
Abstract
Wg/Wnt as a signaling protein binds to Frizzled (Fz) and Arrow (Arr), two Wg co-receptors essential for Wg signaling for cell proliferation, differentiation, and cell survival. Arr has a long extracellular region, a single transmembrane domain and an intracellular region. Here, we report that a new arrm7 mutant is identified in a genetic screen as a suppressor of lethality induced by overexpression of Sol narae (Sona), a secreted metalloprotease in ADAMTS family involved in Wg signaling. arrm7 allele has a premature stop codon, which encodes Arrm7 protein missing the intracellular region. arrm7 clones show cell death phenotype and overexpression of Arrm7 protein also induces cell death. Levels of extracellular Sona were decreased in both arrm7 and arr2 null clones, demonstrating that Arr increases the level of extracellular Sona. Indeed, Arr but not Arrm7, increased levels of Sona in cytoplasm and exosome fraction by inhibiting the lysosomal degradation pathway. Interestingly, Arr itself was identified in the exosome fraction, demonstrating that Arr is secreted to extracellular space. When Sona-expressing S2 cells were treated with exosomal Arr, the extracellular level of active Sona was increased. These results show that exosomal Arr dictates Sona-expressing cells to increase the level of extracellular Sona. This new function of Arr occurred in the absence of Wg because S2 cells do not express Wg. We propose that Arr plays two distinct roles, one as an exosomal protein to increase the level of extracellular Sona in a Wnt-independent manner and the other as a Wg co-receptor in a Wnt-dependent manner.
Similar content being viewed by others


Introduction
Wnt signaling is an important pathway conserved in all metazoans for cell proliferation, cell survival, and differentiation1,2,3,4. Its malfunction causes developmental disorders, metabolic diseases, cancer development, and metastasis5,6,7,8. Wnt signaling is initiated by the binding of extracellular Wnt to the receptor complex containing Frizzled (Fz) as a main receptor and Arrow (Arr)/Lipoprotein receptor protein 6 (LRP6) as a co-receptor. Arr/LRP6 is a type I transmembrane protein that belongs to low density LRP family9. Interaction between Wnt and co-receptors results in a cascade of signaling transduction that leads to gene expression10,11.
Arr/LRP6 consists of a long extracellular region containing four β-propeller/epidermal growth factor repeats (E1–4) crucial for Wnt binding12,13, and three LDL repeats (LDLR) essential for dimerization of Arr/LRP6 proteins14 (Fig. 1). LRP6 without E1–4 activates Wnt signaling without Wnt, establishing that E1–4 negatively regulates Wnt signaling9. On the contrary, dimerization of LRP6 via LDLR domain is prerequisite for activating Wnt signaling14. Upon Wnt binding, the intracellular domain of LRP6 becomes phosphorylated, which eventually leads to stabilization of Armadillo (Arm)/β-catenin for the canonical Wnt signaling9,15.

a Genetic mapping of arrm7. The arr gene is mapped to the region deleted in both deficiencies, Df(2R)BSC273 and Df(2R)BSC274. The exons and introns of the arr gene are shown with the nonsense mutation site (G to A) marked with an asterisk. Wild-type and deletion forms of Arr are shown with domain structures. ΔN-Arr protein lacks the extracellular domain spanning 88–1450th amino acid residues while Arrm7 protein lacks the intracellular domain. b, c Experiments were carried out three times and flies were cultured at 25 °C. Adult progenies from crosses between 30A-Gal4/CyO and Canton S (CS) (n = 137 + 92 + 233 from 3 experiments), UAS-arr RNAi (n = 85 + 129 + 83 from 3 experiments), -sona (n = 103 + 166 + 154 from 3 experiments), -arr RNAi; -sona (n = 97 + 121 + 219 from 3 experiments) were obtained (b). Adult progenies from crosses between 30A-Gal4/CyO and UAS-arr-HA (n = 86 + 81 + 102 from 3 experiments), -sona RNAi (n = 89 + 57 + 137 from 3 experiments), -arr-HA; -sona RNAi (n = 31 + 52 + 53 from 3 experiments) were obtained (c). Data are presented as mean ± S.D.
We identified arrm7 as a suppressor of Sona-induced lethality in a genetic screen16. Other sona suppressors include wntless (wls), pou domain motif 3 (pdm3), and archipelago (ago), which all show genetic interaction with wingless (wg)16,17,18. Sona is a member of a disintegrin and metalloprotease with thrombospondin motif (ADAMTS) family that processes extracellular components. Sona promotes cell survival cell autonomously and increases cell proliferation in a paracrine manner19. Furthermore, extracellular Sona cleaves Wg and generates Wg-CTD that is another active form of Wg17,20. Wg-CTD is less stable than full-length Wg, and is more specialized for cell proliferation than differentiation20. Taken together, sona and sona suppressors are linked to Wg signaling.
Wg and Sona are secreted to extracellular space by both conventional Golgi transport and exosome secretion pathway20. Exosomes are extracellular vesicles (EVs) produced in the endosomal compartment21. Intraluminal vesicles inside of multivesicular body (MVB) are released as exosomes when MVB is fused to the plasma membrane. Exosomes contain mixed populations of EVs that deliver proteins, nucleic acids such as tRNAs and noncoding RNAs, and metabolites to neighboring cells22,23,24,25,26. Multiple Wnt signaling components are also secreted by the exosomal secretion pathway. For instance, Wls-containing exosomes deliver Wg from presynaptic neuron to postsynaptic muscle for development of larval neuromuscular junction27. β-catenin is packaged into exosomes and secreted, which results in downregulation of Wnt signaling28. Given that Sona is also involved in Wg signaling and is secreted by exosomal secretion pathway, Wnt signaling components may be commonly present in exosomes.
We focused on the biochemical relationship between Arr and Sona to understand the underlying mechanism of their genetic interaction. We found that the level of extracellular Sona is decreased in arr clones in wing discs. Consistent with this clonal analysis, overexpressed Arr stabilized Sona by inhibiting the lysosomal degradation pathway, which leads to increased levels of both intracellular and extracellular Sona in fly Schneider 2 (S2) cells. Interestingly, Arr was present in the exosome fraction, raising a possibility that extracellular Arr is also involved in stabilization of Sona. Indeed, more exosomal Sona was secreted from the Sona-expressing S2 cells cultured in the presence of exosomal Arr. This report shows for the first time that exosomal Arr stabilizes intracellular Sona and then increases the level of exosomal Sona.
Materials and methods
Fly lines
arrm7 mutant was derived from m7 sona suppressor as described16. UAS-arrm7-HA, FRT42D arrm7, and FRT42D arr2 are generated with arrm7 and BDSC #3287 for this study. UAS-sona lines are generated in our laboratory17. UAS-arr-HA is a generous gift from Konrad Basler. Other lines are obtained from BDSC and VDRC stock centers.
DNA constructs
To generate pUAST-sona-Myc, the sona cDNA17 was inserted into pUAST-Myc vector. pUAST-GFP-wg was generated by recombining pUAST vector and GFP-wg as described20. pUAST-arr and pUAST-arr-HA were generated by cloning arr cDNA clone from DGRC into pUAST and pUAST-HA vectors, respectively. ΔN-arr-HA was generated by deleting a DNA region encoding 88–1450th amino acid residues from arr-HA.
Cell culture, transfection, and preparation of various fractions
S2 and S2 R+ cell lines (DGRC) were cultured in M3 media (Sigma-Aldrich S8398) containing 10% insect medium supplement (Sigma-Aldrich I7267) at 25 °C. Media for S2 R+ cell lines contained additional yeast extract and bactopeptone. Transfection was performed with effectene transfection reagent (Qiagen). For P100 preparation, conditioned media were sequentially centrifuged at 240×g, 2000×g, 10,000×g and 100,000×g29. After 100,000×g centrifugation, supernatant fraction (SNΔ) was concentrated with a filter column. The obtained P100 fraction contains EVs including exosomes, was washed with PBS, and then pelleted again by additional centrifugation at 100,000×g.
Immunocytochemistry
Sona-Pro and Sona-C antibodies are used to detect Sona protein as described17. The Arr antibody is a generous gift from Suzanne Eaton. For immunocytochemistry, fly larvae were dissected and fixed with PLP solution (2% paraformaldehyde, 0.1 M lysine, 0.25% sodium M-periodate) as described30. Fixed wing discs were blocked in block buffer (50 mM Tris pH 6.8, 150 mM NaCl, 0.2% TritonX-100, 5 mg/ml bovine serum albumin) for 2–6 h at 4 °C and washed in wash buffer (0.2% TritonX-100 in PBS). Antibodies were diluted in wash buffer and incubated overnight at 4 °C or 2 h at room temperature. After washing several times, samples were treated with DAPI and mounted with a Vectashield mounting medium. Sample images were captured with Zeiss LSM laser scanning confocal microscope and processed by Adobe Photoshop. Sona-Pro (rabbit, 1:200), Sona-C (mouse, 1:100), HA (Roche 3F10, rat, 1:200), Arr (guinea pig, 1:150), cleaved caspase 3 (Cell signaling Asp175, rabbit, 1:100), Wg (DSHB 4D4, mouse, 1:500), and β-gal (Abcam ab9361, chicken, 1:100) were used.
For extracellular staining, larvae were dissected in ice-cold M3 medium and incubated in M3 medium containing primary antibody for 2 h at 4 °C as described31. Then, wing discs were washed with M3 and PBS twice each, and fixed in 4% paraformaldehyde in PBS for 50 min. Samples were then processed by the same way as described above but buffers did not contain any detergent. The antibodies were used at ten times higher concentration for extracellular staining than intracellular staining.
Western analysis and co-immunoprecipitation
Western analysis was performed as described with Thermo Scientific West Pico Plus Chemiluminescent Substrate for ECL system17. Sona-Pro (rabbit, 1:5000), HA (Roche 3F10, rat, 1:2000), Arr (guinea pig, 1:3000), Wg (DSHB 4D4, mouse, 1:500) and α-tubulin (Sigma-Aldrich T9026, mouse, 1:5000–10,000) were used.
For co-IP, protein G agarose beads (Merck Millipore) was incubated in primary antibody containing lysis buffer (20 mM HEPES, 150 mM NaCl, 2 mM DTT, 5 mM EDTA, 5 mM EGTA, 0.2% Triton X-100, 10% glycerol and Roche complete protease inhibitor cocktail) for 2 h at room temperature. Pelleted cells were incubated in the lysis buffer on ice for 30 min and cell lysates were precleared with beads for 30 min. This precleared cell lysate was incubated with antibody-conjugated beads overnight at 4 °C. Then, beads were washed with the lysis buffer several times and used for western analysis.
Results
arrm7 is identified as a sona suppressor
We previously identified suppressors that overcome lethality induced by overexpressed Sona in a genetic screen17. A suppressor m7 had two lethal sites and one of them was present in pdm3 as reported16. Pdm3 induces wg transcription and is essential for the development of neuromuscular junctions16. The other lethal site was mapped to the region deleted in two deficiencies, Df(2R)BSC273 and Df(2R)BSC274 (Fig. 1a). Complementation test and whole-genome sequencing determined that the second lethal site is located in the arr gene, and was named arrm7. arrm7 had a nonsense mutation in the seventh exon of the arr gene, which truncates 191 amino acid residues of the intracellular region from 1678 amino acid residues of Arr (Fig. 1a).
Arr functions downstream to Sona
To confirm genetic relationship between arr and sona, we co-expressed UAS-arr RNAi and UAS-sona and checked whether lethality by sona overexpression is suppressed by knockdown of arr (Fig. 1b). Survival rate of 30A-Gal4 > UAS-arr RNAi (30A > arr RNAi) flies was 86%, similar to that of 30A-Gal4/+ control flies. On the other hand, the survival rate of 30A > sona flies was only 5% and that of 30A > GFP; sona was 6%, and both flies showed late pupal lethality (Fig. 1b and S1). Co-expression of arr RNAi and sona increased the survival rate to 40% (Fig. 1b). Furthermore, all 30A > sona flies died in a few hours after eclosion whereas 50% of 30A > arr RNAi; sona files were normal and fertile. Therefore, the lethal phenotype induced by gain of sona was significantly rescued by knockdown of arr.
We then tested whether gain of arr can rescue the lethality induced by knockdown of sona. Survival rates of 30A > arr-HA flies and 30A-Gal4/+ control flies were both 87% (Fig. 1c). The survival rate of 30A > sona RNAi flies was 55% whereas that of 30A > sona RNAi; arr-HA flies was 87% (Fig. 1c). Therefore, the lethal phenotype induced by knockdown of sona was fully rescued by gain of arr. Since the level of Arr affected phenotypes induced by either knockdown or gain of Sona, we concluded that arr functions downstream to sona.
Arrm7 is dominant negative
We utilized UAS-arr-HA and UAS-arrm7-HA flies to compare Arr and Arrm7 proteins in western analysis with the anti-Arr antibody32. The epitope of the anti-Arr antibody was present in the intracellular region of Arr that is missing in Arrm7-HA (Fig. S2A, B). Accordingly, Arr-HA protein was recognized by both anti-HA and anti-Arr antibodies in engrailed (en)>arr-HA wing discs whereas Arrm7-HA protein was recognized by only anti-HA antibody in en > arrm7-HA wing discs (Fig. S2C–F). We also found that en > arr RNAi wings are normal despite substantial reduction in the amount of Arr, indicating that arr RNAi expression still leaves behind a low level of Arr enough for normal development (Fig. 2a, b and S2G). Similar phenomenon with the same UAS-arr RNAi flies has been reported elsewhere33.

a–f en-Gal4; UAS-GFP flies were crossed with UAS lines and wings of their progenies are shown (n = 20, each). Genotypes are written at the lower left without GFP. Extra cross-veins are marked with an arrow in (c). Notched wing is marked with arrowheads in (e). These wing phenotypes showed 100% penetrance. g–i UAS lines were crossed with en-Gal4; UAS-GFP flies and wing discs of their progenies were stained. Images were taken by focusing at the basal position. The en expressing posterior regions are indicated with GFP. The regions marked with yellow squares are magnified and the borders of GFP regions are indicated with dotted lines in (g‴,h‴,i‴). CC3 is marked with an arrow in (h′). j, k Clonal analysis of arrm7 generated at the 2nd larvae stage by heat shock for 45–50 min at 37 °C. Twin spots (+/+) have the highest level of GFP, heterozygous cells (+/m) have the intermediate level of GFP, and arrm7 clones (m/m) have no GFP. Accumulated Arm in arrm7 clones are indicated by arrows. One cross-section at the region marked with a yellow line is shown in (k). Only the apical region is shown. Scale bar, 50 μm.
Extra cross-veins were generated in all en > arr-HA wings examined, and fully rescued in en > arr-HA; arr RNAi wings (Fig. 2a–d). Unlike en > arr-HA wings, notched wings were generated in all en > arrm7-HA wings examined (Fig. 2e). Arrm7 expression by nubbin-Gal4 also induced wing notching, and that by patched-Gal4 resulted in pupal lethality (Fig. S3). When arr-HA and arrm7-HA were co-expressed by en-Gal4, both extra cross-veins and wing notching were fully rescued (Fig. 2f). Similarly, pupal lethality of ptc > arrm7-HA flies was rescued by co-expression with arr-HA (data not shown). Therefore, phenotypes induced by Arr-HA was suppressed by co-expression with Arrm7-HA and vice versa. This suggests that dimerization between Arr and Arrm7 leads to decreased Arr activity due to dominant-negative Arrm7. Our result is consistent with Xenopus LRP6ΔC equivalent to Arrm7, which shows dominant negativity in axis formation34.
arrm7 induces cell death
Wing notching induced by arrm7-HA overexpression may be due to cell death because Sona is important for cell survival19. Indeed, cleaved caspase 3 (CC3) and basally positioned pyknotic nuclei due to chromatin condensation were detected in en > arrm7-HA but not in en-Gal4/+ wing discs35 (Fig. 2g, h). en > arrm7-HA; p35 wing discs had neither CC3 nor pyknotic nuclei, although normal-looking nuclei were still present at the basal position (Fig. 2i). Therefore, the caspase inhibitor p35 partially rescued cell death phenotype induced by arrm7-HA expression.
We also carried out clonal analysis of arrm7 allele. When formation of arrm7 clones was induced during first instar larval stage, no clones but only twin spots were detected in the wing pouch region (Fig. S4). arrm7 clones were detected when induced during second instar larval stage (Fig. 2j). The twin spots were larger than the clones, which is consistent with cell death phenotype of the arrm7 allele. Some arrm7 clones had a spot with higher level of Arm at one edge because arrm7 cells slightly invaginated (Fig. 2k). Similar but severer phenotype was observed with arr2 clones, in which arr2 cells completely invaginate and extruded from the basal side36. Thus, we concluded that the loss of arr induces cell death.
arr clones exhibit higher level of extracellular Wg whereas lower level of extracellular Sona
We tested further whether arrm7 clones show phenotypes similar to arr2 clones. As shown with arr2 clones37,38, the level of extracellular Wg was increased in arr2 MARCM clones (Fig. S5A). We also found that the level of intracellular Wg is slightly increased in arr2 MARCM clones (Fig. S5B). Similarly, arrm7 clones had higher levels of both extra- and intra-cellular Wg than surrounding cells (Fig. S5C, D).
Given the genetic interaction between arr and sona, we reasoned that Arr may affect the level of intracellular or extracellular Sona. arr2 and arrm7 clones as well as control clones showed no obvious change in the level of intracellular Sona detected by Sona-Pro and Sona-C antibodies that recognize Sona pro domain and carboxyl region, respectively (Fig. 3a–c). When control clones were formed in the blade region where the level of extracellular Sona is high, no change was detected in the extracellular level of Sona (Fig. 3d). It was clear, however, that the levels of extracellular Sona in arr2 and arrm7 clones were lower than neighboring wild-type cells (Fig. 3e, f). Therefore, Arr is important for maintaining the level of extracellular Sona in vivo.

Clones were generated at the 2nd larvae stage by heat shock at 37 °C for 45–50 min. Sona was visualized with two antibodies, Sona-Pro and Sona-C. Regions marked with the yellow squares are magnified. Clone boundaries are marked by yellow dots and clones are marked with arrows. Images were taken at the apical position for intracellular Sona as shown by DAPI. a–c Control, arr2 and arrm7 clones with no change in intracellular level of Sona. d A control clone with no change in extracellular level of Sona. e arr2 clones with lower level of extracellular Sona. f arrm7 clones with lower level of extracellular Sona. Scale bar, 50 μm.
Arr increases both intracellular and extracellular levels of Sona in S2 cells
To understand the mechanism by which Arr regulates the level of extracellular Sona in vivo, we utilized S2 cell culture system. S2 cells co-transfected with UAS-sona, UAS-arr and act-Gal4 plasmids were cultured, and then S2 cells were used to prepare cell extract (CX), and conditioned media were used to prepare two fractions, P100 and SNΔ fractions. The P100 fraction containing EVs was obtained as a pellet after centrifuging precleared media at 100,000×g, and the SNΔ fraction containing soluble secreted proteins was obtained as a supernatant29. As previously shown, 70 kDa full-length Sona is processed to a 37 kDa active form of Sona in both intra- and extracellular regions, and 22 kDa fragments processed from the pro domain is co-secreted with the 37 kDa form of active Sona17.
S2 cells do not express Sona or Wg39,40 whereas expressed Arr (Fig. 4a). When S2 cells were co-transfected with a constant amount of UAS-sona plasmid and varying amounts of UAS-arr plasmid together with act-Gal4 plasmid, amounts of 70, 37, and 22 kDa Sona forms were all increased in correlation with the amount of Arr in CX (Fig. 4a). Such increase was not due to increase in the transcription rate of sona by Arr because transcription of transfected sona cDNA in S2 cells is under the control of Gal4 driven by the actin promoter. This established that Arr increases the stability of Sona.

Anti-Arr, Sona-Pro, anti-HA, and anti-α-Tubulin antibodies were used as blotting antibodies to visualize Arr, full-length Sona (SonaFL), active Sona (active), 22 kDa pro domain fragment (pro) and α-Tubulin, respectively. S2 cells were co-transfected with sona and arr cDNAs with actin-Gal4 and cultured for 3 days before sampling. Amounts of transfected DNA in μg are indicated. a–c S2 cells were transfected with a constant amount of sona-HA DNA and increasing amounts of arr DNA. CX, P100, and SNΔ fractions are analyzed in (a–c), respectively. d, e Transfection of a constant amount of sona-Myc and increasing amounts of arrm7-HA. CX and P100 fractions are analyzed. f–h Effects of MG132 (μM) in (f), and chloroquine (μM) in (g, h) on the amount of Sona. The α-Tubulin is a loading control for CX.
We then examined whether Arr increases the level of extracellular Sona in P100 and SNΔ fractions. In fact, the level of active Sona in the P100 fraction was increased whereas that in the SNΔ fraction was not changed with increasing amounts of Arr (Fig. 4b, c). Therefore, Arr specifically increased the level of Sona in P100. One interesting finding was that Arr itself is present in the P100 fraction but not in the SNΔ fraction, which will be addressed later (Fig. 4b, c). We also checked whether Arrm7 changes the level of Sona. The level of all Sona forms in CX and P100 were unchanged when increasing amounts of Arrm7 were co-expressed in S2 cells (Fig. 4d, e). Therefore, the intracellular domain of Arr is essential for stabilization of Sona.
We then asked whether Wg has any effects on this new function of Arr (Fig. S6). When constant amounts of GFP-wg and sona plasmids and increasing amounts of arr plasmid were co-expressed, the levels of all Sona forms were unchanged regardless of Arr. Therefore, Wg somehow eliminated the effect of Arr on the level of Sona so we concluded that Wg is not involved in the stabilization of Sona by Arr.
Arr increases the level of Sona by inhibiting lysosomal degradation pathway
Proteins are degraded by two pathways, proteasomal degradation pathway and lysosomal degradation pathway41,42. To examine which pathway is regulated by Arr for stabilizing Sona, MG132 and chloroquine were used to inhibit proteasomal degradation and lysosomal pathways, respectively43. MG132 was added to culture media to final concentrations of 25 and 50 μM for 6 h before sampling, and the amount of Arm as a positive control was increased in correlation with the amount of MG13244,45 (Fig. 4f). In contrast, MG132 did not change levels of any Sona forms. This showed that proteasomal degradation pathway does not affect stability of Sona.
Treatment of Sona-expressing S2 cells with 100 μM chloroquine for 24 h prior to sampling did not noticeably change the amount of Sona in CX compared to untreated Sona-expressing S2 cells (Fig. 4g). When Sona and Arr were co-expressed in chloroquine-treated S2 cells, however, amounts of Sona forms in CX were slightly increased, and the amount of active Sona in P100 was prominently increased (Fig. 4h). Chloroquine treatment even made the full-length Sona detectable although full-length Sona is usually undetectable in P10020 (Fig. 4h). Therefore, we concluded that Arr increases stability of Sona by inhibiting the lysosomal degradation pathway.
Sona neither cleaves Arr nor changes the level of Arr
It has been proposed that the extracellular region of Arr is processed by an unknown metalloprotease based on the detection of intracellular fragments of Arr46. To test whether Sona processes Arr, the same amount of arr-HA with increasing amounts of sona-Myc were expressed in S2 cells. The amount of full length Arr in CX or P100 was not affected by Sona, indicating that Sona cleaves neither intracellular nor extracellular Arr (Fig. S7A, B). We also tested whether Sona affects the level of Arr in vivo. When Sona was transiently expressed in dpp > sona wing discs for only 24 h using Gal80ts in order to avoid lethality by Sona overexpression, the level of neither Arr-LacZ nor Arr protein changed (Fig. S7C, D). In sum, Sona does not affect transcription, translation and post-translation of arr.
Arr and Sona are present in the same protein complex and co-localize in wing disc cells
The biochemical relationship between Arr and Sona prompted us to examine whether they are present in the same protein complex. We co-expressed sona-Myc and arr-HA in S2 cells, and the CX of these S2 cells was used for co-immunoprecipitation (co-IP) analysis. Arr-HA was detected after precipitation with Sona-Pro antibody, and full-length Sona was detected after precipitation with anti-HA antibody, indicating that intracellular Sona and Arr are present in the same protein complex (Fig. 5a, b). To find which domain of Arr is responsible for co-IP with Sona, Arrm7-HA or ΔN-Arr-HA was co-expressed with Sona in S2 cells (Fig. 1). Arrm7-HA was co-IPed but ΔN-Arr-HA was not with Sona-Myc, showing that the extracellular domain of Arr is involved in interaction with Sona (Fig. 5c, d). We also found that Arr and Sona co-localize in some intracellular vesicular structures (Fig. 5e).

a, b Sona-Myc and Arr-HA are co-IPed. Arr-HA was detected when Sona-Myc was IPed in a, and vice versa in (b). The arrowhead indicates full length Sona above non-specific bands. c, d Co-IP of Sona with Arr-HA requires the extracellular domain of Arr. Arrm7-HA in (c), but not ΔN-Arr-HA in (d) is co-IPed with Sona-Myc. e A wing disc of CS was stained for Arr and Sona and an image of a region near the DV midline was taken. Arrows mark a few vesicular structures with both Arr and Sona. Scale bar, 5 μm.
Arr is present in the exosome fraction
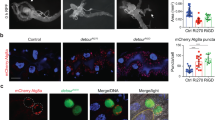
We detected Arr in the P100 fraction prepared from S2 cells transfected with arr cDNA (Fig. 4b). Endogenous Arr in S2 cells was also present in the P100 fraction but not in the SNΔ fraction, demonstrating that Arr is secreted to extracellular space as tethered to EVs (Fig. 6a). To examine which region of Arr is essential for dictating Arr to EVs, S2 R+ cells transfected with arr, arrm7, or arr2 cDNAs were cultured for 3 days, and CX, P100, and SNΔ fractions were prepared. Like endogenous Arr, overexpressed Arr-HA and Arrm7 were detected in the P100 fraction but not in the SNΔ fraction (Fig. 6b). Arr2 protein retaining only the amino terminal half was also detected in P100 fraction with a slight amount in SNΔ fraction (Fig. 1a). Taken together, the extracellular region of Arr is sufficient for secretion of Arr.

a Endogenous Arr expressed in S2 cells are secreted via EVs. Arr is present in the P100 but not in the SNΔ fraction. b Arr-HA, Arrm7-HA, and Arr2-HA are secreted via EVs. S2 cells were transfected with cDNAs encoding different Arr forms and their CX, P100, and SNΔ fractions were analyzed. c A scheme of the experiments in (d–f). S2 cells were transfected with different amounts of arr cDNAs, cultured for 3 days, and then were used to prepare the P100 fraction. Varied amounts of this P100 fraction was added to S2 cells that had been transfected with sona cDNA and cultured for 3 days. After 3 days of culture, S2 cell culture was used to prepare CX, P100, and SNΔ fractions. d–f Amounts of transfected sona-HA and arr DNAs were indicated. The α-Tubulin is a loading control for CX. g A model to explain a new function of Arr for regulating the level of extracellular Sona. Arr is secreted via EVs, and this exosomal Arr acts to send signal to early endosomes to make Sona to take secretion via EVs or channeled to lysosomal degradation pathway.
Exosomal Arr increases the extracellular level of Sona
To examine whether exosomal Arr affects the levels of intracellular and extracellular Sona, we carried out an experiment (Fig. 6c). S2 cells were transfected with arr cDNA and cultured for 3 days to obtain conditioned media, from which the P100 fraction was prepared. This P100 fraction was then added to fresh culture media of S2 cells immediately after sona transfection. After 3 days culture, these cells and conditioned media were used to prepare CX, P100 and SNΔ fractions for comparing the amounts of Sona in these fractions. As a negative control, the P100 fraction prepared from S2 cells transfected with the vector were used.
The addition of the Arr-containing P100 fraction to Sona-expressing cell culture noticeably increased the level of the 22 kDa fragment in CX whereas the amount of other Sona forms was not changed (Fig. 6d). On the other hand, the amount of active Sona in the P100 fraction increased in correlation with that of Arr (Fig. 6e). Such increase was not detected in the SNΔ fraction (Fig. 6f). Thus, exosomal Arr somehow stabilizes Sona in cytoplasm by inhibiting lysosomal pathway, which eventually increases the level of active Sona in EVs.
Discussion
We report here that the loss of arr decreases the level of extracellular Sona in flies, and Arr increases the extracellular level of Sona by stabilizing intracellular Sona in S2 cells through inhibition of lysosomal pathway. Interestingly, Arr was present in the fraction containing exosomes, and the exosomal Arr also increases the level of extracellular Sona by unknown mechanisms. We propose that exosomal Arr inhibits lysosomal degradation pathway and increases the level of exosomal Sona. When Arr is absent, Sona enters the lysosomal pathway to be degraded (Fig. 6g).
The m7 suppressor with both arrm7 and pdm3m7 was identified in the screen using ethyl methanesulfonate (EMS) as a mutagen17. It is worth noting that arrm7 has a G to A transition, whereas pdm3m7 has an insertion of a defective hobo element in the untranslated region of the pdm3 gene16 (Fig. 1a). The point mutation in the arr gene seemed to occur before the insertion of the hobo element in the pdm3 gene, because other suppressors did not have hobo elements in their pdm3 genes. Therefore, both inhibition of wg transcription by pdm3m7 and decrease in the level of extracellular Sona by arrm7 may have been crucial for the m7 suppressor to be identified in the screen.
Arrm7 protein does not have the intracellular domain but still has the LDLR domain involved in dimerization, so dimers formed between Arrm7 and wild-type Arr in arrm7 heterozygotes become inactive. Besides this Arr-Arrm7 dimer, Arr-Arr and Arrm7-Arrm7 dimers are also formed in arrm7 heterozygotes, and the amount of Arr-Arr dimers may be enough to support normal development of arrm7 heterozygotes. This explanation is in line with the finding that en > arr RNAi flies develop normally despite the low level of Arr protein in these flies (Fig. 2b). Although arr RNAi expression induced no visible phenotypes in our experiments, it suppressed phenotypes induced by overexpression of Arr or Sona (Figs. 1b and 2d).
All biochemical experiments in this report were carried out with S2 cells that do not express Wg39,40, indicating that stabilization of Sona by Arr occurs in the absence of Wg (Fig. 4). Similar conclusion was drawn in case of LRP6 and gap junction protein connexin 43 (Cx43) in cardiac gap junction assembly47. Transcription of Cx43 gene is induced by Wnt signaling independent of LRP6, but instead LRP6 post-transcriptionally promotes traffic of Cx43 from endoplasmic reticulum to Golgi apparatus. Reduction of LRP6 leads to retention of Cx43 in ER, which leads to the lysosomal degradation of immature Cx43. Such relationship between LRP6 and Cx43 is remarkably similar to that of Arr and Sona. It has been also reported that LRP6 deficiency results in lethal dilated cardiomyopathy and cardiac dysfunction by activation of dynamin-related protein 1 signaling48. Therefore, Arr/LRP6 seems to play new post-transcriptional roles independent of Wnt signaling.
We have shown here that coexpression of Arr and Sona in S2 cells stabilizes Sona and increases the level of exosomal Sona by inhibiting the lysosomal degradation pathway. In addition, Arr is present in exosomes and these exosomal Arr also stabilizes Sona. It is actually common to find Wg signaling components in EV populations such as Wnts, β-catenin/Arm, Wls, and Fzd-1049. LRP6 is also found in the exosome fraction by proteome profiling, although further confirmation is required50,51. It seems that both Arr/LRP6 and Fz are secreted via EVs, and interaction between the Wnt co-receptors and Wnt in EVs may promote Wnt signaling but may also perform extracellular EV-specific functions independent of Wnt signaling as reported here. Further understanding mechanisms of secreted exosomal Arr in stabilization of Sona will greatly help to reveal Wnt-independent functions of Arr.
References
Masckauchán, T. N. H., Shawber, C. J., Funahashi, Y., Li, C.-M. & Kitajewski, J. Wnt/β-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis 8, 43–51 (2005).
Adamska, M. et al. Wnt and TGF-β expression in the sponge Amphimedon queenslandica and the origin of metazoan embryonic patterning. PLoS ONE 2 e1031 (2007).
Clevers, H. & Nusse, R. Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 (2012).
Baron, R. & Kneissel, M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 19, 179–192 (2013).
Polakis, P. Wnt signaling and cancer. Genes Dev. 14, 1837–1851 (2000).
Grzeschik, K.-H. et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat. Genet. 39, 833–835 (2007).
Nguyen, D. X. et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 138, 51–62 (2009).
Schinner, S. Wnt-signalling and the metabolic syndrome. Horm. Metab. Res. 41, 159–163 (2009).
He, X., Semenov, M., Tamai, K. & Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/β-catenin signaling: arrows point the way. Development 131, 1663–1677 (2004).
Struhl, G. & Basler, K. Organizing activity of wingless protein in Drosophila. Cell 72, 527–540 (1993).
Delanoue, R. et al. The Drosophila wing differentiation factor Vestigial–Scalloped is required for cell proliferation and cell survival at the dorso-ventral boundary of the wing imaginal disc. Cell Death Differ. 11, 110–122 (2004).
Gong, Y. et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107, 513–523 (2001).
Jeon, H. et al. Implications for familial hypercholesterolemia from the structure of the LDL receptor YWTD-EGF domain pair. Nat. Struct. Mol. Biol. 8, 499–504 (2001).
Chen, J. et al. LRP6 dimerization through its LDLR domain is required for robust canonical Wnt pathway activation. Cell. Signal. 26, 1068–1074 (2014).
Logan, C. Y. & Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 (2004).
Kim, Y. & Cho, K.-O. POU domain motif3 (Pdm3) induces wingless (wg) transcription and is essential for development of larval neuromuscular junctions in Drosophila. Sci. Rep. 10, 517 (2020).
Kim, G.-W. et al. Sol narae (Sona) is a Drosophila ADAMTS involved in Wg signaling. Sci. Rep. 6, 31863 (2016).
Nam, S. & Cho, K.-O. Wingless and Archipelago, a fly E3 ubiquitin ligase and a homolog of human tumor suppressor FBW7, show and antagonistic relationship in wing development. BMC Dev. Biol. 20, 14 (2020).
Tsogtbaatar, O. et al. An ADAMTS Sol narae is required for cell survival in Drosophila. Sci. Rep. 9, 1–15 (2019).
Won, J.-H. et al. ADAMTS Sol narae cleaves extracellular Wingless to generate a novel active form that regulates cell proliferation in Drosophila. Cell Death Dis. 10, 564 (2019).
Théry, C., Zitvogel, L. & Amigorena, S. Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2, 569–579 (2002).
Laulagnier, K. et al. Characterization of exosome subpopulations from RBL-2H3 cells using fluorescent lipids. Blood Cells Mol. Dis. 35, 116–121 (2005).
Yáñez-Mó, M. et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 4, 27066–27066 (2015).
Willms, E. et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 6, 22519 (2016).
Théry, C. et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7, 1535750–1535750 (2018).
Lee, S.-S. et al. A novel population of extracellular vesicles smaller than exosomes promotes cell proliferation. Cell Commun. Signal. 17, 1–15 (2019).
Korkut, C. et al. Trans-synaptic transmission of vesicular Wnt signals through Evi/Wntless. Cell 139, 393–404 (2009).
Chairoungdua, A., Smith, D. L., Pochard, P., Hull, M. & Caplan, M. J. Exosome release of β-catenin: a novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 190, 1079–1091 (2010).
Gross, J. C., Chaudhary, V., Bartscherer, K. & Boutros, M. Active Wnt proteins are secreted on exosomes. Nat. Cell Biol. 14, 1036–1045 (2012).
McLean, I. W. & NAKANE, P. K. Periodate-lysine-paraformaldehyde fixative a new fixative for immunoelectron microscopy. J. Histochem. Cytochem. 22, 1077–1083 (1974).
Strigini, M. & Cohen, S. M. Wingless gradient formation in the Drosophila wing. Curr. Biol. 10, 293–300 (2000).
Marois, E., Mahmoud, A. & Eaton, S. The endocytic pathway and formation of the Wingless morphogen gradient. Development 133, 307–317 (2006).
Tan, Y., Yu, D., Busto, G. U., Wilson, C. & Davis, R. L. Wnt signaling is required for long-term memory formation. Cell Rep. 4, 1082–1089 (2013).
Tamai, K. et al. LDL-receptor-related proteins in Wnt signal transduction. Nature 407, 530–535 (2000).
Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 (2007).
Widmann, T. J. & Dahmann, C. Wingless signaling and the control of cell shape in Drosophila wing imaginal discs. Dev. Biol. 334, 161–173 (2009).
Piddini, E., Marshall, F., Dubois, L., Hirst, E. & Vincent, J.-P. Arrow (LRP6) and Frizzled2 cooperate to degrade Wingless in Drosophila imaginal discs. Development 132, 5479–5489 (2005).
Rives, A. F., Rochlin, K. M., Wehrli, M., Schwartz, S. L. & DiNardo, S. Endocytic trafficking of Wingless and its receptors, Arrow and DFrizzled-2, in the Drosophila wing. Dev. Biol. 293, 268–283 (2006).
Brown, J. B. et al. Diversity and dynamics of the Drosophila transcriptome. Nature 512, 393–399 (2014).
Cherbas, L. et al. The transcriptional diversity of 25 Drosophila cell lines. Genome Res. 21, 301–314 (2011).
Wojcik, S. Crosstalk between autophagy and proteasome protein degradation systems: possible implications for cancer therapy. Folia Histochem. Cytobiol. 51, 249–264 (2013).
Lecker, S. H., Goldberg, A. L. & Mitch, W. E. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 17, 1807–1819 (2006).
Gonzalez-Noriega, A., Grubb, J. H., Talkad, V. & Sly, W. S. Chloroquine inhibits lysosomal enzyme pinocytosis and enhances lysosomal enzyme secretion by impairing receptor recycling. J. Cell Biol. 85, 839–852 (1980).
Song, D. H. et al. CK2 phosphorylation of the armadillo repeat region of β-catenin potentiates Wnt signaling. J. Biol. Chem. 278, 24018–24025 (2003).
Swarup, S. & Verheyen, E. M. Drosophila homeodomain-interacting protein kinase inhibits the Skp1-Cul1-F-box E3 ligase complex to dually promote Wingless and Hedgehog signaling. Proc. Natl Acad. Sci. USA 108, 9887–9892 (2011).
Mi, K. & Johnson, G. V. Regulated proteolytic processing of LRP6 results in release of its intracellular domain. J. Neurochem. 101, 517–529 (2007).
Li, J. et al. LRP6 acts as a scaffold protein in cardiac gap junction assembly. Nat. Commun. 7, 1–11 (2016).
Chen, Z. et al. Cardiomyocyte-restricted low density lipoprotein receptor-related protein 6 (LRP6) deletion leads to lethal dilated cardiomyopathy partly through Drp1 signaling. Theranostics 8, 627 (2018).
Principia, S. M. et al. Frizzled-10 extracellular vesicles plasma concentration is associated with tumoral progression in patients with colorectal and gastric cancer. J. Oncol. 2019, 2715968–2715968 (2019).
Hurwitz, S. N. et al. Proteomic profiling of NCI-60 extracellular vesicles uncovers common protein cargo and cancer type-specific biomarkers. Oncotarget 7, 86999–87015 (2016).
Demory Beckler, M. et al. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol. Cell. Proteom. 12, 343 (2013).
Acknowledgements
We are grateful to Dr. K.-W. Choi and colleagues in our lab for discussion and comments on this paper. We are in debt to K. Basler and S. Eaton for the fly line and the anti-Arr antibody. We thank Bloomington Drosophila Stock Center, Vienna Drosophila Resource Center, Drosophila Genomics Resource Center, Developmental Studies Hybridoma Bank and Korea Drosophila Resource Center for fly lines, antibodies and embryo injection. This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Education, 2017R1A2B4009254 and 2019R1H1A2039726.
Author information
Authors and Affiliations
Contributions
J.-H.H., Y.K. and K.-O.C. designed the experiments and analyzed data. J.-H.H. and Y.K. conducted the experiments. J.-H.H. and K.-O.C. wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by E. Baehrecke
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, JH., Kim, Y. & Cho, KO. Exosomal arrow (Arr)/lipoprotein receptor protein 6 (LRP6) in Drosophila melanogaster increases the extracellular level of Sol narae (Sona) in a Wnt-independent manner. Cell Death Dis 11, 944 (2020). https://doi.org/10.1038/s41419-020-02850-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-02850-x

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}