon H, Norris JR. The photoexcited triplet state and photosynthesis. Chem Rev. 1978;78:185–98.
Schmidt D, Son M, Lim JM, Lin M-J, Krummenacher I, Braunschweig H, Kim D, Würthner F. Perylene bisimide radicals and biradicals: synthesis and molecular properties. Angew Chem Int Ed. 2015;54:13980–4.
Lakowicz JR. Principles of fluorescence spectroscopy. New York, Springer US, 2006.
Dexter DL. A theory of sensitized luminescence in solids. J Chem Phys. 1953;21:836.
Giacobbe EM, Mi Q, Colvin MT, Cohen B, Ramanan C, Scott AM, Yeganeh S, Marks TJ, Ratner MA, Wasielewski MR. Ultrafast intersystem crossing and spin dynamics of photoexcited perylene-3,4:9,10-bis(dicarboximide) covalently linked to a nitroxide radical at fixed distances. J Am Chem Soc. 2009;131:3700–12.
Ito A, Shimizu A, Kishida N, Kawanaka Y, Kosumi D, Hashimoto H, Teki Y. Excited-state dynamics of pentacene derivatives with stable radical substituents. Angew Chem Int Ed. 2014;53:6715–9.
Wang Z, Zhao J, Barbon A, Toffoletti A, Liu Y, An Y, Xu L, Karatay A, Yaglioglu HG, Yildiz EA, Hayvali M. Radical-enhanced intersystem crossing in new bodipy derivatives and application for efficient triplet-triplet annihilation upconversion. J Am Chem Soc. 2017;139:7831–42.
Colvin MT, Giacobbe EM, Cohen B, Miura T, Scott AM, Wasielewski MR. Competitive electron transfer and enhanced intersystem crossing in photoexcited covalent TEMPO−perylene-3,4:9,10-bis(dicarboximide) dyads: unusual spin polarization resulting from the radical−triplet interaction. J Phys Chem A. 2010;114:1741–8.
Kawanaka Y, Shimizu A, Shinada T, Tanaka R, Teki Y. Using stable radicals to protect pentacene derivatives from photodegradation. Angew Chem Int Ed. 2013;52:6643–7.
Shimizu A, Ito A, Teki Y. Photostability enhancement of the pentacene derivative having two nitronyl nitroxide radical substituents. Chem Commun. 2016;52:2889–92.
Chernick ET, Casillas R, Zirzlmeier J, Gardner DM, Gruber M, Kropp H, Meyer K, Wasielewski MR, Guldi DM, Tykwinski RR. Pentacene appended to a TEMPO stable free radical: the effect of magnetic exchange coupling on photoexcited pentacene. J Am Chem Soc. 2015;137:857–63.
Dyar SM, Margulies EA, Horwitz NE, Brown KE, Krzyaniak MD, Wasielewski MR. Photogenerated quartet state formation in a compact ring-fused perylene-nitroxide. J Phys Chem B. 2015;119:13560–9.
Chernick ET, Mi Q, Kelley RF, Weiss EA, Jones BA, Marks TJ, Ratner MA, Wasielewski MR. Electron donor−bridge−acceptor molecules with bridging nitronyl nitroxide radicals: influence of a third spin on charge- and spin-transfer dynamics. J Am Chem Soc. 2006;128:4356–64.
Colvin MT, Carmieli R, Miura T, Richert S, Gardner DM, Smeigh AL, Dyar SM, Conron SM, Ratner MA, Wasielewski MR. Electron spin polarization transfer from photogenerated spin-correlated radical pairs to a stable radical observer Spin. J Phys Chem A. 2013;117:5314–25.
Horwitz NE, Phelan BT, Nelson JN, Krzyaniak MD, Wasielewski MR. Picosecond control of photogenerated radical pair lifetimes using a stable third radical. J Phys Chem A. 2016;120:2841–53.
Rugg BK, Phelan BT, Horwitz NE, Young RM, Krzyaniak MD, Ratner MA, Wasielewski MR. Spin-selective photoreduction of a stable radical within a covalent donor–acceptor–radical triad. J Am Chem Soc. 2017;139:15660–3.
Matsuda K, Matsuo M, Irie M. Photoswitching of intramolecular magnetic interaction using diarylethene with oligothiophene π-conjugated chain. J Org Chem. 2001;66:8799–803.
Yapici NB, Jockusch S, Moscatelli A, Mandalapu SR, Itagaki Y, Bates DK, Wiseman S, Gibson KM, Turro NJ, Bi L. New rhodamine nitroxide based fluorescent probes for intracellular hydroxyl radical identification in living cells. Org Lett. 2012;14:50–53.
Miao F, Lim ZL, Hu P, Dong S, Qi Q, Zhang X, Wu J. BODIPY blocked anthroxyl radicals: the substituent effect on reactivity and fluorescence turn-on detection of a hydroxyl radical. Org Biomol Chem. 2017;15:3188–91.
Hattori Y, Kusamoto T, Nishihara H. Luminescence, stability, and proton response of an open-shell (3,5-dichloro-4-pyridyl)bis(2,4,6-trichlorophenyl)methyl radical. Angew Chem Int Ed. 2014;53:11845–8.
Peng Q, Obolda A, Zhang M, Li F. Organic light-emitting diodes using a neutral π radical as emitter: the emission from a doublet. Angew Chem Int Ed. 2015;54:7091–5.
Rajca A. Organic diradicals and polyradicals: from spin coupling to magnetism? Chem Rev. 1994;94:871–93.
Zeng Z, Shi X, Chi C, López Navarrete JT, Casado J, Wu J. Pro-aromatic and anti-aromatic π-conjugated molecules: an irresistible wish to be diradicals. Chem Soc Rev. 2015;44:6578–96.
Thiele J, Balhorn H. Ueber einen chinoïden Kohlenwasserstoff. Berichte der Dtsch. Chem Ges. 1904;37:1463–70.
Tschitschibabin AE. Über einige phenylierte Derivate desp,p-Ditolyls. Berichte der Dtsch. Chem Ges. 1907;40:1810–9.
Montgomery LK, Huffman JC, Jurczak EA, Grendze MP. The molecular structures of Thiele’s and Chichibabin’s hydrocarbons. J Am Chem Soc. 1986;108:6004–11.
Tan G, Wang X. Isolable bis(triarylamine) dications: analogues of thiele’s, chichibabin’s, and müller’s hydrocarbons. Acc Chem Res. 2017;50:1997–2006.
Zeng Z, Sung YM, Bao N, Tan D, Lee R, Zafra JL, Lee BS, Ishida M, Ding J, López Navarrete JT, Li Y, Zeng W, Kim D, Huang K-W, Webster RD, Casado J, Wu J. Stable tetrabenzo-chichibabin’s hydrocarbons: tunable ground state and unusual transition between their closed-shell and open-shell resonance forms. J Am Chem Soc. 2012;134:14513–25.
Zhang K, Huang K-W, Li J, Luo J, Chi C, Wu J. A soluble and stable quinoidal bisanthene with nir absorption and amphoteric redox behavior. Org Lett. 2009;11:4854–7.
Yui K, Aso Y, Otsubo T, Ogura F. Novel electron acceptors bearing a heteroquinonoid system. i. synthesis and conductive complexes of 5,5′-bis(dicyanomethylene)-5,5′-dihydro-δ2,2′-bithiophene and related compounds. Bull Chem Soc Jpn. 1989;62:1539–46.
Heeger AJ. Semiconducting and metallic polymers: the fourth generation of polymeric materials (nobel lecture). Angew Chem Int Ed. 2001;40:2591–611.
MacDiarmid AG. ‘Synthetic metals’: a novel role for organic polymers (nobel lecture). Angew Chem Int Ed. 2001;40:2581–90.
Müllen K, Wegner G. Electronic materials: the oligomer approach. Weinheim, Wiley-VCH, 1998.
Ten Hoeve W, Wynberg H, Havinga EE, Meijer EW. Substituted 2,2’:5 ’,2’’:5’’,2’’’:5’ ’’,2’’’’:5 ’’’’,2’’ ’’’: 5’’’ ’’,2 ’ ’’’’ ’:5’’’ ’’ ’,2’ ’’’’ ’’:5’’ ’’ ’’ ’,2’’’ ’’ ’’ ’:5’ ’’ ’’’’ ’,2’’’ ’’ ’’ ’’:5’’ ’’’’ ’’ ’,2’ ’’’’ ’’’’’- undecithiophenes, the longest characterized oligothiophenes. J Am Chem Soc. 1991;113:5887–9.
Casado J, Ponce Ortiz R, López Navarrete JT. Quinoidal oligothiophenes: new properties behind an unconventional electronic structure. Chem Soc Rev. 2012;41:5672.
Takahashi T, Matsuoka K, Takimiya K, Otsubo T, Aso Y. Extensive quinoidal oligothiophenes with dicyanomethylene groups at terminal positions as highly amphoteric redox molecules. J Am Chem Soc. 2005;127:8928–9.
Ponce Ortiz R, Casado J, Hernández V, López Navarrete JT, Viruela PM, Ortí E, Takimiya K, Otsubo T. On the biradicaloid nature of long quinoidal oligothiophenes: experimental evidence guided by theoretical studies. Angew Chem Int Ed. 2007;46:9057–61.
González SR, Ie Y, Aso Y, López Navarrete JT, Casado J. The frontiers of quinoidal stability in long oligothiophenes: raman spectra of dicationic polaron pairs. J Am Chem Soc. 2011;133:16350–3.
Lu X, Lee S, Kim JO, Gopalakrishna TY, Phan H, Herng TS, Lim Z, Zeng Z, Ding J, Kim D, Wu J. Stable 3,6-linked fluorenyl radical oligomers with intramolecular antiferromagnetic coupling and polyradical characters. J Am Chem Soc. 2016;138:13048–58.
Das S, Herng TS, Zafra JL, Burrezo PM, Kitano M, Ishida M, Gopalakrishna TY, Hu P, Osuka A, Casado J, Ding J, Casanova D, Wu J. Fully Fused quinoidal/aromatic carbazole macrocycles with poly-radical characters. J Am Chem Soc. 2016;138:7782–90.
Zeng Z, Lee S, Zafra JL, Ishida M, Zhu X, Sun Z, Ni Y, Webster RD, Li R-W, López Navarrete JT, Chi C, Ding J, Casado J, Kim D, Wu J. Tetracyanoquaterrylene and tetracyanohexarylenequinodimethanes with tunable ground states and strong near-infrared absorption. Angew Chem Int Ed. 2013;52:8561–5.
Zeng Z, Ishida M, Zafra JL, Zhu X, Sung YM, Bao N, Webster RD, Lee BS, Li R-W, Zeng W, Li Y, Chi C, Navarrete JTL, Ding J, Casado J, Kim D, Wu J. Pushing extended p-quinodimethanes to the limit: stable tetracyano-oligo(n-annulated perylene)quinodimethanes with tunable ground states. J Am Chem Soc. 2013;135:6363–71.
Anthony JE. The larger acenes: versatile organic semiconductors. Angew Chem Int Ed. 2008;47:452–83.
Anthony JE. Functionalized acenes and heteroacenes for organic electronics. Chem Rev. 2006;106:5028–48.
Sun Z, Ye Q, Chi C, Wu J. Low band gap polycyclic hydrocarbons: from closed-shell near infrared dyes and semiconductors to open-shell radicals. Chem Soc Rev. 2012;41:7857.
Narita A, Wang X-Y, Feng X, Müllen K. New advances in nanographene chemistry. Chem Soc Rev. 2015;44:6616–43.
Eric C. The aromatic sextet. in Rondia D et al., editors. Mobile source emissions including policyclic organic species. Dordrecht, Springer Netherlands, 1983; p. 49–58.
Kubo T, Shimizu A, Sakamoto M, Uruichi M, Yakushi K, Nakano M, Shiomi D, Sato K, Takui T, Morita Y, Nakasuji K. Synthesis, intermolecularinteraction, and semiconductive behavior of a delocalized singlet biradical hydrocarbon. Angew Chem Int Ed. 2005;44:6564–8.
Shimizu A, Kubo T, Uruichi M, Yakushi K, Nakano M, Shiomi D, Sato K, Takui T, Hirao Y, Matsumoto K, Kurata H, Morita Y, Nakasuji K. Alternating covalent bonding interactions in a one-dimensional chain of a phenalenyl-based singlet biradical molecule having kekulé structures. J Am Chem Soc. 2010;132:14421–8.
Shimizu A, Hirao Y, Matsumoto K, Kurata H, Kubo T, Uruichi M, Yakushi K. Aromaticity and π-bond covalency: prominent intermolecular covalent bonding interaction of a Kekulé hydrocarbon with very significant singlet biradical character. Chem Commun. 2012;48:5629.
Li Y, Heng W-K, Lee BS, Aratani N, Zafra JL, Bao N, Lee R, Sung YM, Sun Z, Huang K-W, Webster RD, López Navarrete JT, Kim D, Osuka A, Casado J, Ding J, Wu J. Kinetically blocked stable heptazethrene and octazethrene: closed-shell or open-shell in the ground state? J Am Chem Soc. 2012;134:14913–22.
Lukman S, Richter JM, Yang L, Hu P, Wu J, Greenham NC, Musser AJ. Efficient singlet fission and triplet-pair emission in a family of zethrene diradicaloids. J Am Chem Soc. 2017;139:18376–85.
Streifel BC, Zafra JL, Espejo GL, Gómez-García CJ, Casado J, Tovar JD. An Unusually small singlet-triplet gap in a quinoidal 1,6-methano[10]annulene resulting from baird’s 4n π-electron triplet stabilization. Angew Chem Int Ed. 2015;54:5888–93.
Barker JE, Frederickson CK, Jones MH, Zakharov LN, Haley MM. Synthesis and properties of quinoidal fluorenofluorenes. Org Lett. 2017;19:5312–5.
Maekawa T, Ueno H, Segawa Y, Haley MM, Itami K. Synthesis of open-shell ladder π-systems by catalytic C–H annulation of diarylacetylenes. Chem Sci. 2016;7:650–4.
Chase DT, Rose BD, McClintock SP, Zakharov LN, Haley MM. Indeno[1,2-b]fluorenes: fully conjugated antiaromatic analogues of acenes. Angew Chem Int Ed. 2011;50:1127–30.
Chase DT, Fix AG, Rose BD, Weber CD, Nobusue S, Stockwell CE, Zakharov LN, Lonergan MC, Haley MM. Electron-accepting 6,12-diethynylindeno[1,2-b]fluorenes: synthesis, crystal structures, and photophysical properties. Angew Chem Int Ed. 2011;50:11103–6.
Frederickson CK, Rose BD, Haley MM. Explorations of the indenofluorenes and expanded quinoidal analogues. Acc Chem Res. 2017;50:977–87.
Rudebusch GE, Zafra JL, Jorner K, Fukuda K, Marshall JL, Arrechea-Marcos I, Espejo GL, Ponce Ortiz R, Gómez-García CJ, Zakharov LN, Nakano M, Ottosson H, Casado J, Haley MM. Diindeno-fusion of an anthracene as a design strategy for stable organic biradicals. Nat Chem. 2016;8:753–9.
Rudebusch GE, Espejo GL, Zafra JL, Peña-Alvarez M, Spisak SN, Fukuda K, Wei Z, Nakano M, Petrukhina MA, Casado J, Haley MM. A biradical balancing act: redox amphoterism in a diindenoanthracene derivative results from quinoidal acceptor and aromatic donor motifs. J Am Chem Soc. 2016;138:12648–54.
Liu J, Ma J, Zhang K, Ravat P, Machata P, Avdoshenko S, Hennersdorf F, Komber H, Pisula W, Weigand JJ, Popov AA, Berger R, Müllen K, Feng X. π-extended and curved antiaromatic polycyclic hydrocarbons. J Am Chem Soc. 2017;139:7513–21.
Dong S, Herng TS, Gopalakrishna TY, Phan H, Lim ZL, Hu P, Webster RD, Ding J, Chi C. Extended bis(benzothia)quinodimethanes and their dications: from singlet diradicaloids to isoelectronic structures of long acenes. Angew Chem Int Ed. 2016;55:9316–20.
Huang R, Phan H, Herng TS, Hu P, Zeng W, Dong S, Das S, Shen Y, Ding J, Casanova D, Wu J. Higher order π-conjugated polycyclic hydrocarbons with open-shell singlet ground state: nonazethrene versus nonacene. J Am Chem Soc. 2016;138:10323–30.
Konishi A, Okada Y, Nakano M, Sugisaki K, Sato K, Takui T, Yasuda M. Synthesis and characterization of dibenzo[a,f]pentalene: harmonization of the antiaromatic and singlet biradical character. J Am Chem Soc. 2017;139:15284–7.
Li Y, Huang K-W, Sun Z, Webster RD, Zeng Z, Zeng W, Chi C, Furukawa K, Wu J. A kinetically blocked 1,14:11,12-dibenzopentacene: a persistent triplet diradical of a non-Kekulé polycyclic benzenoid hydrocarbon. Chem Sci. 2014;5:1908.
Ni Y, Lee S, Son M, Aratani N, Ishida M, Samanta A, Yamada H, Chang Y-T, Furuta H, Kim D, Wu J. A diradical approach towards bodipy-based dyes with intense near-infrared absorption around λ =1100 nm. Angew Chem Int Ed. 2016;55:2815–9.
Shi X, Burrezo PM, Lee S, Zhang W, Zheng B, Dai G, Chang J, López Navarrete JT, Huang K-W, Kim D, Casado J, Chi C. Antiaromatic bisindeno-[n]thienoacenes with small singlet biradical characters: syntheses, structures and chain length dependent physical properties. Chem Sci. 2014;5:4490–503.
Shi X, Quintero E, Lee S, Jing L, Herng TS, Zheng B, Huang K-W, López Navarrete JT, Ding J, Kim D, Casado J, Chi C. Benzo-thia-fused [n]thienoacenequinodimethanes with small to moderate diradical characters: the role of pro-aromaticity versus anti-aromaticity. Chem Sci. 2016;7:3036–46.
Tan G, Li S, Chen S, Sui Y, Zhao Y, Wang X. Isolable diphosphorus-centered radical anion and diradical dianion. J Am Chem Soc. 2016;138:6735–8.
Swager TM. 50th anniversary perspective: conducting/semiconducting conjugated polymers. a personal perspective on the past and the future. Macromolecules. 2017;50:4867–86.
Yuen JD, Wang M, Fan J, Sheberla D, Kemei M, Banerji N, Scarongella M, Valouch S, Pho T, Kumar R, Chesnut EC, Bendikov M, Wudl F. Importance of unpaired electrons in organic electronics. J Polym Sci Part A Polym Chem. 2015;53:287–93.
Li Y, Li L, Wu Y, Li Y. A review on the origin of synthetic metal radical: singlet open-shell radical ground state? J Phys Chem C. 2017;121:8579–88.
Zeng Z, Lee S, Son M, Fukuda K, Burrezo PM, Zhu X, Qi Q, Li R-W, Navarrete JTL, Ding J, Casado J, Nakano M, Kim D, Wu J. Push–pull type oligo(n-annulated perylene)quinodimethanes: chain length and solvent-dependent ground states and physical properties. J Am Chem Soc. 2015;137:8572–83.
Acknowledgements
This work was supported by NSF grant CHE-1821863 (EE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Huang, Y., Egap, E. Open-shell organic semiconductors: an emerging class of materials with novel properties. Polym J 50, 603–614 (2018). https://doi.org/10.1038/s41428-018-0070-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-018-0070-6
This article is cited by
-
Electronic properties and optical spectra of donor–acceptor conjugated organic polymers
Scientific Reports (2023)
-
Synthesis of medium-sized benzo[b]azocines and benzo[b]azonines by photoinduced 8-/9-endo sulfonyl-cyclization
Science China Chemistry (2023)
-
Design of an open-shell nitrogen-centered diradicaloid with tunable stimuli-responsive electronic properties
Communications Chemistry (2022)
-
Evolution of the electronic structure in open-shell donor-acceptor organic semiconductors
Nature Communications (2021)
