
Abstract
Adult mammalian cardiomyocytes (CM) are postmitotic, differentiated cells that cannot re-enter the cell cycle after any appreciable injury. Therefore, understanding the factors required to induce CM proliferation for repair is of great clinical importance. While expression of muscle pyruvate kinase 2 (Pkm2), a cytosolic enzyme catalyzing the final step in glycolysis, is high in end-stage heart failure (HF), the loss of Pkm2 promotes proliferation in some cellular systems, in vivo. We hypothesized that in the adult heart CM proliferation may require low Pkm2 activity. Thus, we investigated the potential for Pkm2 to regulate CM proliferation in a mouse model of myocardial infarction (MI) employing inducible, cardiac-specific Pkm2 gene knockout (Pkm2KOi) mice. We found a lack of cardiac hypertrophy or expression of the fetal gene program in Pkm2KOi mice post MI, as compared to vehicle control animals (P < 0.01), correlating with smaller infarct size, improved mitochondrial (mt) function, enhanced angiogenesis, reduced degree of CM apoptosis, and reduced oxidative stress post MI. There was significantly higher numbers of dividing CM in the infarct zone between 3–9 days post MI (P < 0.001). Mechanistically, we determined that Pkm2 interacts with β-catenin (Ctnnb1) in the cytoplasm of CM, inhibiting Ctnnb1 phosphorylation at serine 552 and tyrosine 333, by Akt. In the absence of Pkm2, Ctnnb1 translocates to the nucleus leading to transcriptional activation of proliferation-associated target genes. All these effects are abrogated by genetic co-deletion of Pkm2 and Ctnnb1. Collectively, this work supports a novel antiproliferative function for Pkm2 in CM through the sequestration of Ctnnb1 in the cytoplasm of CM whereas loss of Pkm2 is essential for CM proliferation. Reducing cardiac Pkm2 expression may provide a useful strategy for cardiac repair after MI in patients.
Similar content being viewed by others


Introduction
Heart failure (HF) occurs when the heart is unable to adequately pump blood to meet the demands of the body. It is the leading cause of morbidity and mortality in North America [1] with myocardial infarction (MI) being the leading cause [2, 3]. As adult CM are postmitotic, differentiated cells that cannot re-enter the cell cycle, the heart has a limited regenerative capacity after ischemic injury [4,5,6]. Therefore, identifying mechanisms that can induce cardiomyocyte (CM) proliferation is of great clinical importance.
The muscle pyruvate kinase (Pkm) gene encodes two isoforms, Pkm1 and Pkm2, that catalyze the conversion of phosphoenolpyruvate to pyruvate, the final step in glycolysis. Both isoforms are generated through alternative splicing (Supplementary Fig. 1) [7,8,9]. Pkm1 is the predominant isoform in tissues with high ATP demand (heart, brain, skeletal muscle) and in non-proliferating tumor cells. In contrast, Pkm2 is predominantly expressed during embryonic development, in adult organs (liver, lung, spleen) and in some tumor cell lines [10, 11]. Ectopic overexpression of Pkm1 inhibits tumor proliferation [12], whereas Pkm2 shifts glucose metabolism to promote cancer cell proliferation [11]. Also, loss of Pkm2 promotes tumor proliferation in breast cancer cells [13]. Notably, Pkm2 protein expression is upregulated in end-stage human HF [14].
Glycogen synthase kinase-3β (Gsk-3β) is an inhibitor of the canonical Wnt/β-catenin (Ctnnb1) pathway [9], suppressing downstream signal pathways regulating many cellular processes including glycogen biosynthesis and cell cycle [15]. The Wnt effector, Ctnnb1, is predominantly associated with CM’s intercalated disks [16]. Casein kinase-1 and Gsk-3β phosphorylate Ctnnb1 to target Ctnnb1 for proteosomal degradation [9]. In contrast, activation of Wnt signaling inhibits Gsk-3β-dependent Ctnnb1 phosphorylation, leading to stabilization of Ctnnb1 and translocation to the nucleus to transactivate genes, such as Myc and Cyclin D1 [9].
Here we demonstrate that loss of Pkm2 induces CM proliferation with restoration of cardiac structure and function, post MI. Mechanistically, this occurs through Pkm2’s ability to sequester Ctnnb1 in the cytoplasm. With the loss of Pkm2, Ctnnb1 shuttles to the nucleus post MI, to activate the transcription of genes critical for cell-cycle progression. The effect is lost in the absence of both Pkm2 and Ctnnb1.
Materials and methods
Animal housing
All animal usage in this study was in accordance with approved institutional animal care guidelines of the University Health Network (AUP 1379; Canadian Council in Animal Care). All animals used in this study were male and female at 10–16 weeks old (22–27 g) at the beginning of experimentation. All experiments used isogenic littermate controls of matched age and sex. After weaning, experimental male mice were housed in groups of 3–5 animals in mechanically ventilated cages (600 cm2; changed fortnightly) environmentally enriched by bedding and nesting materials and crawl tubes. Animals were held in a temperature-controlled environment at 19–22 °C on a diurnal 12 h light cycle. Mice were provided free access to standard non-medicated pelleted laboratory rodent chow containing minutes levels of phytoestrogens (Tekland irradiated LM-485 mouse/rat Diet 7912; Envigo, Madison WI, USA). Tap water was supplied ad libitum from a portable water faucet. Animals were not randomized. No blinding was performed.
Generation of cardiac-specific conditional knockout mice
The Pkm2fl/fl strain (stock no. 024048) mice were obtained from Jackson (Bar Harbor, ME 04609 USA) [13]. During generation of this strain, mice were crossed to mice expressing FLP1 recombinase to excise the neomycin selection cassette. In our lab, this strain was backcrossed to C57BL/6J mice (stock no. 000664) at least four times. The alpha-MHC-MerCreMer transgene (mcm) strain (stock no. 005657) was from Jackson (Bar Harbor, ME 04609 USA). In these mice, the cardiac muscle α-myosin heavy chain 6 promoter constitutively drives the expression of cytoplasmic Cre (c) recombinase fused to two mutant (m) estrogen-receptor ligand-binding domains that translocates into the nuclei when exposed to tamoxifen (Tam). Mice were backcrossed to C57BL/6J for more than ten generations to generate this congenic strain. We then crossed mcm transgenic mice on a C57BL/6J background with mice carrying the conditional allele Pkm2fl/fl to obtain Pkm2fl/fl;mcm animals. After Tam administration, these mice had exon 10 from Pkm2 deleted in adult CM (Pkm2KOi) [13]. The Ctnnb1fl/fl strain (stock no. 004152) mice on a C57BL/6J background were obtained from Jackson (Bar Harbor, ME 04609 USA) [17]. We crossed mcm transgenic mice with mice carrying the conditional alleles Ctnnb1fl/fl to obtain Ctnnb1fl/fl;mcm animals. After Tam administration, these mice had exons 2–6 of Ctnnb1 deleted in adult CM (Ctnnb1KOi). This deletion contained the ATG translational start codon and rendered the recombined Ctnnb1 allele unable to generate a functional Ctnnb1 protein [17]. Since the Pkm2 and Cnnb1 gene loci are both located on chromosome 9 in the mouse genome [13, 17], we crossed Pkm2fl/fl mice with Ctnnb1fl/fl animals to obtain the Pkm2fl/fl;Ctnnb1fl/fl double homozygous strain. We then crossed Pkm2fl/fl;Ctnnb1fl/fl mice with mcm animals to obtain Pkm2fl/+;Ctnnb1fl/+;mcm animals. Pkm2fl/+;Ctnnb1fl/+;mcm mice were bred to Pkm2fl/fl;Ctnnb1fl/fl animals to generate homozygous Pkm2fl/fl;Ctnnb1fl/fl;mcm double knockout mice. After Tam administration, these mice did not express functional forms of either Pkm2 or Ctnnb1 (DKOi).
Isolation of DNA and genotyping
DNA isolated from fresh tail snips (<5 mm) was employed for genotyping. Samples were incubated in 300 μl of 50-mM NaOH for 2 h, 80 °C while rocking. Then, samples were neutralized with 25 μl 1.0-M HCl, 700-μl H2O, vigorously vortexed, centrifuged for 13,000 rpm for 10 min, and stored at 4 °C. We used 0.5 μl of DNA solution per PCR reaction and the following primers: Pkm2 forward 5′-GTCTGGAGAAACAGCCAAGG-3′; Pkm2 reverse 5′-TCTTCAAACAGCA GACGGTG-3′ [13]. We performed PCR analysis with Quanta Accustart Geltrac with GelDye (no. 95136-04K; VWR) and Platinum Blue Supermix (no. 12580-023; Invitrogen): Pkm2 wild-type allele, 578 bp; Pkm2 floxed allele, 680 bp. Genomic recombination efficiency at the Pkm2 gene locus was measured by post-Cre PCR employing the following primers spanning exon 7–9: forward 5′-GTCTGGAGAAACAGCCAAGG-3′; reverse 5′-CGGAGTTCCTCGAATAGCTG-3′. Amplicon size: 195 bp [13]. Mcm forward 5′-AGGTGGACCTGATCATGGAG-3′; mcm reverse 5′-ATACCGGAGATCATGCAAGC-3. Amplicon size: mcm wild type; no band since this assay did not distinguish hemizygous from homozygous transgenic animals; mcm transgene: 4410 bp (Supplementary Table 1). We used 0.5 μl of DNA solution per PCR reaction and the following primers: Ctnnb1 forward 5′-AAGGTAGAGTGATGAAAGTTGTT-3′; Ctnnb1 reverse 5′-CACCATGTCCTCT GTCTATTC-3′ [17]. Ctnnb1 wild-type allele, 223 bp; Ctnnb1 floxed allele, 300 bp. Genomic recombination efficiency at the Ctnnb1 gene locus was measured by post-Cre PCR employing the following primers spanning exon 1–7: forward 5′-AATCACAGGGACTTCCAT ACCAG-3′; reverse 5′-GCCCAGCCTTAGCCCAACT-3′. Amplicon size: 631 bp (4).
Tamoxifen injections
Because Tam and Cre recombinase expression can be toxic to cells, we included vehicle- and Tam-injected wild-type C57BL/6J, mcm, and Pkm2f/f mice in all initial analyses of the corresponding mutants. We found that mice of these experimental groups were phenotypically indistinguishable from vehicle-injected Pkm2fl/fl;mcm (Veh-control) animals used in this study, as judged by heart body weight ratios, fibrosis, and fractional shortening (FS).
Tam was prepared and injected as described previously [18]. Briefly, two vials (100-mg total) of Tam (hydroxy-tamoxifen; H6278; Sigma-Aldrich) were dissolved in 5-ml pure ethanol and vortexed for 3–5 min or until the solution became clear. Then, 32-ml peanut oil were added, and the suspension was first vortexed for 2 min and then sonicated for 20–60 s until the emulsion became clear. Aliquots were stored at −20 °C. We injected 200-μl Tam per mouse (25-g body weight) daily for 4 consecutive days (80-μg/g body weight cumulative dosage). The injected volume of Tam was adjusted to body weight (160–240-μl Tam). Animals at 10 weeks of age were injected daily between 4 and 6 p.m. for 4 consecutive days (80-μg/g body weight cumulative dosage). Veh-control mice were intraperitoneally injected with an ethanol–peanut oil emulsion lacking Tam. Homologous recombination was completed at 7-day (d) post-Tam. At this time point, mice were 13 weeks old. The day of the MI surgery was arbitrarily set as day 0. At 4-d post MI, mice were categorized as 14 weeks old. At 21-d post MI, mice were 16 weeks old.
Coronary artery ligation
To create a MI, permanent ligation of the left descending coronary artery was performed in 13 weeks old knockout strains at 7-d post-Tam or post-Veh injections, as described previously [19]. A single subcutaneous dose (30 mg/g body weight) of buprenorphine (B9275; Sigma-Aldrich) was given at the time of surgery and at 24 h post MI. Animals were matched for both age and time post MI.
Echocardiography
Echocardiography in anesthetized mice was performed pre-MI and at 21-d post MI as described previously [19].
Immunofluorescence microscopy, morphometric analyses, apoptosis, and DNA synthesis assays
For EDU-labeling experiments, we intraperitoneally injected the modified thymidine analog EDU (160-mg/kg body weight cumulative dosage) twice at 4-d post MI and harvested cardiac samples 2 h after the second injection. Detection of incorporated EDU was done employing the Click-iT™ EdU Cell Proliferation Kit for Imaging (Alexa Fluor™ 488 dye; C10337; Thermo Fisher) according to the manufacturer’s specifications. Detection of fragmented genomic DNA (apoptosis assay) was performed by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling according to the manufacturer’s instructions (no. 1684795910; Roche). For immunofluorescence measurements of apoptosis, angiogenesis, cell-cycle distribution, and CM nuclei content, three independent fields of ~0.25 mm2 per left ventricular (LV) sample were counted on three consecutive sections. Confocal immunofluorescence microscopy and 3D-reconstruction of digital microphotographs were performed as described previously [20].
Preparation of protein extracts from LV heart tissue extracts
Individual LV specimens were rapidly cut into 4–6 pieces, briefly rinsed in 40-ml ice-cold PBS, immediately snap frozen and stored at −80 °C. Since CM proliferation was restricted to the LV, we only used LV tissue to prepare the different extracts and excluded the interventricular septum and the right ventricular in the analysis. To prepare total protein extracts, LV tissue (80–100-mg wet weight) was placed in a petri dish, kept on ice and was minced into 1–2 mm cubes for 60 s using a scalpel. The minced tissue was transferred to a 1.5-ml reaction tube and resuspended in 1.5-ml ice-cold RIPA Buffer (no. 9806; Cell Signaling) composed of 20-mM Tris-HCl (pH 7.5), 150-mM NaCl, 1.0-mM EGTA, 1.0% NP-40, 1.0% sodium deoxy cholate, 2.5-mM sodium pyrophosphate, 1.0-mM β-glycerophosphate, 1.0-mM Na3VO4, and 1.0-mg/ml leupeptin, that was further supplemented with 1,4-dithiothreitol (4-mM final concentration; no. 10197777001; Sigma) and protease/phosphatase inhibitors (HALTTM Protease and Phosphatase Inhibitor Cocktail; no. 78440; Thermo Fisher Scientific). Cells were lysed at 4 °C for 3 min at a frequency of 30/s using the TissueLyser II (Qiagen). Samples were incubated on ice for 30 min, vortexing every 5 min for 30 s. Samples were then subjected to ten cycles of 3 s on/1 s off sonification at 30% amplitude on ice, and incubated for 10 min, vortexing every 5 min for 30 s. After centrifugation at 12,700 rpm for 30 min at 4 °C, the supernatant was mixed with 10% glycerol, aliquoted, immediately snap frozen on dry ice, and stored at −80 °C. Total protein concentration was determined employing the Qubit™ Protein Assay Kit (no. 33211; Thermo Fisher Scientific) and a Qubit™ 2.0 Fluorometer (Thermo Fisher Scientific).
Western blotting
Protein samples were mixed with equal amounts of sample buffer (Novex Bolt LDS sample buffer, 4×; no. 2107345; Thermo Fisher Scientific) and boiled for 7 min. Samples (60-mg total protein per lane) were resolved (Novex Bolt MES SDS-Running Buffer, 20×; no. 2122845; Thermo Fisher Scientific) by SDS-PAGE employing 4–12% pre-cast gradient gels (Novex Bolt 4–12% Bis-Tris Plus; 1.0 mm × 10 wells; no. NW04120BOX; Thermo Fisher Scientific). To monitor protein migration, protein transfer onto membranes and sizing of protein, prestained protein markers (Seeblue Plus2 Prestained Protein Standard; LC5925; Thermo Fisher Scientific) were run simultaneously with the samples during SDS-polyacrylamide gel electrophoresis.
Samples were electrotransferred for 7 min employing semi-dry iBlot Gel transfer Stacks (PVDF, regular; no. 2020-07-16; Thermo Fisher Scientific) and an iBlot system (Thermo Fisher Scientific). Detection was performed with Luminata Crescendo (WBLUR0100, Millipore) on a digital imaging system (Molecular Imager ChemiDoc Imaging System; BioRad). Primary antibodies are listed in Supplementary Table 2. The following secondary antibodies were employed for chemiluminescence detection of proteins: horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (no. 7074; Cell Signaling), HRP-conjugated anti-mouse IgG (no. 7076; Cell Signaling).
Immunoprecipitation assays and the immunocomplex kinase assay
For immunoprecipitation (IP) assays, LV tissue (80–100-mg wet weight; stored at −80 °C) was placed in a petri dish and was minced into 1–2-mm cubes for 60 s on ice using a scalpel. To prepare total protein extracts, the minced tissue was transferred to a 2.0-ml reaction tube and resuspended in 1.5-ml ice-cold, nondenaturing Cell Lysis Buffer (no. 9803; Cell Signaling) composed of 20-mM Tris-HCl (pH 7.5), 150-mM NaCl, 1-mM Na2EDTA, 1.0-mM EGTA, 1.0% Triton X-100, 2.5-mM sodium pyrophosphate, 1.0-mM β-glycerophosphate, 1.0-mM Na3VO4, and 1.0-mg/ml leupeptin, that was further supplemented with 1,4-dithiothreitol (4-mM final concentration; no. 10197777001; Sigma) and protease/phosphatase inhibitors (HALTTM Protease and Phosphatase Inhibitor Cocktail; no. 78440; Thermo Fisher Scientific). Cells were lysed at 4 °C for 3 min at a frequency of 30/s using the TissueLyser II (Qiagen). Samples were incubated on ice for 30 min, vortexing every 5 min for 30 s. After centrifugation at 12,700 rpm for 30 min at 4 °C, the supernatant was mixed with 10% glycerol, aliquoted, immediately snap frozen on dry ice, and stored at −80 °C.
For IP assays, we used 1.5-ml reaction tubes pre-blocked with 5% BSA in TBST (0.1% Tween-20). Five hundred microliter cellular extracts corresponding to 8.0 mg total protein were incubated with antibodies to Pkm2, Ctnnb1, or normal rabbit IgG and incubated for 16 h at 4 °C while gently rotating (Supplementary Table 2). Immunocomplexes were collected with 50-μl magnetic beads (Mag Sepharose Xtra; no. 28-9670-10; GE Healthcare) for 3 h at 4 °C while gently rotating. Samples were subsequently washed 2× with ice-cold Cell Lysis Buffer, mixed with an equal volume of SDS-sample buffer (40 ml), and boiled for 7 min. Samples (20 ml) were then subjected to immunoblotting with antibodies to Pkm2 and Ctnnb1. The following secondary antibodies were employed for chemiluminescence detection of proteins: Protein A-conjugated HRP (Protein A-HRP; no. 101023; Thermo Fisher Scientific) diluted 40,000-fold.
Immunocomplex kinase assay
Cellular extracts (500 ml corresponding to 4.0-mg total protein) in supplemented Cell Lysis Buffer were incubated with antibodies to Cdk2 or normal rabbit IgG. Immunocomplex kinase assays were performed as described previously [20]. Protein A-conjugated HRP (Protein A-HRP; no. 101023; Thermo Fisher Scientific) diluted 40,000-fold was employed for chemiluminescence detection of proteins in the Western blot procedure.
Total RNA isolation, reverse transcription, and quantitative real time PCR assays
Total RNA isolation, reverse transcription, and quantitative real time PCR assays were performed as described previously [18].
Identification of Ctnnb1 promoter binding elements in target genes and ChIP assays
Nuclear Ctnnb1 associates with TCF/Lef sequence-specific transcription factors and activates expression of target genes involved in the regulation of cell growth and proliferation. Potential Ctnnb1 core consensus binding elements (BE) containing the sequence CTTTG(A/T)(A/T) or the complementary sequence (A/T)(A/T)CAAAG in selected gene promoters were identified employing the internet applications Ensembl (http://useast.ensembl.org/index.html) and TFBIND (http://tfbind.hgc.jp/). We identified potential Ctnnb1 BE in the Axin2, Ccnd1, Myc, Sox2, and Tnnt3 gene loci mouse gene promoters by in silico promoter analysis employing Ensembl (http://useast.ensembl.org/index.html) and TFSEARCH software (version 1.3; http://www.cbrc.jp/research/db/TFSEARCH.html). The threshold score in TFSEARCH was set to 65%. The primer sequences used for chromatin immunoprecipitation (ChIP) detection are listed in Supplementary Table 3. The nucleotide sequence of the Ctnnb1 BE in the mouse Axin2 gene promoter is located at −75 and −176 bp in intron 1 upstream of the transcriptional start site located upstream of exon 2. The nucleotide sequence of the Ctnnb1 BE in the mouse Ccnd1 gene promoter is located at −402 and −581 bp in the 5′UTR region upstream of the transcriptional start site in exon 1. The nucleotide sequence of the Ctnnb1 BE in the mouse Myc gene promoter is located at +276 and +363 bp downstream of the transcriptional start site in exon 1. The nucleotide sequence of the Ctnnb1 BE in the mouse Sox2 gene promoter is located at −318 and −441 bp upstream of the transcriptional start site upstream of exon 1. The nucleotide sequence of the Ctnnb1 BE in the mouse Tnnt3 gene promoter, that served as negative control, is located at −322 and −490 bp in intron 1 upstream of the transcriptional start site upstream of exon 2. ChIP analysis was performed as described previously [21].
Mitochondrial biogenesis and capacity
mt assays were performed as described previously [22, 23].
Detection of oxidative damage, antioxidants levels, and metabolism assays
Individual LV specimens were rapidly cut into 4–6 pieces, briefly rinsed in 40-ml ice-cold PBS, immediately snap frozen in liquid nitrogen, and stored at −80 °C. Assays for determination of small metabolic molecules (4-hydoxyalkenals (4-HAE), ATP, GSH/GSSG, NADH/NAD+, NADPH/NADP+) were deproteinized prior to analysis using HCl/NaOH-based precipitation. Final values were normalized for the total protein concentration for each sample determined prior to precipitation. All assays were performed according to the manufacturer’s instructions with minor modifications as described previously [22]: ATP/ADP (ATP Assay Kit, no. MAK190, Sigma; ADP Assay Kit, no. MAK033, Sigma); GSH/GSSG (GSH/GSSG Assay Kit, no. K264-100, Biovision); 4-HAE (Lipid Peroxidation Assay Kit, no. ab238538, Abcam), L-Lactate (Lactate Assay Kit, no. MAK064; Sigma); NAD+/NADH (NADPH/NADP Quantification Kit, no. MAK037; Sigma), NADPH/NADP+ (NADPH/NADP Quantification Kit, no. MAK038; Sigma).
Statistical analyses
All statistical analyses were performed employing GraphPad InStat (version 3.1) and GraphPad Prism (GraphPad Software, version 8.3.1; La Jolla, CA 92037 USA). All data are reported as means ± s.e.m. We considered two-tailed P values of <0.05 as significant. When groups passed the normality test, we performed data evaluation between two groups by an unpaired Student t-test. The statistical significance of four groups was calculated using one-way analysis of variance (ANOVA) and Tukey–Kramer multiple comparison post test.
We calculated experimental power with GraphPad Statmate (GraphPad Software, version 1 La Jolla, CA 92037 USA). For example, we used for the FS data (Fig. 1L) the standard deviation (S.D.) value of 1 for Pkm2KOi versus Veh-controls post MI, a significance level alpha = 0.01 (two-tailed), and 80% power, we needed at least n = 4 per group in each experiment, which we increased to n = 6 per group. Briefly, tests with 80% power and higher are considered to be statistically powerful. In 80% (the power) of those experiments, the P value will be <0.01 (two-tailed) so the results will be deemed “statistically significant.” In the remaining 20% of the experiments, the difference between means will be deemed “not statistically significant,” so we will have made a Type II (beta) error.

A PCR genotyping of mouse tail DNA from Pkm2KOi mice (top). PCR genotyping of DNA from Pkm2KOi hearts (bottom). B Administration of Tam markedly decreased endogenous cytoplasmic Pkm2 protein expression at 4-d post MI, as analyzed by confocal immunofluorescence microscopy. Sections of LV were stained with anti-Pkm2 (red), WGA to stain the ECM (green) and co-stained with Dapi (genomic DNA; blue). Pkm2KOi, +Tam. Veh-Control, −Tam. One representative example of three independent experiments using biological replicates is shown. C Quantification of Pkm2 mRNA levels in hearts from Pkm2KOi mice as analyzed by RT-qPCR at 4-d post MI. Data are means ± s.e.m. n = 4. *P < 0.001 vs. sham/Veh-control. D Western blot analysis of Pkm1 and Pkm2 expression in LV extracts from Pkm2KOi mice at 4-d post MI employing antibodies as indicated on the left. For normalization, membranes were re-probed with anti-nucleophosmin (Npm1). Western blots were done twice with similar results using two independent biological replicates. E Heart weight corrected for body weight at 21-d post MI. Data are means ± s.e.m. n = 6. F Heart weight corrected for tibia length at 21-d post MI. Data are means ± s.e.m. n = 6. G Quantification (left) of cross-sectional area of CM in the border zone at 21-d post MI as analyzed by confocal immunofluorescence microscopy (right). Data are means ± s.e.m. n = 6. *P < 0.01 vs. sham/Veh #P < 0.01 vs. sham/Pkm2KOi.-control. #P < 0.05 vs. sham/Pkm2KOi. H Quantification of infarct sizes at 21-d post MI. n = 6 (left). Representative micrographs of Masson-stained cardiac cross-sections (right). I Quantification of mRNA levels of hypertrophic marker genes as analyzed by RT-qPCR at 4-d post MI. Data are mean ± s.e.m. n = 4. *P < 0.01 vs. sham/Veh-control. #P < 0.01 vs. sham/Pkm2KOi. K Immunoblot of protein expression of the mTor pathway at 4-d post MI employing specific antibodies as shown on the left. Western blots were done twice employing two independent biological replicates yielding similar results. Echocardiographic assessment of cardiac function: L FS, M LVEDD, and N LVESD in Pkm2KOi mice at 21-d post MI. Data are mean ± s.e.m. n = 6. *P < 0.01 vs. sham/Veh-control. #P < 0.01 vs. sham/Pkm2KOi. O Representative examples of M-mode utilized in echocardiographic analyses shown in Fig. 1L–N. P Lung weight corrected for body weight at 21-d post MI. Data are means ± s.e.m. n = 6. *P < 0.01 vs. sham/Veh-control. Q Mantel–Cox test shows significant increased survival in Pkm2KOi at 21-d post MI in comparison to Veh-controls (P < 0.009). MI/Pkm2KOi: n = 20; MI/Veh-control: n = 24; Sham/Pkm2KOi: n = 6; Sham/Veh-control: n = 6.
Survival analysis was performed with using a conservative log-rank test (Mantel–Cox) and GraphPad Prism. For calculation of experimental power, we expected a proportion of event-free (surviving) subjects of 20% for the Pkm2KOi versus Veh-controls. Thus, a sample size of n = 20 for Pkm2KOi and 24 Ctrl for Veh-controls post MI in each group provided 80% power to detect an increase in survival proportion of 0.115 with a significance level alpha of 0.01 (two-tailed).
We used for the proliferation data (Fig. 5A, B) the S.D. value of 3.5 and n = 6 per group in each experiment for Pkm2KOi versus Veh-controls post MI. This experiment had an 80% power to detect a difference between means of 5.78 with a significance level (alpha) of 0.01 (two-tailed).
Results
Ablation of Pkm2 improves heart function post MI
To determine the physiological consequence of Pkm2 ablation (Fig. 1A, B and and Supplementary Fig. 1), cardiac structure and function in Pkm2KOi [24, 25] mice was assessed. In Veh-control, we observed increased Pkm2 mRNA and protein levels with a concomitant decrease in Pkm1 expression at 4-d post MI (Fig. 1C, D). This effect was restricted to CM residing in the infarct and border zones (BZs) in the Veh-control animals. Induction of ischemia caused a progressive increase in Pkm2 protein levels over the first 7d (Supplementary Fig. 2), followed by a decline to near baseline by 21d in Veh-control animals. In contrast, Pkm2 protein was not detected in non-cardiomyocytes (NCM). As expected, there was little expression of Pkm2 in CM of Pkm2KOi mice (Fig. 1B).
Increases in heart weight/body weight (HBW) and heart weight/tibia length ratios in Pkm2KOi mice were significantly less post MI (Fig. 1E, F and Supplementary Table 4). CM cross-sectional area (peri-infarct zone) and infarct size were significantly decreased in Pkm2KOi animals, 21-d post MI (Fig. 1G, H). RT-qPCR analysis revealed decreased transcript levels of canonical hypertrophic marker genes (ANP, BNP, β-MHC) in Pkm2KOi mice post MI (Fig. 1I).
The mechanistic target of rapamycin (mTor) is an important regulator of protein synthesis in CM [26, 27], associated with hypertrophy in response to ischemia [28, 29]. To determine whether reduced protein synthesis is responsible for the decrease in CM size in Pkm2KOi post MI, western blot analysis was performed. mTor was equally activated in Pkm2KOi and Veh-controls at 4-d post MI, as evidenced by immunoreactivity with an antibody to mTor.Pi-Ser2448 (Fig. 1K). In addition, Ribosomal Protein S6 Kinase B1 (p70S6K) and Eukaryotic Translation Initiation Factor 4B (eIF4B), two downstream effectors of the mTor signaling, were also activated in both Pkm2KOi and Veh-control mice post MI (Fig. 1K). Our finding suggests that the smaller CM size in the Pkm2KOi mice was not caused by limitations in de novo protein synthesis.
As assessed by echocardiography, cardiac function was improved with no evidence of LV dilation at 21-d post MI in the Pkm2KOi post MI (Fig. 1L–O and Supplementary Table 5). Moreover, lung weight/body weight ratios, an index of pulmonary congestion, was markedly elevated in Veh-controls but not in Pkm2KOi mice, post MI (Fig. 1P). We also observed a significant impact on survival for the Pkm2KOi mice post MI (Fig. 1Q). Collectively, these findings indicate that Pkm2 ablation decreases the probability of developing HF with early death, post MI.
Quantitative analysis of neoangiogenesis detected a significantly increase in coronary microvessel density in the peri-infarct zone (Fig. 2A), exclusively in Pkm2KOi mice post MI with reduced degree of CM apoptosis (P < 0.01) (Fig. 2B) [30], detected as early as 2-d post MI (Supplementary Fig. 3). This data suggest that the reduction in CM apoptosis was an early contributor to the beneficial effect of Pkm2 ablation on cardiac structure and function.

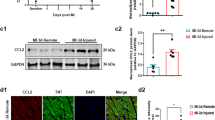
A Quantification of capillary density at 4-d post MI (left), as determined in myocardial LV cross-sections employing anti-Von Willebrand factor (Vwf) staining (right; green). Data are mean ± s.e.m. n = 6. *P < 0.01 vs. sham/Veh-control. #P < 0.01 vs. sham/Pkm2KOi. B Quantification of cardiomyocyte apoptosis (left) in LV sections (right) at 2-d post MI. White asterisks, CM. n = 6. Data are mean ± s.e.m. *P < 0.01 vs. Sham/Veh-control. #P < 0.01 vs. sham/Pkm2KOi. C Quantification of cardiac-specific gene expression in LV specimen at 21-d post MI as analyzed by RT-qPCR. Data are means ± s.e.m. n = 4. *P < 0.01 vs. Sham/Veh-control. #P < 0.01 vs. sham/Pkm2KOi. D Immunoblot analysis of cardiac-specific gene expression in LV extracts at 21-d post MI employing antibodies as indicated on the left. Western blots were repeated at least once with similar results employing two independent biological replicates. E Loss of sarcomeric Z-disk structure as indicated by loss of a striated actinin pattern in Veh-control animals at 4-d post MI as assessed using α-actinin antibodies specific to cardiac Z-disks. One representative result of three independent experiments is shown.
Consistent with reports that impaired calcium handling is associated with HF [31], mRNA and protein levels of Ryr2, Serca2a, Mlc2v, and PKA were significantly lower in Veh-controls compared to Pkm2KOi animals at 21-d post MI (P < 0.01) (Fig. 2C, D). In addition, fetal α-SMC was highly expressed in Veh-controls but absent in Pkm2KOi mice post MI. Transcript levels of key sarcomeric proteins [31], (tropomyosin (Tpm1), troponin (Tnnc1)) were downregulated (Fig. 2D) with a loss of the striated actinin pattern in Veh-control mice post MI. These observations could explain the lower FS in Veh-control animals (Fig. 2E) [32]. Transcript levels of other contractile factors (Tpm2, Tnni3, Tnnt3, titin (Ttn)) were significantly increased in both Pkm2KOi and Veh-controls post MI, as compared to shams. However, the quantitative differences in transcript levels is consistent with a lower degree of hypertrophy in the Pkm2KOi as compared to Veh-control animals post MI.
Loss of Pkm2 protects against ischemic oxidative stress
Inhibition of Pkm2 by reactive oxygen species (ROS) can divert glycolytic intermediates toward the pentose phosphate pathway (PPP) to increase the production of NADPH required to maintain the reduced form of glutathione, a key element in the detoxification of ROS [33, 34]. We found higher NADPH/NADP+ ratios in Pkm2KOi mice at 4-d post MI (Fig. 3A). This was accompanied with decreased transcription of members of the phase-2 antioxidant system [35] including Nrf2-target genes [36] (heme oxygenase (HO-1) [37] and Nqo1 [38]) in the Pkm2KOi mice (Fig. 3B). In contrast, mRNA levels of the other anti-oxidative factors (Gsta4, Gpx4, Sod2, and Pink1 [22]) were significantly higher with enhanced Nrf2 protein levels in both Veh-controls and Pkm2KOi animals, at 4-d post MI (Fig. 3C). This is consistent with studies reporting Nrf2 stabilization by Keap1 in models of ischemic stress [39, 40]. In addition, protein expression of Nrf2-regulated genes (Cat, Nqo1, Sod2) was found to be increased in Pkm2KOi mice post MI only (Fig. 3C). This is in keeping with the lower observed levels of NADPH oxidase (Nox4) [41] in Pkm2KOi post MI. Therefore, transcriptional transactivation of Nrf2-target genes is less sensitive to ischemic stress, in the absence of Pkm2. We also observed lower levels of cytotoxic 4-HAE, an indicator of ROS-dependent lipid peroxidation (Fig. 3D) [42, 43] with higher glutathione/oxidized glutathione (GSH/GSSG) ratios in Pkm2KOi mice post MI (Fig. 3E). Collectively, Pkm2 deficiency correlated with an enhanced ability of the heart to limit oxidative stress after MI.

A Higher NADPH/NADP+ ratios in LV extracts obtained from Pkm2KOi mice at 4-d post MI, when compared to Veh-controls. Data are means ± s.e.m. n = 4. *P < 0.001 vs. sham/Veh-control. #P < 0.05 vs. sham/Pkm2KOi. B Expression levels of selected factors that play an important role in detoxification processes of reactive oxygen species (ROS) analyzed by RT-qPCR at 4-d post MI. Data are means ± s.e.m. n = 4. *P < 0.001 vs. sham/Veh-control. #P < 0.001 vs. sham/Pkm2KOi. C Immunoblot analysis of key genes involved in ROS detoxification processes. Western blots were repeated at least once with similar results employing two independent biological replicates. D Levels of 4-HAE, a biomarker for lipid peroxidation, in hearts of Pkm2KOi mice at 4-d post MI. Data are means ± s.e.m. n = 4. *P < 0.001 vs. sham/Veh-control. #P < 0.01 vs. sham/Pkm2KOi. E Levels of GSSG (left) and GSH (right), indicators of cardiac oxidative stress, in Pkm2KOi mice in comparison to MI/Veh-controls at 4-d post MI. Data are means ± s.e.m. n = 4. *P < 0.001 vs. sham/Veh-control. #P < 0.01 vs. sham/Pkm2KOi.
Loss of Pkm2 protects mitochondrial energetics
Cardiac oxidative stress is associated with mt dysfunction [22, 44]. To examine this we measured mRNA (Fig. 4A) and protein expression (Fig. 4B) for several mt factors regulating energy metabolism and found Pgc-1α, Sdhc, Tfam, citrate synthase, Ucp3, ATP5a, Mfn2, Opa1, and Sod2 were significantly higher in Pkm2KOi mice post MI. Reductions in mt ATP production is detrimental to contractile function and viability of CM [44,45,46,47]. We noted an improvement in mt copy number and respiratory capacity in Pkm2KOi post MI (Fig. 4C, D) with concomitant improvement in ATP/ADP ratios (Fig. 4E).

A Quantification of mRNA expression levels of various factors involved in the maintenance of mitochondrial (mt) function at 4-d post MI. Data are means ± s.e.m. n = 4. *P < 0.01 vs. sham/Veh-control. #P < 0.001 vs. sham/Pkm2KOi. B Immunoblot of mt protein expression in Pkm2KOi mice at 4-d post MI vs. MI/Veh-controls employing antibodies as indicated on the left. Western blots were done twice with similar results employing independent biological replicates. C Mt biogenesis, defined as relative DNA copy number of the mt encoded Cytb gene normalized to the copy number of the nuclear gene B2m, was determined by qPCR. Data are means ± s.e.m. n = 4. *P < 0.001 vs. sham/Veh-control. #P < 0.01 vs. sham/Pkm2KOi. D Mt capacity, defined as relative mRNA levels of the nuclear gene cytochrome b (Cytb), a constituent of OxPhos complex III, normalized to B2m transcript expression, was determined by RT-qPCR at 4-d post MI. Data are means ± s.e.m. n = 4. *P < 0.001 vs. sham/Veh-control. #P < 0.01 vs. sham/Pkm2KOi. E Quantification of ATP (left), ADP (middle), and ATP/ADP ratios (right) in Pkm2KOi mice vs. Veh-controls at 4-d post MI. Data are means ± s.e.m. n = 4. *P < 0.001 vs. sham/Veh-control. #P < 0.05 vs. sham/Pkm2KOi. F Quantification of NAD+ (left) NADH (middle) and NAD+/NADPH ratios (right) in LV extracts obtained from Pkm2KOi mice compared to Veh-controls at 40d post MI. Data are means ± s.e.m. n = 4. *P < 0.01 vs. sham/Veh-control. G Cardiac lactate levels at 4-d post MI in LV extract from Pkm2KOi and Veh-control animals. Data are means ± s.e.m. n = 4. *P < 0.01 vs. sham/Veh-control.
NADH is an electron donor to mt NADH dehydrogenase in complex I and is the major entry point for NADH-derived electrons to the electron transport chain [48]. We found a significant decrease in the NAD+/NADH ratios in Pkm2KOi hearts (Fig. 4F). Lactate accumulation has also been observed in HF in association with alterations in NADH metabolism [49, 50]. Indeed, we found significant increases in lactate levels in Veh-controls but not in Pkm2KOi mice post MI indicating that Pkm2 ablation tempers oxidative energy depletion (Fig. 4G).
Loss of Pkm2 induces cytokinesis of adult CMs post MI
To define the role of Pkm2 in cardiac remodeling post MI, we needed to consider changes throughout the LV. Early in the process (4-d post MI), the area at risk in Pkm2KOi and Veh-control groups was of equal size and involved >80% of the cross-sectional LV area, at the level of the papillary muscles (Fig. 5A). We also noted a decreased α-actinin expression and lower extracellular matrix content (wheat germ agglutinin (WGA); Fig. 5A). The BZ showed α-actinin expression comparable to that of the area at risk with a markedly higher level of WGA reactivity. In contrast, the remote region exhibited levels of α-actinin and WGA comparable to both sham control groups. Thus, there is reduced remodeling observed early in Pkm2KOi animals.

A Quantitative analysis of CM (left) and non-cardiomyocytes (NCM; middle) in S-phase in Pkm2KOi mice at 4-d post MI. Confocal immunofluorescence microscopic analysis (right) of CM in S-phase. Fixed tissue sections were subjected to immunostaining using anti-α actinin (red) in conjunction with the Click-iT Edu assay to detect S-phase nuclei (green), wheat germ agglutinin (WGA) to detect the ECM (cyan) and DAPI to stain genomic DNA (blue). White asterisks, CM. AR area at risk, BZ border zone, RA remote area, TN total number. The yellow dotted line represents the border between AR and BZ. Data are means ± s.e.m. n = 6. *P < 0.001 vs. sham/Veh-control. #P < 0.001 vs. sham/Pkm2KOi. B Quantification of CM (left) and NCM (middle) in M-phase in Pkm2KOi mice at 4-d post MI. Confocal immunofluorescence microscopic analysis (right) of M-phase CM (white stars) in fixed LV specimen employing antibodies to Histone H3Pi-Ser28 (green) and α-actinin (red). Data are means ± s.e.m. n = 6. *P < 0.001 vs. sham/Veh-control. #P < 0.001 vs. sham/Pkm2KOi. C Mitotic CM in Pkm2KOi mice at 4-d post MI. Top row: CM in anaphase (left), CM in early telophase (middle), CM in prophase (right). Bottom row: binucleated CM in prophase (left). NCM in anaphase (right). 3D-reconstruction of serial confocal microphotographs was performed employing IMARIS software to unambiguously distinguish mitotic CM from mitotic NCM. Representative examples of six independent experiments is shown. D Quantitative analysis of cytokinesis in Pkm2KOi CM at 4-d post MI (left). Representative immunofluorescence micrographs of LV tissue samples from Pkm2KOi mice using antibodies recognizing AurkB (green) during cytokinesis (right). 3D reconstitution of typical confocal micrographs visualizes AurkB positive midbody structure (white arrowhead) between two dividing daughter CM. Fixed LV tissue sections were stained for indirect immunofluorescence microscopy analysis with antibodies to α-actinin (red), and AurkB (green). Data are means ± s.e.m. n = 6. #P < 0.001 vs. sham/Pkm2KOi. E Analysis of remodeling in the area at risk in the LV free wall in Pkm2KOi mice at 21-d post MI. Confocal immunofluorescence microscopy was employed using co-staining of the ECM. Representative examples of six independent experiments are shown. F Assessment of mono- and binucleated CM in the AR at 10-d post MI. Quantification of CM ploidy in adult Pkm2KOi mice (left) by confocal immunofluorescence microscopy and 3D-reconstruction analysis (right). 3D-reconstruction of serial confocal microphotographs was performed employing IMARIS software to unambiguously distinguish mononucleated CM from binucleated NCM. Fifty CM located in three different regions in the infarct zone were counted per sample. Data are means ± s.e.m. n = 6. #P < 0.001 vs. sham/Pkm2KOi.
We also noted the presence of smaller CM in Pkm2KOi mice compared to Veh-controls at 21-d post MI (Fig. 1G) and speculated that Pkm2 may potentially regulate CM cell division. Thus, the degree of DNA synthesis was investigated with EDU injection. We found the following numbers of CM in S-phase in Pkm2KOi mice at 4-d post MI: (i) area at risk (AR) 76 ± 9.6 nuclei/mm2, (ii) BZ 37 ± 8.3 CM nuclei/mm2, (iii) remote area (RA) zero CM nuclei/mm2, and (iv) total number (TN) 113 ± 8.3 CM nuclei/mm2 (P < 0.01) (Fig. 5A).
Assessment of numbers of CM in Pkm2KOi mice in M-phase by immunofluorescence microscopy employing anti-phospho-histone 3 phosphorylated at Ser28 (PiH3) showed: (i) AR 21.5 ± 4.8 CM nuclei/mm2, (ii) BZ 10.1 ± 4.6 CM nuclei/mm2, (iii) RA zero CM nuclei/mm2, and (iv) TN 31.6 ± 5.4 CM nuclei/mm2 (P < 0.01) (Fig. 5B, C). In addition, there were no sex-specific differences noted (Supplementary Fig. 4). In all these experiments, EDU-positive and PiH3-positive CM were absent from Veh-control mice.
We next assessed the importance of Pkm2 in driving cardiac remodeling through CM proliferation, post MI. We expected that the relative number of replicating CM/mm2 in Pkm2KOi mice would remain constant post MI. Indeed, there was no observable difference in the number of CM/mm2 in the infarct zone of Pkm2KOi mice with small infarcts versus large infarcts (Supplementary Fig. 5), implying that CM proliferation due to Pkm2 ablation is indeed germane to its cardioprotective effect.
Adult CM can be binucleated or polyploid, implying that M-phase can occur independently of cell division [51]. Thus we investigated whether CM completed cytokinesis by detecting midbody structures with anti-AurkB antibodies in the final phase of daughter cell separation. Ablation of Pkm2 induced cytokinesis in 15.2 ± 4.6 CM/mm2 in the AR and minimally in the BZ (Fig. 5D). This was not observed in Veh-controls. This supports the notion that Pkm2 ablation endows a restricted effect on CM that are specifically exposed to ischemia to acquire a proliferative potential contributing to cardiac repair (Fig. 5E). This proliferative window for CM in Pkm2KOi extended only during the period of 3–10d post MI. Of note, we did not observe any degree of CM proliferation in hearts from sham mice. All these findings lend support to our hypothesis that replication of CM due to the loss of Pkm2 protects the myocardium against ischemic injury.
Finally, we examined whether Pkm2 deficiency post MI influenced the ploidy of CM. We found a significant increase (3.8-fold) in the percentage of mononucleated CM, and a decrease (−43.1%) in binucleated CM in the combined AR/BZ in Pkm2KOi post MI providing additional evidence that ablation of Pkm2 can lead to CM proliferation and not polyploidy (P < 0.001) (Fig. 5F).
Infarct “wound healing” is characterized by initiation of the proliferative phase of NCM that comprises mainly endothelial cells and fibroblasts [52]. There was a significant increase in NCM in S- and M-phase in the Pkm2KOi and Veh-control mice at 4d following MI (P < 0.001 vs. sham/Pkm2KOi and sham/Veh-control mice) (Fig. 5A, B). Numbers of proliferating NCM were markedly lower (−47%) in hearts of Pkm2KOi mice compared to Veh-controls probably explaining higher levels of extracellular matrix deposition in control mice versus Pkm2KOi at 21-d post MI (P < 0.01) (Fig. 5E).
Differential regulation of cell-cycle factors with loss of Pkm2
Transition of adult CM through the cell cycle requires elimination of multiple inhibitory factors [53]. We found that transcript and protein levels of cell-cycle inhibitors (E2f6, p15, p18, p21, p27, p57) were specifically downregulated in Pkm2KOi hearts, but not in Veh-controls at 4-d post MI (Fig. 6A, B). Interestingly, we observed a concomitant increase in transcript levels of Skp2, an E3 ubiquitin ligase for p21 and p27, and expression of key factors driving mitosis specifically in Pkm2KOi mice (P < 0.01) (Fig. 6C, D). There was also an increase, albeit lower, in transcription levels of these factors in Veh-controls post MI, likely reflective of NCM replication (Fig. 6C).

A Expression of cell-cycle inhibitory factors in the LV of Pkm2KOi mice at 4-d post MI as analyzed by RT-qPCR. Data are means ± s.e.m. n = 4. *P < 0.001 vs. sham/Veh-control. #P < 0.001 vs. sham/Pkm2KOi. B Immunoblot of protein expression of cell-cycle inhibitors at 4-d post MI employing specific antibodies as shown on the left. Western blots were done twice yielding similar results using two independent biological replicates. C Transcript levels of cell-cycle-activating genes in LV samples derived from Pkm2KOi mice at 4-d post MI as analyzed by RT-qPCR. Data are means ± s.e.m. n = 4. *P < 0.01 vs. sham/Veh-control. #P < 0.001 vs. sham/Pkm2KOi. D Immunoblot of protein expression of cell-cycle-activating factors in Pkm2KOi mice at 4-d post MI using antibodies as indicated on the left. Western blots were repeated once with similar results using an independent biological replicate. E Quantification of mRNA expression of genes required for cytokinesis in Pkm2KOi mice at 4-d post MI as analyzed by RT-qPCR. Data are means ± s.e.m. n = 4. *P < 0.01 vs. sham/Veh-control. #P < 0.001 vs. sham/Pkm2KOi.
Initiation of DNA replication in adult CM is tightly regulated by Cdk2 [20]. At 4-d post MI, Veh-control hearts did not show an induction of Cdk2-associated activity relative to sham/Veh-controls (Fig. 6D). In contrast, Cdk2 kinase activity was markedly induced 8.2-fold in Pkm2KOi mice, post MI. This gain in Cdk2 kinase activity is consistent with extremely low protein levels of inhibitory p21, p27, and p57 (Fig. 6B), and sustained induction of Cyclin D2 and Cyclin A (Fig. 6D). Intriguingly, ablation of Pkm2 also increased transcription of factors that are required for proper execution of cytokinesis [54] (AurkB, Cdc20, Ect2, Itgb1, RacGap1) (Fig. 6E).
Pkm2 interacts with Ctnnb1 in CMs in vivo
Reportedly, Egfr mediates Pkm2/Ctnnb1-dependent proliferation of brain tumors [55] and Ctnnb1 can induce cell-cycle re-entry in immature CM [56, 57]. Therefore, we investigated the possibility that proliferation occurs through the Pkm2–Ctnnb1 axis. The loss of Ctnnb1 (Supplementary Fig. 6A–D) did not result in changes in HBW and FS in Ctnnb1KOi mice by 21-d post MI (Supplementary Fig. 6E, F). There was also no evidence of the number of mitotic CM in Ctnnb1KOi mice 4-d post MI (Supplementary Fig. 6G). However, we wanted to investigate whether the combined loss of Pkm2 and Ctnnb1 in double knockout (DKOi) mice affected CM proliferation (Supplementary Fig. 7A–C). We found no significant improvement in FS in DKOi mice at 3 weeks post MI (Fig. 7A–C and Supplementary Fig. 7D) with no evidence of mitotic CM in DKOi mice at 4-d post MI (Fig. 7D). Combined, Ctnnb1 activity is indispensable for proliferation of Pkm2-deficient CM.

Echocardiographic assessment of cardiac function: A FS, B LVEDD, and C LVESD in Pkm2/Ctnnb1 double-knockout (DKOi) mice at 21-d post MI. Data are means ± s.e.m. n = 6. D Quantification of CM (left) and NCM (middle) in M-phase in DKOi mice at 4-d post MI. Confocal immunofluorescence microscopic analysis (right panel) of mitotic CM in fixed LV specimen employing antibodies to Histone H3.Pi-Ser28 (green) and α-actinin (red). Data are means ± s.e.m. n = 6. *P < 0.001 vs. sham/Veh-control. #P < 0.001 vs. sham/Pkm2KOi. ns not significant. E, F Total cardiac extracts from Pkm2KOi mice were subjected to immunoprecipitation with antibodies specific to Pkm2 or Ctnnb1 and Ctrl IgG as indicated (top). Immunoblotting was performed with antibodies specific to Ctnnb1 (e) or Pkm2 (f). Experiments were repeated at least once with similar results employing independent biological replicates. G Localization of Ctnnb1 4-d post MI. In the presence of Pkm2, Ctnnb1 resides in the cytoplasm of unstimulated CM in the infarct zone of Pkm2KOi mice at 4-d post MI (left panel: bottom). In contrast, in the absence of Pkm2, a substantial fraction of endogenously expressed Ctnnb1 protein is found in the nucleus of CM in Pkm2KOi mice at 4-d post MI as analyzed by 3D-confocal immunofluorescence microscopy (right panel: bottom). LV specimen was stained with antibodies against Ctnnb1 (green) and α-actinin (red). One representative example of three independent experiments using biological replicates is shown. H Intracellular localization of Ctnnb1 protein analyzed by immunoblotting. Mechanically and chemically fractionated cardiac cytoplasmic and nuclear extracts from Pkm2KOi mice at 4-d post MI were analyzed by immunoblotting employing antibodies as indicated on the left. Western blots were repeated once with similar results employing a independent biological replicates. I Chromatin IP (ChIP) experiments illustrating the binding of Ctnnb1 to promoters of Axin2, Ccnd1, Myc, and Sox2. LV tissue was obtained from Pkm2KOi at 4-d post MI. Tnnt3 ChIPs served as negative control. Normal rabbit IgG was used for control ChIPs. Input lanes of chromatin levels used for the immunoprecipitation step demonstrate equal loading. One representative result of three experiments employing independent biological replicates is shown. K Quantification of transcript levels of Ctnnb1 target genes in the hearts of Pkm2KOi at 4-d post MI by RT-qPCR. n = 4. L Immunoblot of the Gsk-3β–Pi3K-Akt-Ctnnb1 signal transduction pathway. Protein expression of LV samples from Pkm2KOi mice at 4-d post MI was analyzed by Western blotting with antibodies as indicated on the left. Immunoblotting was repeated at least once with similar results employing two independent biological replicates.
To analyze whether Pkm2 exists as an endogenous complex with Ctnnb1 in the heart, we performed co-IP analysis (Fig. 7E, F). We found that Pkm2/Ctnnb1 complexes were present only in hearts from Veh-control mice post MI, providing evidence that Pkm2 can bind Ctnnb1 and vice versa. There was no evidence of Pkm2/Ctnnb1 interaction in the Pkm2KOi mice.
Gsk-3β is an important inhibitor of cell division in proliferating cellular systems [9, 15], including CM [58,59,60,61]. The hallmark of Ctnnb1’s ability to direct gene transcription is its nuclear translocation [62]. In addition, Ctnnb1 is a Pkm2 target in tumors [63]. Therefore, we investigated whether Pkm2 regulates the subcellular localization of Ctnnb1 in CM. In the presence of Pkm2, Ctnnb1 protein expression was exclusively cytoplasmic at 4-d post MI (Fig. 7G). In contrast, Ctnnb1 accumulated in the nucleus of Pkm2-deficient CM post MI (Fig. 7G) indicating that Pkm2 is involved in the regulation of Ctnnb1 subcellular localization. In addition, cytoplasmic Gsk-3β is known to phosphorylate Ctnnb1 to target it for proteasomal degradation [9, 15, 51]. We found that Gsk-3β-specific phosphorylation of Ctnnb1-Pi (Thr-41, Ser-37, Ser-33) was present in cytoplasmic fractions at baseline and post MI, independent of Tam (Fig. 7H). Ctnnb1.Pi-Thr-45/Ser-37/Ser-33 was never observed in nuclear fractions [9, 15]. Notably, Akt-dependent phosphorylation of Ctnnb1 (Ser-552, Tyr-333) was confined to nuclear fractions and solely observed in Pkm2KOi mice post MI (Fig. 7H). Thus, these residues of Ctnnb1 are critical for the regulation of its subcellular localization. In addition, Pkm2 is dispensable for Ctnnb1.Pi-Tyr-333 in the heart (Fig. 7H) [52].
Next, we next analyzed Ctnnb1 binding to established target promoters [9, 15] by ChIP assays. We were able to identify enhanced binding of Ctnnb1 to target gene promoters of Axin2, Ccnd1, Myc, and Sox2 exclusively in Pkm2KOi mice post MI (lane 12; Fig. 7I), confirmed by RT-qPCR (Fig. 7K). These findings support the concept that nuclear translocation of Ctnnb1 is a consequence of Ctnnb1 phosphorylation, which is inhibited by Pkm2.
Stimulation of proliferation by the Pi3K/Akt/Erk signal transduction pathway in undifferentiated cells involves inhibition of Gsk-3β by Akt [9, 15]. However, Akt generally fails to activate Ctnnb1 in adult CM, suggesting that Gsk-3β is not accessible to Akt [64]. Immunoblot analysis revealed higher protein levels of Akt, increased Akt.Pi-Thr308, an activating phosphorylation site, and markedly elevated levels of Erk1/2 phosphorylation of Thr202/Tyr204 (Fig. 7L), solely in Pkm2KOi mice at 4-d post MI (Fig. 7L). Next, we analyzed Akt-dependent phosphorylation of Gsk-3β on Ser9 and found that phosphorylation of this site was also confined to Pkm2KOi mice at 4-d post MI (Fig. 7L). In addition, total Gsk-3β protein levels were drastically lower in the Pkm2KOi extracts compared to Veh-controls. We conclude from all these findings, that the presence of Pkm2 inhibits adult CM proliferation, at least in part, by inhibition of Akt-mediated phosphorylation of the Gsk-3β/Ctnnb1 pathway.
Discussion
Canonical Wnt/Ctnnb1 signaling regulates regeneration in the developing heart and remodeling after injury [16, 65, 66]. However, experimental evidence is lacking to define Ctnnb1’s role in adult CM proliferation post MI. Our study demonstrates that Pkm2 binds to Ctnnb1 and inhibits Ctnnb1 phosphorylation by Akt. However, Pkm2 ablation enables nuclear accumulation of Ctnnb1 and induction of Ctnnb1-dependent gene transcription. Genetic deletion of both Pkm2 and Ctnnb1 abrogates the protective effect of CM proliferation on cardiac function post MI (Fig. 8). This role of cytoplasmic Pkm2 as an intrinsic inhibitor of CM proliferation is novel.

Pkm2 inhibits Akt-dependent activation of cytoplasmic Ctnnb1. Gsk3-b mediated phosphorylation targets Ctnnb1 for proteasomal degradation (MI – Tam; top left). In the absence of Pkm2, Akt prevents Gsk3-b dependent Ctnnb1 inactivation post MI. Akt phosphorylation of Ctnnb1 contributes to its nuclear translocation (MI + Tam; top right). Nuclear Ctnnb1 transactivates genes necessary for cell cycle re-entry and proliferation of adult cardiomyocytes (bottom panel). Blue circles, components of the Ctnnb1 destruction complex. Green circles, activated factors. Red circles, inactivated factors. Green Pi, activating phosphorylation. Red Pi, inactivating phosphorylation. Red block arrow, inactivation. Red arrow, inactive gene transcription. Green arrow, activated gene transcription.
Our data support the paradigm that cell-cycle activation is executed by nuclear Ctnnb1. Akt signaling activates the cell cycle through inactivating phosphorylation of Gsk-3β. In its inactive form, Gsk3b cannot “mark” Ctnnb1 for proteasomal degradation and Ctnnb1 translocates to the nucleus [67] to form complexes with several transcription factors (TCF and LEF). This leads to the expression of numerous genes, including cyclin D1 and c-Myc, to initiate G1/S-phase transition [68]. Cyclin D1-Cdk4/6 complexes then phosphorylate and inactivate the retinoblastoma protein, leading to an increase in Cdk2/cyclin E levels needed for DNA synthesis [69, 70] and the activation of transcription by E2f family members (E2F1–3) to synergistically promote cell-cycle progression through induction of their targets Cdk2 and cyclin A [71, 72]. Relief of Cdk4/6-Cyclin D1 complexes from inhibition by p21 and p27 allows CM to re-enter the cell cycle [73, 74]. In this regard, the transcription factor c-Myc exerts a dual role in G1 phase, by inducing Cyclin D1 and repressing p21 and p27 [75,76,77].
Phosphorylation of Ctnnb1 can serve two different purposes: proteosomal degradation versus nuclear import [62, 64]. After MI, we observed the activation of Akt in both the Veh-controls and Pkm2KOi animals with cytoplasmic and nuclear Ctnnb1, respectively. We believe that the presence of Pkm2 interferes with the inactivation of Gsk-3β by Akt. In this study, Akt-dependent phosphorylation of Gsk-3β on Ser9 was confined to Pkm2KOi mice at 4-d post MI. This coincided with Pi.Ctnnb1-Ser-552/Tyr-333 and CM proliferation. Therefore, phosphorylation of Ser-552 on and Tyr-333 in Ctnnb1 is critical for its nuclear translocation.
Ctnnb1 lacks a nuclear localization signal (NLS) and this raises the question how is the nuclear transport of Ctnnb1 regulated, in addition to its phosphorylation status? Recently, the guanine nucleotide exchange factor RAPGEF5 was identified to regulate Ctnnb1 nuclear translocation independent of the cytoplasmic retention of Ctnnb1 and the importin nuclear transport system [78]. It would be of interest to investigate this further.
Is the cardioprotective effect observed in the absence of Pkm2 dependent on CM proliferation? We think that it is likely a combination of effects that couple with the ability of adult CM to proliferate, resulting in improved cardiac function [53, 79]. CM proliferation in Pkm2KOi mice does occur in the presence of decreased cardiac remodeling at 4-d post MI. Interestingly, both fetal and neonatal murine hearts contain proliferating CM that participate in cardiac regeneration after extensive injury [80]. In contrast, the adult heart consist of differentiated postmitotic CM, devoid of any meaningful regenerative capacity [81, 82]. Clearly, CM replication is not the only driving force in the repair of cardiac function after ischemic injury, since hypoxia [83,84,85], neoangiogenesis [86], ECM remodeling [87, 88], oxidative restoration [89], and improved energy substrate utilization [90] also play critical roles in this process.
Reportedly, Akt signaling promotes fetal CM proliferation [56]. The cooperation between downstream effectors of this pathways in adult CM, however, is incompletely understood. Our data suggest that Ctnnb1 nuclear translocation relies on Akt-mediated phosphorylation. Genetic deletion of Akt1 in mice causes neonatal lethality due to insufficient CM proliferation and is in agreement with this view [91]. Moreover, transgenic overexpression of Erbb2, in the presence of Nrg1 post MI, induced adult CM proliferation in an Akt and Gsk-3β/Ctnnb1-dependent manner [92].
There is also functional cooperativity between Wnt/Ctnnb1 and Hippo/Yap signal transduction pathways in CM proliferation [93,94,95,96]. Ctnnb1 or Yap knockout mice die at E12.5 and E10, respectively, due to reduced CM proliferation [57, 96]. Overexpression of Yap, in the absence of ischemic injury, resulted in inactivation of Gsk-3β and activation of Ctnnb1 gene transcription [56]. Moreover, constitutively active Yap induced CM proliferation and regeneration in neonatal mouse hearts [55]. Pi3K is an important direct target of Yap, through which Yap can activate Akt and CM proliferation in the adolescent mouse hearts [97]. Thus, Yap provides a mechanistic link between Pi3K/Akt signaling and the Gsk-3β/Ctnnb1 axis, in embryonic and neonatal CM. In the developing fetal heart, Hippo signaling inhibits a pro-proliferative Wnt/Ctnnb1-Yap interaction as the heart transitions from rapid cell cycling to moderate proliferation kinetics in the maturing heart. In adult CM, Yap is restricted to the cytoplasm by Hippo signaling, resulting in transcription of Yap/Ctnnb1-regulated genes [98]. Yap’s potential role in adult CM proliferation under stress conditions remains to be investigated.
Pkm2, but not Pkm1, alters glucose metabolism in dividing and cancer cells by mechanisms that are not explained by its enzymatic activity. There is a large body of literature describing a nuclear role of Pkm2 in tumorigenesis [8]. Structurally, Pkm2 carries a carboxyl-terminal NLS, and nuclear Pkm2 translocation can induce caspase-independent apoptosis [99], stimulate cell proliferation [100], and activate transcription by acting as a protein kinase for histone H3 or Mek5 [101, 102]. Nuclear Pkm2 can also activate Oct-4 thereby regulating the self-renewal capacity of undifferentiated embryonic stem cells [55]. In addition, Egfr mediates nuclear Pkm2 translocation, where it forms a heterodimeric complex with Ctnnb1 leading to cyclin D1 activation [55]. In contrast, our data have identified a critical role for Pkm2 in the cytoplasmic sequestration of Ctnnb1. We also present evidence that cytoplasmic Ctnnb1, complexed with Pkm2, is refractory to phosphorylation by Akt. This suggests that there is a second Pkm2-mediated barrier, isolating mitogenic Akt signaling from nuclear Ctnnb1 translocation and cell-cycle re-entry.
While the induction of CM proliferation post MI in Pkm2KOi mice is a sex-independent process, further research is needed that addresses the impact of Pkm2 as it relates to estrogen status. Independent of age, more women than men will die or present with cardiovascular complications such as HF and stroke, after a MI [103,104,105,106]. Numerous pathways have been described by which estrogen might influence inflammation and wound healing after MI, including Tgf-β [107] and nitric oxide synthase pathways [108,109,110]. It was recently demonstrated that estrogen can increase the phosphorylation of Pkm2 at Ser-37, thereby inducing its nuclear translocation [111]. However, a more direct link between Pkm2 and estrogen status has yet to be described and would be an interesting area of future investigation.
A recent publication by the Zangi group reported contrary consequences through Pkm2 but with similar outcomes [112]. This work showed that Pkm2 was present in CM in the fetal heart and in the early postnatal period, but not during adulthood. Ablation of Pkm2 in fetal CM in vivo inhibited CM proliferation with a resultant decrease in heart size. CM-specific overexpression employing a modified Pkm2-endcoing RNA induced adult CM proliferation with a reduction in oxidative stress and enhancement of heart function, post MI. Mechanistically, ectopic Pkm2 regulated CM nuclear translocation of Pkm2/Ctnnb1 complexes enabling transcriptional transactivation of Ctnnb1-target genes. The authors concluded that Pkm2 is important for CM proliferation and ROS defense, providing a novel strategy for gene delivery in a clinical setting. However, Pkm2 expression was predominantly observed in NCM (including CD45+ leukocytes) in this study whereas we detected Pkm2 exclusively in adult CM post MI. The major distinction between the two studies relates to a single injection of the modified Pkm2 RNA reagent into the heart, as opposed to permanent removal of Pkm2 by a classic knockout strategy in our study. Both strategies led to an induction of CM proliferation, post MI. While the basis of the disparity between these findings is unclear, this may be explained by the fact that cell fate decisions in overexpression studies depend on the abundance of the ectopically expressed factor [113, 114]. More specifically, ectopically delivered Pkm2 formed complexes with Ctnnb1 to trigger increased nuclear translocation and thereby induce CM division [112]. Our findings support the view that Pkm2 functions as a cytoplasmic anchor preventing nuclear translocation of Ctnnb1 in the heart. This situation is reminiscent of the observations from the tumor literature showing the bimodal function for Pkm2, either as an activator or inhibitor of cellular division [13, 63].
There are aspects deserving further consideration to critically evaluate loss of Pkm2 with structural remodeling in the heart, that occurs independently of CM proliferation. For example, Pkm2 and Gsk-3β are also involved in glucose metabolism and energy homeostasis. In this regard, our data also support the importance of Pkm2 activity that is independent of its impact on mitosis. In particular, Pkm2KOi mice display higher degrees in capillary density post MI such that the mismatch between capillary density and CM is much lower. Development of cardiac hypertrophy contributes to myocardial hypoxia [115]. When oxygen tension is low, Phd3 stabilizes Hif1α, an important mediator of the hypoxic response, thereby activating glycolysis through stimulation of Glut1, Hk1, and Ldha [116]. Importantly, HIF1α mediates a switch to the Pkm2 isoform, post MI [117]. Pkm2 can also directly bind to Phd3, stimulating its function as a Hif1α coactivator in the activation of Hif1α-target genes [118].
Our study provides a conceptual change in the understanding of the complex regulation of CM proliferation in the adult heart. Loss of Pkm2 in ischemic CM creates an opportunity for Ctnnb1 to translocate to the nucleus and transactivate critical genes mediating cell-cycle progression. The combined activities associated with Pkm2 loss result in restoration of cardiac structure and function in response to cardiac injury. Therefore, strategies to inhibit Pkm2 in patients, who present with MI, could be beneficial. The delivery of modified siRNAs targeting Pkm2 in an ischemia-reperfusion animal model of injury may better mimic the clinical scenario. However, an important limitation to this strategy relates to finding effective Pkm2-specific siRNAs due to the sequence similarity present in both Pkm2 and Pkm1. Other potential limitations of a siRNA-based therapy include potential off-target tumorigenic or immunological effects. Therefore, we will need to explore the potential for detrimental effects of Pkm2 loss in the long term. Despite these limitations, the ability to provide timely revascularization, combined with CM proliferation, restricted remodeling and maintenance of heart function, could provide an exciting avenue for future treatments to come.
References
Farmakis D, Stafylas P, Giamouzis G, Maniadakis N, Parissis J. The medical and socioeconomic burden of heart failure: a comparative delineation with cancer. Int J Cardiol. 2016;203:279–81.
Mincucci MF, Azevedo PS, Polegato BF, Paiva SA, Zornoff LA. Heart failure after myocardial infarction: clinical implications and treatment. Clin Cardiol. 2011;34:410–4.
Anderson JL, Morrow DA. Acute myocardial infarction. N Engl J Med. 2017;376:2053–64.
Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26.
Foglia MJ, Poss KD. Building and re-building the heart by cardiomyocyte proliferation. Development. 2016;143:729–40.
Cahill TJ, Choudhury RP, Riley PR. Heart regeneration and repair after myocardial infarction: translational opportunities for novel therapeutics. Nat Rev Drug Discov. 2017;16:699–17.
Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci U S A. 2010;107:1894–9.
Chaneton B, Gottlieb E. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem Sci. 2012;37:309–16.
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26.
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33.
Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–80.
Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397–409.
Rees ML, Subramaniam J, Li Y, Hamilton DJ, Frazier OH, Taegtmeyer H. A PKM2 signature in the failing heart. Biochem Biophys Res Commun. 2015;459:430–6.
Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–99.
Zelarayan L, Gehrke C, Bergmann MW. Role of beta-catenin in adult cardiac remodeling. Cell Cycle. 2007;6:2120–6.
Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–64.
Hauck L, Stanley-Hasnain S, Fung A, Grothe D, Rao V, Mak TW, et al. Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death. PLoS ONE. 2017;12:e0189861.
Billia F, Hauck L, Grothe D, Konecny F, Rao V, Kim RH, et al. Parkinson-susceptibility gene DJ-1/PARK7 protects the murine heart from oxidative damage in vivo. Proc Natl Acad Sci U S A. 2013;110:6085–90.
Stanley-Hasnain S, Hauck L, Grothe D, Aschar-Sobbi R, Beca S, Butany J, et al. p53 and Mdm2 act synergistically to maintain cardiac homeostasis and mediate cardiomyocyte cell cycle arrest through a network of microRNAs. Cell Cycle. 2017;16:1585–600.
Dadson K, Hauck L, Hao Z, Grothe D, Rao V, Mak TW, et al. The E3 ligase Mule protects the heart against oxidative stress and mitochondrial dysfunction through Myc-dependent inactivation of Pgc-1α and Pink1. Sci Rep. 2017;7:41490.
Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. 2011;108:9572–7.
Mak TW, Hauck L, Grothe D, Billia F. p53 regulates the cardiac transcriptome. Proc Natl Acad Sci U S A. 2017;114:2331–6.
Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–79.
Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–5.
McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–5.
Yang Z, Ming XF. mTOR signalling: the molecular interface connecting metabolic stress, aging and cardiovascular diseases. Obes Rev. 2012;13 Suppl 2:58–68.
Kemi OJ, Ceci M, Wisloff U, Grimaldi S, Gallo P, Smith GL, et al. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J Cell Physiol. 2008;214:316–21.
Zhang D, Contu R, Latronico MVG, Zhang J, Rizzi R, Catalucci D, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805–16.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest. 2013;123:46–52.
Hamdani N, Kooij V, van Dijk S, Merkus D, Paulus WJ, Remedios CD, et al. Sarcomeric dysfunction in heart failure. Cardiovasc Res. 2008;77:649–58.
Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012;23:560–6.
Anastasiou D, Poulogiannis G, Asara JM, Gu Y. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–83.
Kim HJ, Nel AE. The role of phase II antioxidant enyzmes in protecting memory T cells from spontaneous apoptosis in young and old mice. J Immunol. 2005;175:2948–59.
Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29:1727–45.
Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, et al. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest. 1999;103:R23–9.
Ross D, Siegel D. Functions of NQO1 in cellular protection and CoQ 10 metabolism and its potential role as a redox sensitive molecular switch. Front Physiol. 2017;8:595.
Chen QM, Maltagliati AJ. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol Genom. 2018;50:77–97.
Zhang X, Hu C, Kong CY, Song P, Wu HM, Xu SC, et al. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020;27:540–55.
Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–70.
Dalleau S, Baradat M, Guéraud F, Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013;20:1615–30.
Santin Y, Fazal L, Sainte-Marie Y, Sicard P, Maggiorani D, Tortosa F, et al. Mitochondrial 4-HNE derived from MAO-promotes mitoCa2+ overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2019. https://doi.org/10.1038/s41418-019-0470-y.
Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181–90.
Neuber S. The failing heart—an engine out of fuel. N Engl J Med. 2007;356:1140–51.
Fan F, Duan Y, Yang F, Trexler C, Wang H, Huang L, et al. Deletion of heat shock protein 60 in adult mouse cardiomyocytes perturbs mitochondrial protein homeostasis and causes heart failure. Cell Death Differ. 2020;27:587–600.
Ventura-Clapier R, Garnier A, Veksler V. Energy metabolism in heart failure. J Physiol. 2004;555:1–13.
Berthiaume JM, Kurdys JG, Muntean DM, Rosca MG. Mitochondrial NAD+/NADH redox state and diabetic cardiomyopathy. Antioxid Redox Signal. 2019;30:375–98.
Lazzeri C, Valente S, Chiostri M, Gensini GF. Clinical significance of lactate in acute cardiac patients. World J Cardiol. 2015;7:483–9.
Benjaminutes D, Robay D, Hindupur SK, Pohlmann J, Colombi M, El-Shemerly MY, et al. Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep. 2018;25:3047–58.
Gandarillas A, Molinuevo R, Sanz-Gómez N. Mammalian endoreplication emerges to reveal a potential developmental timer. Cell Death Differ. 2018;25:47–6.
Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol. 2003;163:2433–40.
Yuan X, Braun T. Multimodal regulation of cardiac myocyte proliferation. Circ Res. 2017;121:293–309.
González-Rosa JM, Sharpe M, Field D, Soonpaa MH, Field LJ, Burns CE, et al. Myocardial polyploidization creates a barrier to heart regeneration in Zebrafish. Dev Cell. 2018;44:433–46.
Lee J, Kim HK, Han YM, Kim J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int J Biochem Cell Biol. 2008;40:1043–54.
Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, et al. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4:ra70.
Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. 2013;110:13839–44.
Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest. 2008;118:3609–18.
Woulfe KC, Gao E, Lal H, Harris D, Fan Q, Vagnozzi R, et al. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res. 2010;106:1635–45.
Gomez L, Thiebaut PA, Paillard M, Ducreux S, Abrial M, Crola Da Silva C, et al. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ. 2016;23:313–22.
Zhou J, Ahmad F, Parikh S, Hoffman NE, Rajan S, Verma VK, et al. Loss of adult cardiac myocyte GSK-3 leads to mitotic catastrophe resulting in fatal dilated cardiomyopathy. Circ Res. 2016;118:1208–22.
Krieghoff E, Behrens J, Mayr B. Nucleo-cytoplasmic distribution of beta-catenin is regulated by retention. J Cell Sci. 2006;119:1453–63.
Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011;480:118–22.
Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, et al. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–9.
Ozhan G, Weidinger G. Wnt/β-catenin signaling in heart regeneration. Cell Regen. 2015;4:3.
Baurand A, Zelarayan L, Betney R, Gehrke C, Dunger S, Noack C, et al. Beta-catenin downregulation is required for adaptive cardiac remodeling. Circ Res. 2007;100:1353–62.
Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24.
Lecarpentier Y, Schussler O, Hébert JL, Vallée A. Multiple targets of the canonical WNT/β-catenin signaling in cancers. Front Oncol. 2019;9:1248.
Herwig S, Strauss M. The retinoblastoma protein: a master regulator of cell cycle, differentiation and apoptosis. Eur J Biochem. 1993;246:581–601.
Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11.
Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci U S A. 1974;71:1286–90.
Bertoli C, Skotheim JM, de Bruin RAM. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14:518–28.
Xiao G, Mao S, Baumgarten G, Serrano J, Jordan MC, Roos KP, et al. Inducible activation of c-Myc in adult myocardium in vivo provokes cardiac myocyte hypertrophy and reactivation of DNA synthesis. Circ Res. 2001;89:1122–9.
von Harsdorf R, Hauck L, Mehrhof F, Wegenka U, Cardoso MC, Dietz R. E2F-1 overexpression in cardiomyocytes induces downregulation of p21CIP1 and p27KIP1 and release of active cyclin-dependent kinases in the presence of insulin-like growth factor I. Circ Res. 1999;85:128–36.
Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–511.
García-Gutiérrez L, Delgado MD, León J. MYC oncogene contributions to release of cell cycle brakes. Genes. 2019;10:244.
Cui N, Yang WT, Zheng PS. Slug inhibits the proliferation and tumor formation of human cervical cancer cells by up-regulating the p21/p27 proteins and down-regulating the activity of the Wnt/β-catenin signaling pathway via the trans-suppression Akt1/p-Akt1 expression. Oncotarget. 2016;7:26152–67.
Griffin JN, Del Viso F, Duncan AR, Robson A, Hwang W, Kulkarni S, et al. RAPGEF5 regulates nuclear translocation of β-catenin. Dev Cell. 2018;44:248–60.
Nguyen NUN, Canseco DC, Xiao F, Nakada Y, Li S, Lam NT, et al. A calcineurin-Hoxb13 axis regulates growth mode of mammalian cardiomyocytes. Nature. 2020;58:271–6.
Mohamed TMA, Ang YS, Radzinsky E, Zhou P, Huang Y, Elfenbein A, et al. Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell. 2018;173:104–6.e12.
Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wan M, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–6.
Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, et al. Dynamics of cell generation and turnover in the human heart. Cell. 2015;161:1566–75.
Kimura W, Xiao F, Canseco DC, Muralidhar S, Thet S, Zhang HM, et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature. 2015;523:226–30.
Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, et al. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541:222–7.
Ahmed MS, Sadek HA. Hypoxia induces cardiomyocyte proliferation in humans. JACC Basic Transl Sci. 2020;5:461–2.
Tao Z, Chen B, Tan X, Zhao Y, Wang L, Tiebing Zhu T, et al. Coexpression of VEGF and angiopoietin-1 promotes angiogenesis and cardiomyocyte proliferation reduces apoptosis in porcine myocardial infarction (MI) heart. Proc Natl Acad Sci U S A. 2011;108:2064–9.
Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature. 2017;547:179–84.
Morikawa Y, Heallen T, Leach J, Xiao Y, Martin JF. Dystrophin-glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature. 2017;547:227–31.
Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–79.
Cardoso AC, Lam NT, Savla JJ, Nakada Y, Pereira AHM, Elnwasany A, et al. Mitochondrial substrate utilization regulates cardiomyocyte cell cycle progression. Nat Metab. 2020;2:167–78.
Chang Z, Zhang Q, Feng Q, Xu J, Teng T, Luan Q, et al. Deletion of Akt1 causes heart defects and abnormal cardiomyocyte proliferation. Dev Biol. 2010;347:384–91.
D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. 2015;17:627–38.
Zhou Q, Li L, Zhao B, Guan KL. The hippo pathway in heart development, regeneration, and diseases. Circ Res. 2015;116:1431–47.
Furth N, Aylon Y. The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death Differ. 2017;24:1488–501.
Wang X, Ha T, Liu L, Hu Y, Kao R, Kalbfleisch J, et al. TLR3 mediates repair and regeneration of damaged neonatal heart through glycolysis dependent YAP1 regulated miR-152 expression. Cell Death Differ. 2018;25:966–82.
von Gise A, Lin Z, Schlegelmilch K. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109:2394–9.
Lin Z, Zhou P, von Gise A. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ Res. 2015;116:35–45.
Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–61.
Steták A, Veress R, Ovádi J, Csermely P, Kéri G, Ullrich A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer Res. 2007;67:1602–8.
Hoshino A, Hirst JA, Fujii H. Regulation of cell proliferation by interleukin-3-induced nuclear translocation of pyruvate kinase. J Biol Chem. 2007;282:17706–11.
Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–96.
Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609.
Mozaffarian D, Benjaminutes EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–322.
Mehta LS, Beckie TM, DeVon HA, Grines CL, Krumholz HM, Johnson MN, et al. Acute myocardial infarction in women: a scientific statement from the American Heart Association. Circulation. 2016;133:916–47.
Vaccarino V, Parsons L, Every NR, Barron HV, Krumholz HM. Sex-based differences in early mortality after myocardial infarction. National registry of myocardial infarction 2 participants. N Engl J Med. 1999;341:217–25.
Alexander KP, Newby LK, Hellkamp AS, Harrington RA, Peterson S, Kopecky S, et al. Initiation of hormone replacement therapy after acute myocardial infarction is associated with more cardiac events during follow-up. J Am Coll Cardiol. 2001;38:1–7.
Ashcroft GS, Dodsworth J, van Boxtel E, Tarnuzzer RW, Horan MA, Schultz GS, et al. Estrogen accelerates cutaneous wound healing associated with an increase in TGF-beta1 levels. Nat Med. 1997;3:1209–15.
Binko J, Murphy TV, Majewski H. 17Beta-oestradiol enhances nitric oxide synthase activity in endothelium-denuded rat aorta. Clin Exp Pharm Physiol. 1998;25:120–7.
Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, et al. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res. 2000;87:677–82.
Ashcroft GS, Greenwell-Wild T, Horan MA, Wahl SM, Ferguson MW. Topical estrogen accelerates cutaneous wound healing in aged humans associated with an altered inflammatory response. Am J Pathol. 1999;155:1137–46.
Lu Y, Liu X, Zhang E, Kopras EJ, Smith EP, Astreinidis A, et al. Estrogen activates pyruvate kinase M2 and increases the growth of TSC2-deficient cells. PLoS ONE. 2015;20:e0228894.
Magadum A, Singh N, Kurian AA, Munir I, Mehmood T, Brown K, et al. Pkm2 regulates cardiomyocyte cell cycle and promotes cardiac regeneration. Circulation. 2020. https://doi.org/10.1161/CIRCULATIONAHA.119.043067.
Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013;20:576–88.
Shats I, Deng M, Davidovich A, Zhang C, Kwon JS, Manandhar D, et al. Expression level is a key determinant of E2F1-mediated cell fate. Cell Death Differ. 2017;24:626–37.
Tomanek RJ. Response of the coronary vasculature to myocardial hypertrophy. J Am Coll Cardiol. 1990;15:528–33.
Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56.
Williams AL, Khadka V, Tang M, Avelar A, Schunke KJ, Menor M, et al. HIF1 mediates a switch in pyruvate kinase isoforms after myocardial infarction. Physiol Genom. 2018;50:479–94.
Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–44.
Acknowledgements
This work was supported by grants awarded by the Canadian Institute of Health Research to FB. FB is the recipient of the Canadian Institute of Health Research Phase II Clinician-Scientist Award.
Author information
Authors and Affiliations
Contributions
FB and LH designed research; DG, SC, KD, and LH performed research; FB and LH analyzed data; FB and LH wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by T. Mak
Rights and permissions
About this article

Cite this article
Hauck, L., Dadson, K., Chauhan, S. et al. Inhibiting the Pkm2/b-catenin axis drives in vivo replication of adult cardiomyocytes following experimental MI. Cell Death Differ 28, 1398–1417 (2021). https://doi.org/10.1038/s41418-020-00669-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00669-9
This article is cited by
-
Inhibition of Pyruvate kinase M2 (PKM2) by shikonin attenuates isoproterenol-induced acute myocardial infarction via reduction in inflammation, hypoxia, apoptosis, and fibrosis
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Targeting cardiomyocyte cell cycle regulation in heart failure
Basic Research in Cardiology (2024)
-
M-type pyruvate kinase 2 (PKM2) tetramerization alleviates the progression of right ventricle failure by regulating oxidative stress and mitochondrial dynamics
Journal of Translational Medicine (2023)
-
Role of Pyruvate Kinase M2 (PKM2) in Cardiovascular Diseases
Journal of Cardiovascular Translational Research (2023)
-
The role of glycolytic metabolic pathways in cardiovascular disease and potential therapeutic approaches
Basic Research in Cardiology (2023)
