
Abstract
The receptor activator of nuclear factor-κB (RANK) protein activates various protein kinase signaling cascades, including those involving NF-κB, mitogen-activated protein kinase (MAPK), and Bruton tyrosine kinase (Btk)/tyrosine-protein kinase Tec. However, the mechanism underlying the negative regulation of RANK by downstream signaling molecules remains unclear. Here, we report that Src homology 3 domain and cysteine-rich domain-containing protein 2 (STAC2) is a novel RANK ligand-inducible protein that negatively regulates RANK-mediated osteoclast formation. STAC2 physically interacts with RANK and inhibits the formation of the RANK signaling complex, which contains Grb-2-associated binder 2 (Gab2) and phospholipase Cγ2 (PLCγ2), thus leading to the suppression of RANK-mediated NF-κB and MAPK activation. Furthermore, STAC2 overexpression limits Btk/Tec-mediated PLCγ2 phosphorylation via the interaction between STAC2 and Btk/Tec. Taken together, our results reveal a novel mechanism whereby RANK signaling is restricted by its physical interaction with STAC2.
Similar content being viewed by others


Introduction
Skeletal homeostasis depends on a balance between bone-forming osteoblasts and bone-resorbing osteoclasts. An imbalance between bone resorption and formation causes various bone diseases, such as rheumatoid arthritis and osteoporosis [1, 2]. Osteoclasts are multinucleated cells (MNCs) that can degrade the mineralized bone matrix. Macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB (RANK) ligand (RANKL) are considered to be essential cytokines for osteoclastogenesis [3, 4]. The binding of RANKL to RANK initiates the recruitment of tumor necrosis factor receptor-associated factor 6 (TRAF6), which induces the synthesis of multiple polyubiquitin chains that target TGFβ-activated kinase 1 (TAK1), leading to the activation of mitogen-activated protein kinase (MAPK) and NF-κB pathways and influencing the formation, survival, and resorptive activity of osteoclasts [5,6,7,8]. Recent studies reported that the Grb-2-associated binder 2 (Gab2) protein is an important molecular adaptor for the RANK-mediated activation of the c-Jun N-terminal kinase (JNK) and NF-κB pathways. Moreover, Gab2 is recruited to RANK and forms a complex with phospholipase Cγ2 (PLCγ2) to modulate osteoclastogenic signals [9,10,11].
RANKL activates the expression of transcription factors, such as c-Fos, microphthalmia transcription factor, and nuclear factor of activated T cells c1 (NFATc1). Among these, NFATc1 is a key transcription factor in osteoclastogenesis. Its expression is dependent on TRAF6 and c-Fos pathways during RANKL-induced osteoclastogenesis [3, 6, 12]. The induction of NFATc1 is also dependent on the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM)-containing adaptors, such as the γ-chain of the Fc receptor and DNAX-activating protein 12, which mediate calcium signaling in association with the costimulatory receptor, osteoclast-associated receptor (OSCAR) [13, 14]. The activation of ITAM-containing adaptors results in the phosphorylation of the spleen tyrosine kinase, which in turn recruits Btk/Tec kinases. Btk and Tec kinases link RANK to ITAM signals with adaptor molecules like the B-cell linker, leading to the activation of PLCγ2 and subsequent calcium mobilization [10, 13, 15, 16]. The mechanism by which RANK and ITAM signals regulate these downstream signaling pathways remains unclear.
The Src homology 3 and cysteine-rich domain 2 (STAC2) gene is a member of the STAC gene family. Recent studies have shown that the protein encoded by another member of this family, STAC3, is essential for skeletal muscle development and myoblast differentiation. Furthermore, STAC3 is involved in the excitation-contraction (EC) coupling that mediates muscle contractility and voltage-induced Ca2+ release [17, 18]. Unlike STAC3, STAC1 and STAC2 proteins are expressed in the central nervous system [19]. However, little is known about the role of STAC2 in osteoclasts.
In this study, we identified that STAC2 is strongly induced by RANKL-stimulated NFATc1 during osteoclastogenesis. The overexpression of STAC2 inhibited osteoclast differentiation by attenuating NF-κB and MAPK signaling pathways. Furthermore, RANKL-induced STAC2 abolished RANK complex formation, which is composed of PLCγ2, Gab2, and Btk/Tec kinases, and impaired RANKL-stimulated PLCγ2 activation. Thus, our data suggest that STAC2 plays an important role in the negative regulation of osteoclastogenesis.
Results
STAC2 is upregulated by RANKL during osteoclast differentiation
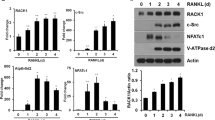
To identify RANKL-induced molecules that are potentially involved in osteoclastogenesis, we performed a genome-wide screen of mRNAs expressed during the differentiation of bone marrow-derived macrophages (BMMs) into osteoclasts [20]. We found that STAC2 mRNA expression levels increased by 4.58-fold, 72 h after RANKL stimulation (Fig. 1a). Both protein and mRNA levels of STAC2 significantly increased during osteoclast formation in response to RANKL (Fig. 1b, c). Unlike STAC2, the mRNA levels of STAC1 and STAC3, other members of the STAC gene family, barely changed (Supplementary Fig. S1). Next, we examined whether RANKL-induced STAC2 expression was regulated by NFATc1, a key transcription factor involved in RANK signaling. We found that STAC2 protein levels were strongly decreased by cyclosporin A (CsA), a known calcineurin/NFAT inhibitor (Fig. 1d). Conversely, overexpression of a constitutively active form of NFATc1 (CaNFATc1) increased STAC2 levels in the absence of RANKL stimulation (Fig. 1e). Consistently, NFATc1 downregulation by short-interfering RNA (siRNA) markedly decreased RANKL-induced STAC2 expression (Supplementary Fig. S2). Furthermore, a 2.5-kb STAC2 promoter fragment (−2500 to + 90), linked to a luciferase reporter construct, was activated by CaNFATc1 overexpression compared with a 0.1-kb promoter fragment (−100 to +90) (Supplementary Fig. S3a, b). We further examined which of the two NFATc1-binding sites (BS1 and BS2) was critical for NFATc1 binding. The BS1 mutant did not affect luciferase activity in the presence of NFATc1. However, the BS2 single and double mutants each significantly decreased luciferase activity in the presence of NFATc1 compared to the wild-type controls (Supplementary Fig. S3b). Thus, these findings indicate that RANKL induces the expression of STAC2 via NFATc1 during osteoclast differentiation.

Identification of STAC2 as a novel RANKL-induced gene during osteoclastogenesis. a Gene chip analysis of STAC2 mRNA after RANKL stimulation. b BMMs were stimulated with RANKL for the indicated times, followed by lysis and western blot analysis with antibodies to STAC2, NFATc1, TRAF6, Atp6v0d2, and GAPDH. c Quantitative real-time PCR of the mRNA expression levels of STAC2, NFATc1, and Atp6v0d2 relative to β-actin in RANKL-stimulated BMMs for the indicated times. Data are presented as the mean ± S.D. *P < 0.005, **P < 0.001. d BMMs treated with DMSO (Mock) or cyclosporin A (CsA, 2 μM) were cultured with M-CSF in the absence or presence of RANKL for 3 days. Total cell lysates were subjected to western blot analysis with the indicated antibodies. e BMMs transduced with retroviral vector pMX-puro (EV) or constitutively active NFATc1 (CaNFATc1) were cultured with M-CSF for 5 days. Cells were lysed and subjected to western blot analysis with the indicated antibodies. Data are representative of at least three independent experiments b, c or at least two independent experiments in d–e
STAC2 is a negative regulator of osteoclastogenesis
To investigate whether STAC2 is involved in osteoclast differentiation, BMMs were infected with empty or STAC2-expressing retrovirus. Transduced BMMs were cultured with M-CSF and RANKL, and stained for tartrate-resistant acid phosphatase (TRAP). STAC2 overexpression greatly decreased the number of TRAP-positive MNCs (≥10 nuclei) after RANKL stimulation (Fig. 2a). We examined the protein expression levels of the osteoclast formation marker genes NFATc1 and Atp6v0d2. Compared with control cells, BMMs overexpressing STAC2 had markedly decreased NFATc1 and Atp6v0d2 protein levels after RANKL stimulation (Fig. 2b). The blockade of osteoclast formation was further confirmed in pre-osteoclasts infected with STAC2 retrovirus (Supplementary Fig. S4a, b). We then used siRNA-mediated knock down of STAC2 to examine whether STAC2 inhibits RANKL-mediated osteoclastogenesis. STAC2 siRNA-transfected BMMs displayed significantly enhanced osteoclast formation compared to controls (Fig. 2c and Supplementary Fig. S5). In addition, the downregulation of STAC2 increased NFATc1 expression (Fig. 2d). Likewise, the area of bone resorption pits was decreased significantly by STAC2 overexpression (Fig. 2e). This decrease in resorptive activity possibly reflected decreased large osteoclast formation under differentiation conditions in vitro. Unlike STAC2, STAC1, and STAC3 overexpression had no effect on osteoclast differentiation or resorption (Supplementary Fig. S6a, b). In contrast to osteoclasts, the overexpression of STAC2 had little effect on the bone morphogenetic protein-induced differentiation of osteoblasts (Supplementary Fig. S7a, b). Collectively, these results indicate that STAC2 plays a critical role in suppressing the differentiation of osteoclasts but not that of osteoblasts.

STAC2 negatively regulates osteoclast differentiation. a, b BMMs infected with retroviral vector pMX-puro (EV) or pMX-puro-Flag-STAC2 (STAC2) were cultured with M-CSF or with both M-CSF and RANKL for 4 days (a) or 3 days (b). a Cells were fixed and stained for TRAP, and the number of TRAP-positive MNCs ( ≥ 10 nuclei per cell) per well was counted. Scale bar, 200 μm. Data are presented as the mean ± S.D. **P < 0.001. b Cells were harvested, and the lysates were subjected to western blot analysis with antibodies to NFATc1, Flag, Atp6v0d2, and GAPDH. c, d After incubating BMMs with control siRNAs or STAC2 siRNAs, cells were cultured with M-CSF in the absence or presence of RANKL for 4 days (c) or 3 days (d). c Osteoclasts were fixed and stained for TRAP, and the number of TRAP-positive MNCs (≥10 nuclei per cell) per well was counted. Scale bar, 200 μm. Data are presented as the mean ± S.D. **P < 0.001. d Protein expression levels were determined by western blot analysis with the indicated antibodies. e BMMs transduced with retrovirus (EV and STAC2) were cultured with M-CSF and RANKL on dentine slices. TRAP-positive MNCs (≥3 nuclei per cell) were generated and stained with hematoxylin to visualize pit formation. Right panel shows pooled data from three experiments normalized to the number of osteoclasts. Data are representative of at least five independent experiments a, b or at least three independent experiments c–e
STAC2 attenuates RANK signaling pathways
Given our data that overexpression of STAC2 inhibits RANKL-induced osteoclast formation, we explored the effects of STAC2 on the activation of RANKL-dependent NF-κB and MAPK pathways. BMMs that had been transduced with retroviral vectors expressing empty or STAC2 were stimulated with RANKL and the activation of JNK, p38, ERK, and IκBα were measured. Notably, these pathways were remarkably attenuated in STAC2-overexpressing BMMs (Fig. 3a). However, unlike RANKL, STAC2 overexpression had no effect on the M-CSF- and LPS-induced NF-κB and MAPK signaling pathways (Fig. 3b and Supplementary Fig. S8), suggesting that STAC2 acts specifically in response to RANKL to inhibit NF-κB and MAPK activation.

STAC2 inhibits the RANKL-stimulated activation of MAPK and NF-κB. a, b BMMs were transduced with the pMX-puro retrovirus (EV) or retrovirus expressing Flag-STAC2 (STAC2). The transduced BMMs were then serum-starved and incubated with RANKL (200 ng/ml) (a) or M-CSF (100 ng/ml) (b) for the indicated times, and subjected to western blot analysis with antibodies to p-JNK, JNK, p-p38, p38, p-ERK, ERK, IκBα, Flag, and tubulin. Data are representative of at least three independent experiments a, b
STAC2 interacts with RANK after RANKL stimulation
Because STAC2 acts as a negative regulator of osteoclast formation by inhibiting RANKL-initiated signaling pathways, we wondered whether STAC2 might physically interact with RANK to block these pathways. We found that STAC2 interacted with RANK after co-transfection in 293 T cells (Fig. 4a). To further examine the interaction between RANK and STAC2, BMMs were incubated with M-CSF, with or without RANKL, for 3 days, and endogenous RANK was immunoprecipitated with anti-RANK antibodies. The result of these immunoprecipitation experiments showed that endogenous STAC2 interacts with RANK following RANKL stimulation (Fig. 4b). We isolated membrane rafts after RANKL stimulation and observed that RANK and STAC2 were recruited to the lipid rafts, which are specialized domains in RANK signal transduction [21, 22] (Supplementary Fig. S9).

STAC2 interacts with the IVVY motif of RANK. a The 293T cells were transfected with the indicated combinations of plasmids. Whole-cell lysates were prepared and subjected to immunoprecipitation with anti-Flag antibodies and analyzed by western blotting. b BMMs were cultured with M-CSF in the absence or presence of RANKL for 3 days. Cells were lysed and immunoprecipitated with anti-RANK antibodies. The resulting immunoprecipitated samples and total whole-cell lysates were subjected to western blot analysis with antibodies to RANK and STAC2. c Schematic diagrams of wild-type and mutant constructs of GST-RANK. Constructs that can interact with STAC2 are indicated by + . d The 293T cells were co-transfected with Flag-STAC2 and various GST-RANK mutants. Cell extracts were precipitated with Glutathione-Sepharose beads and subjected to western blot analysis using the indicated antibodies. Data are representative of at least three independent experiments (a–d). e Schematic diagrams of wild-type and deletion mutant constructs of Flag-STAC2. Constructs that can interact with RANK are indicated by + . f The 293T cells were co-transfected with GST-RANK and various Flag-STAC2 mutants. Cell extracts were precipitated with Glutathione-Sepharose beads and subjected to western blot analysis using the indicated antibodies. Data are representative of at least three independent experiments (a–f)
In osteoclast precursor cells, the highly conserved region (HCR) within the RANK cytoplasmic domain is known as an important platform for the RANK signaling complex. Gab2 and PLCγ2 associate with RANK through the HCR, which is distinct from TRAF6-binding sites, to mediate RANK signaling pathways [11]. On the basis of previous studies showing that the IVVY sequence (amino acids 535–538) in the HCR is critical for osteoclast formation [20, 23], we further investigated whether STAC2 interacts with RANK through the IVVY motif. We used several RANK mutants, in which certain amino acids were mutated to alanine, and examined their ability to bind STAC2. Interestingly, mutants spanning the IVVY motif, 535–537 (IVV) and 538–540 (YVS), dramatically abolished the RANK-STAC2 interaction (Fig. 4c, d). These results demonstrate that the IVVY motif within the HCR of RANK is crucial for the STAC2-RANK interaction, which likely plays a regulatory role in osteoclast formation.
To identify specific domain in STAC2 that interacts with RANK, we generated a series of STAC2 deletion mutants lacking proline-rich, zinc finger, or SH3 domain. By overexpressing RANK and STAC2 mutants in 293T cells, we observed that multiple domains, including the proline-rich, zinc finger, and SH3 of STAC2, are required for RANK interaction (Fig. 4e, f). To further determine the functional domain of STAC2 that regulates osteoclast formation, we retrovirally infected BMMs with the wild-type or deletion mutants of STAC2. Interestingly, only proline-rich deletion mutant showed inhibitory effects same as those showed by wild-type STAC2 on osteoclast formation, whereas the deletion mutants, such as zinc finger and SH3 of STAC2, abolished their inhibitory effects (Supplementary Fig. S10). Therefore, we conclude that zinc finger and/or SH3 domains, but not proline-rich domain of STAC2, are important for the negative regulation of RANKL-mediated osteoclastogenesis.
STAC2 abolishes the RANK signaling complex
Previous studies reported that RANK associates with PLCγ2 and Gab2 after RANKL stimulation [9,10,11]. Because our results indicated that STAC2 also interacts with RANK, we questioned how STAC2 participates in the RANK signaling complex after RANKL stimulation. Interestingly, STAC2 overexpression in 293T cells abrogated the association of Gab2 and PLCγ2 with RANK in a dose-dependent manner (Fig. 5a). Furthermore, retroviral overexpression of STAC2 in BMMs significantly inhibited the association of PLCγ2 and Gab2 with RANK (Fig. 5b). These data suggest that RANKL-induced STAC2 expression interrupts the formation of the RANK signaling complex, which is composed of PLCγ2 and Gab2.

Effects of STAC2 on the RANK-Gab2-PLCγ2 complex. a The 293T cells were co-transfected with GST-RANK, PLCγ2, Gab2, and Flag-STAC2. Protein complexes were precipitated with Glutathione-Sepharose beads and subjected to western blot analysis using the indicated antibodies. b BMMs were infected with pMX-puro retrovirus (EV) and pMX-puro retrovirus expressing STAC2 (STAC2). The transduced BMMs were cultured with M-CSF and RANKL for 3 days and serum-starved. Cells were then stimulated with RANKL (300 ng/ml) for 15 min and lysed for immunoprecipitation with anti-RANK antibodies. Data are representative of at least three independent experiments a, b
Because the Btk/Tec kinases are known to link the RANK and ITAM pathways to calcium signaling via PLCγ2 activation [15], we investigated whether overexpression of STAC2 could affect the relationship between PLCγ2 and Btk/Tec. Consistent with results from previous reports, the phosphorylation of PLCγ2 increased in the presence of Btk and Tec. However, Btk-/Tec-mediated PLCγ2 phosphorylation was decreased by STAC2 overexpression in 293T cells (Fig. 6a, b). In addition, when STAC2 was overexpressed, the interaction of PLCγ2 with Btk/Tec was significantly decreased, but STAC2 still interacted with Btk/Tec (Fig. 6c, d). To further confirm the underlying mechanism of RANKL signaling, we performed a similar experiment in RANKL-induced pre-osteoclasts. The result of this immunoprecipitation experiment confirmed that the endogenous interaction between PLCγ2 and Btk was inhibited by STAC2 in pre-osteoclasts. We also found that STAC2 infection decreased PLCγ2 phosphorylation in pre-osteoclasts (Fig. 6e). Taken together, these results suggest that STAC2 abolishes the RANK signaling complex containing Gab2 and PLCγ2 as well as blocks the association between PLCγ2 and Btk/Tec.

STAC2 suppresses Btk/Tec-mediated PLCγ2 activation. a, b The 293T cells were transfected with PLCγ2, Flag-STAC2, and either Btk or Tec. Total whole-cell lysates were analyzed using the indicated antibodies. c, d The 293T cells were co-transfected with GST-Btk (c) or GST-Tec (d) together with PLCγ2 and Flag-STAC2 as indicated. Protein complexes were precipitated with Glutathione-Sepharose beads and subjected to western blot analysis using the corresponding antibodies. e BMMs transduced with retroviruses (EV and STAC2) were cultured with M-CSF and RANKL for 3 days. After serum starvation, BMMs were stimulated with RANKL (300 ng/ml) for 15 min. Cells were then lysed and immunoprecipitated with antibodies to Btk. Data are representative of at least three independent experiments a–d, or at least two independent experiments (e)
Discussion
The negative regulation of osteoclast formation is important to control healthy skeletal remodeling and prevent the development of bone-related diseases. Initiation and termination of the signaling pathways underlying the formation of mature osteoclasts from mononuclear precursors is a critical checkpoint of this control. The complicated network of transcriptional repressors, including IRF8, Bcl6, and MafB [24,25,26], and extracellular secreted molecules, such as osteoprotegerin and interferon-β, are tightly regulated to control the osteoclast formation process [27]. In addition, previous studies have demonstrated the inhibitory roles of SHIP-1 and IRAK-M during osteoclastogenesis. These proteins are abundantly expressed in macrophages and contribute to the suppression of innate immune signaling [28, 29]. SHIP-1 targets M-CSF receptor signaling, whereas IRAK-M inhibits the interleukin-1 receptor pathway, thereby regulating osteoclast differentiation and activation [30,31,32]. In this regard, our results suggest that STAC2, unlike SHIP-1 or IRAK-M, targets RANK receptor signaling for the suppression of osteoclast differentiation.
The temporal regulation of signaling downstream of the RANK receptor can be divided into several phases, including initiation, commitment, and termination, depending on the signaling processes of osteoclast formation. During the initiation phase, RANKL stimulation leads to the rapid activation of MAPK and canonical NF-κB pathways through TRAF6 within 1 h of culture [33,34,35]. Next, during the commitment phase, NFATc1 begins to accumulate approximately 1–2 days after RANKL stimulation, as cytosolic Ca2+ levels begin to oscillate [12]. The concerted actions of RANK as well as the Ig-like receptors OSCAR and TREM-2 drive this phase [36, 37]. Finally, during the termination phase, RANK signaling regulates cell fusion and bone resorption, mainly through NFATc1, which orchestrates the transcription of osteoclast-specific genes together with AP-1, PU.1, and the microphthalmia transcription factor [38, 39]. Considering the expression patterns of STAC2 during osteoclast formation, it is probable that STAC2 participates in signaling pathways that mediate the commitment or termination phase, which is consistent with our observations that STAC2 targets the RANK signaling complex and destabilizes the interaction between RANK and Gab2/PLCγ2.
The adaptor protein, Gab2, a crucial component of the RANK signaling pathway, is required for the activation of NF-κB and JNK, but not of NFATc1 [9]. PLCγ2 associates with Gab2 in immune cells [10, 40], suggesting that this same complex regulates RANK signaling in osteoclasts. PLCγ2 binds Gab2 in response to RANKL, mediates Gab2 recruitment to RANK, and is required for Gab2 phosphorylation, thereby activating AP-1 and NF-κB. Moreover, Btk/Tec kinases activate the PLCγ2-Ca2+-NFATc1 cascade. This dual activity of PLCγ2 to upregulate NFATc1 and activate NF-κB and AP-1 pathways is likely because of its enzymatic activity and adaptor function, respectively [10]. Thus, it appears that STAC2 inhibits NF-κB and MAPK activation through the dissociation of Gab2/PLCγ2 from the RANK complex. In addition, STAC2 might prevent Btk/Tec-mediated PLCγ2 phosphorylation by abolishing the interaction between PLCγ2 and Btk/Tec.
Presently, there are no reports of any STAC2 function in vitro or in vivo. Among the members of the STAC family, STAC3 was recently reported as a novel regulator of skeletal muscle development. Although Stac3-/- mice are perinatal lethal, these mice show skeleton abnormalities accompanied by a dramatic reduction in bone ridge formation and abnormal myofiber morphology [18, 41]. Furthermore, Polster et al. [42] reported that STAC3 is required for both the Ca2+ channel and EC coupling functions of CaV1.1, implying that STAC3 is important for modulating Ca2+ currents. Such evidence supports the possibility that STAC2 could modulate the PLCγ2-activated Ca2+ signaling required for the induction and activation of NFATc1 during osteoclast formation and play a crucial role in skeletal biology.
In conclusion, by identifying STAC2 as a negative regulator of RANK signaling in osteoclasts, the present study provides novel insight into the mechanism of RANK-dependent signaling. After RANKL stimulation, STAC2 directly associates with RANK, resulting in the suppression of Gab2/PLCγ2-mediated NF-κB and MAPK signaling pathways as well as the inhibition of Btk/Tec-mediated PLCγ2-NFATc1 pathways. These results demonstrate that STAC2 negatively regulates RANK-mediated osteoclastogenic signaling, at least in part by inhibiting PLCγ2-mediated signal transduction. Notably, negative regulators that restrain osteoclastogenesis help maintain bone homeostasis under physiological conditions and limit the amount of bone loss in pathological conditions, such as inflammatory states. Further research into pathophysiological roles of STAC2 will be required to understand whether STAC2 truly can serve as a key negative feedback loop that limits RANK signaling. These efforts will provide us with a deeper understanding of bone homeostasis, and they might lead to new approaches for therapeutic intervention in bone diseases.
Materials and methods
Reagents and plasmids
Recombinant human M-CSF was purchased from R&D Systems (Minneapolis, MN, USA), and RANKL was obtained from Peprotech EC (London, UK). Antibodies used in the study include anti-phospho-PLCγ2, anti-Btk, anti-phospho-ERK1/2, anti-ERK1/2, anti-phospho-JNK, anti-JNK, anti-phospho-p38, anti-p38, and anti-IκBα (Cell Signaling Technology, Beverly, MA, USA); anti-GST, anti-PLCγ2, anti-NFATc1, anti-Tec, anti-Tubulin, and anti-GAPDH (Santa Cruz Biotechnology Inc.); monoclonal anti-FLAG (Sigma-Aldrich), anti-RANK (Abcam Corporation, Cambridge, UK), anti-Gab2 (Millipore), and rabbit polyclonal anti-STAC2 (Novus Biologicals). Anti-Atp6v0d2 antibody was kindly provided by Y. Choi (University of Pennsylvania, Philadelphia PA, USA). Mouse STAC2 and various deletion mutants were generated by polymerase chain reaction (PCR) and cloned into the pFLAG-CMV2 and pMX-puro expression vectors. The pCMV6-Entry-mSTAC1 and pCMV6-Entry-mSTAC3 vectors were purchased from OriGene Technologies. The retroviral vector containing a constitutively active form of NFATc1 (CaNFATc1) has been previously described [20].
Bone marrow macrophage and osteoclast cultures
BMMs and osteoclasts were obtained from murine bone marrow precursors of 4- to 6-week-old C57BL/6 mice (The Jackson Laboratory). BMMs were cultured for 3 days in α -minimum essential medium (α -MEM; HyClone, South Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; HyClone) and antibiotics containing M-CSF (30 ng/ml). After 3 days, adherent BMMs were harvested to obtain osteoclast precursor cells of the monocyte/ macrophage lineage. For osteoclasts, BMMs were cultured in the presence of M-CSF (30 ng/ml) and RANKL (50 ng/ml) for 3–5 days as described above. For early time point signaling, infected BMMs were serum-starved for 6 h with 0.1% FBS in the absence of M-CSF. Next, cells were stimulated with RANKL (200 ng/ml), M-CSF (100 ng/ml), or LPS (1 μg/ml) for the indicated time periods. All primary cell experiments were approved by the Institutional Animal Care and Use Committee of Ewha Laboratory Animal Genomics Center and were conducted according to the approved guidelines.
Pit formation assay
Pre-osteoclasts were plated on dentin slices in 96-well culture plates with α -MEM. The cells were cultured for 3 days with RANKL (50 ng/ml) in the presence of M-CSF (30 ng/ml). The dentin slices were washed with water, and pits formed by mature osteoclasts were stained with hematoxylin solution (Sigma-Aldrich). Individual pits were observed and evaluated using Image-Pro Plus version 4.5 software (Media-Cybernetics, Rockville MD, USA) as previously described [43].
Reverse transcription and real-time quantitative PCR
Total RNA was extracted from cultured BMMs using RNAiso plus (Takara), according to the manufacturer’s instruction. A DiaStarTM Reverse Transcription kit (SolGent, Seoul, Korea) was used for the reverse transcription of purified RNA. Real-time quantitative PCR was performed in triplicate on an ABI PRISM 7300 unit (Applied Biosystems, Foster City, CA, USA) and the SYBR Green Master kit (Kapa Biosystems, Wilmington, MA, USA). Primers specific for STAC1, STAC2, STAC3, Nfatc1, Atp6V0d2, and Actin were as follows: STAC1, 5′-AAAGATTCAGGACAGGGTTGG-3′ and 5′-GAGGTCACGCATATCTGGTTC-3′; STAC2, 5′-GAGAATGAACCGGATGACG-3′ and 5′-TTCGAAGGATGGTTTTGAGG-3′; STAC3, 5′- CGGATTCTTCCCTCCAAACT-3′ and 5′- CCACCAGCTTCATCTCCTTT-3′; Nfatc1, 5′-CCAGAAAATAACATGC-3′ and 5′-GTGGGATGTGAACTCG-3′; Atp6v0d2, 5′-CAGAGATGGAAGCTGT-3′ and 5′-TGCCAAATGAGTTCAG-3′; and Actin, 5′-GCTTCTTCTTTGCAGCTCCT-3′ and 5′-ATCGTCATCCATGGCGA-3′.
Retroviral infection
PLAT-E retrovirus packaging cells were transfected with pMX-puro empty, pMX-puro-Flag-STAC1, pMX-puro-Flag-STAC2, pMX-puro-Flag-STAC2 deletion mutants, pMX-puro-Flag-STAC3, or pRC-NFATc1-CA retroviral vector using polyethylenimine (Sigma-Aldrich) reagent, and the supernatant was collected 36-48 h after transfection. BMMs were infected with the retroviral supernatant in the presence of M-CSF (30 ng/ml) and polybrene (10 μg/ml) for 5 h. After a 24 h infection, M-CSF (30 ng/ml) and puromycin (2 μg/ml) were added for 2 days to select the infected cells. Puromycin-resistant BMMs were used for osteoclast differentiation and incubated in the presence of M-CSF (30 ng/ml) and RANKL (50 ng/ml) for an additional 3–5 days.
siRNA transfection
The siRNA targeting mouse STAC2 and NFATc1 was designed and purchased from Genolution (Seoul, Korea). The corresponding target mRNA sequences for the siRNAs were as follows: si-STAC2, 5′-TGTAAGACTTGCAAAGTCAuu-3′; si-NFATc1, 5′- GGACTCCAAGGTCATTTTCuu-3′; scrambled non-targeting siRNA (negative control), 5′-ACGTGACACGTTCGGAGAAuu-3′. BMMs were transfected with the siRNAs (20 nmol) using Lipofectamine RNAiMAX reagent (Invitrogen), according to the manufacturer’s protocol. After 1 day, the BMMs were cultured with M-CSF (30 ng/ml) and RANKL (100 ng/ml) for osteoclast generation or protein analyses.
GST pull-down assay
The 293T cells were cultured in Dulbecco's modified Eagle's medium (HyClone) containing 10% FBS and antibiotics. Cells were re-plated 24 h prior to transfection and transfected with the indicated combinations of expression vectors using polyethylenimine. After 48 h, cell extracts were obtained after treatment of cells with 1% NP-40 lysis buffer (20 mM HEPES, pH 7.0, 10 mM EDTA, 150 mM NaCl, 150 mM KCl, 10% glycerol, and 1% Nonidet P-40) containing protease inhibitors. Whole-cell lysates were precipitated by Glutathione-Sepharose 4B (GE Healthcare Life Science) for 3 h at 4 °C and analyzed by western blotting as previously described [44].
Western blot analysis and immunoprecipitation
BMMs were incubated with M-CSF (30 ng/ml) in the presence or absence of RANKL (50 ng/ml) for the indicated periods. Cells were lysed in cell extraction buffer (20 mM HEPES, pH 7.9, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100, and 10% glycerol) containing protease inhibitors. The cell lysates were harvested and subjected to western blot analysis using the indicated antibodies. Immunoprecipitation was performed on whole-cell extracts that were incubated with specific antibodies overnight, followed by incubation with protein G-Sepharose (GE Healthcare Life Science) for 2 h at 4 °C. The immunoprecipitates were then subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and western blot analysis. The 293T cells were harvested 48 h after transfection and lysed in 1% NP-40 lysis buffer. Whole-cell lysates were subjected to western blotting or immunoprecipitation as described above [45].
References
Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–8.
Walsh MC, Kim N, Kadono Y, Rho J, Lee SY, Lorenzo J, et al. Osteoimmunology: interplay between the immune system and bone metabolism. Annu Rev Immunol. 2006;24:33–63.
Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42.
Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345–57.
Josien R, Wong BR, Li HL, Steinman RM, Choi Y. TRANCE, a TNF family member, is differentially expressed on T cell subsets and induces cytokine production in dendritic cells. J Immunol. 1999;162:2562–8.
Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251–64.
Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. 2006;440:692–6.
Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–51.
Wada T, Nakashima T, Oliveira-dos-Santos AJ, Gasser J, Hara H, Schett G, et al. The molecular scaffold Gab2 is a crucial component of RANK signaling and osteoclastogenesis. Nat Med. 2005;11:394–9.
Mao D, Epple H, Uthgenannt B, Novack DV, Faccio R. PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J Clin Invest. 2006;116:2869–79.
Taguchi Y, Gohda J, Koga T, Takayanagi H, Inoue J. A unique domain in RANK is required for Gab2 and PLCgamma2 binding to establish osteoclastogenic signals. Genes Cells. 2009;14:1331–45.
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901.
Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, et al. The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci USA. 2004;101:6158–63.
Kaifu T, Nakahara J, Inui M, Mishima K, Momiyama T, Kaji M, et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J Clin Invest. 2003;111:323–32.
Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132:794–806.
Ishiai M, Kurosaki M, Pappu R, Okawa K, Ronko I, Fu C, et al. BLNK required for coupling Syk to PLC gamma 2 and Rac1-JNK in B cells. Immunity. 1999;10:117–25.
Ge X, Zhang Y, Park S, Cong X, Gerrard DE, Jiang H. Stac3 inhibits myoblast differentiation into myotubes. PLoS ONE. 2014;9:e95926.
Reinholt BM, Ge X, Cong X, Gerrard DE, Jiang H. Stac3 is a novel regulator of skeletal muscle development in mice. PLoS ONE. 2013;8:e62760.
Legha W, Gaillard S, Gascon E, Malapert P, Hocine M, Alonso S, et al. stac1 and stac2 genes define discrete and distinct subsets of dorsal root ganglia neurons. Gene Expr Patterns. 2010;10:368–75.
Kim H, Choi HK, Shin JH, Kim KH, Huh JY, Lee SA, et al. Selective inhibition of RANK blocks osteoclast maturation and function and prevents bone loss in mice. J Clin Invest. 2009;119:813–25.
Choi HK, Kang HR, Jung E, Kim TE, Lin JJ, Lee SY. Early estrogen-induced gene 1, a novel RANK signaling component, is essential for osteoclastogenesis. Cell Res. 2013;23:524–36.
Ha H, Kwak HB, Le SW, Kim HH, Lee ZH. Lipid rafts are important for the association of RANK and TRAF6. Exp Mol Med. 2003;35:279–84.
Xu D, Wang S, Liu W, Liu J, Feng X. A novel receptor activator of NF-kappaB (RANK) cytoplasmic motif plays an essential role in osteoclastogenesis by committing macrophages to the osteoclast lineage. J Biol Chem. 2006;281:4678–90.
Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292–304.
Zhao B, Takami M, Yamada A, Wang X, Koga T, Hu X, et al. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat Med. 2009;15:1066–71.
Park SJ, Huh JE, Shin J, Park DR, Ko R, Jin GR, et al. Sirt6 cooperates with Blimp1 to positively regulate osteoclast differentiation. Sci Rep. 2016;6:26186.
Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416:744–9.
Sly LM, Rauh MJ, Kalesnikoff J, Song CH, Krystal G. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity. 2004;21:227–39.
Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr., Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202.
Takeshita S, Namba N, Zhao JJ, Jiang Y, Genant HK, Silva MJ, et al. SHIP-deficient mice are severely osteoporotic due to increased numbers of hyper-resorptive osteoclasts. Nat Med. 2002;8:943–9.
Li H, Cuartas E, Cui W, Choi Y, Crawford TD, Ke HZ, et al. IL-1 receptor-associated kinase M is a central regulator of osteoclast differentiation and activation. J Exp Med. 2005;201:1169–77.
Zhou P, Kitaura H, Teitelbaum SL, Krystal G, Ross FP, Takeshita S. SHIP1 negatively regulates proliferation of osteoclast precursors via Akt-dependent alterations in D-type cyclins and p27. J Immunol. 2006;177:8777–84.
Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y. The TRAF family of signal transducers mediates NF-kappaB activation by the TRANCE receptor. J Biol Chem. 1998;273:28355–9.
Galibert L, Tometsko ME, Anderson DM, Cosman D, Dougall WC. The involvement of multiple tumor necrosis factor receptor (TNFR)-associated factors in the signaling mechanisms of receptor activator of NF-kappaB, a member of the TNFR superfamily. J Biol Chem. 1998;273:34120–7.
Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. 2001;20:1271–80.
Kim N, Takami M, Rho J, Josien R, Choi Y. A novel member of the leukocyte receptor complex regulates osteoclast differentiation. J Exp Med. 2002;195:201–9.
Cella M, Buonsanti C, Strader C, Kondo T, Salmaggi A, Colonna M. Impaired differentiation of osteoclasts in TREM-2-deficient individuals. J Exp Med. 2003;198:645–51.
Matsumoto M, Kogawa M, Wada S, Takayanagi H, Tsujimoto M, Katayama S, et al. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J Biol Chem. 2004;279:45969–79.
Luchin A, Purdom G, Murphy K, Clark MY, Angel N, Cassady AI, et al. The microphthalmia transcription factor regulates expression of the tartrate-resistant acid phosphatase gene during terminal differentiation of osteoclasts. J Bone Miner Res. 2000;15:451–60.
Nishida K, Wang L, Morii E, Park SJ, Narimatsu M, Itoh S, et al. Requirement of Gab2 for mast cell development and KitL/c-Kit signaling. Blood. 2002;99:1866–9.
Nelson BR, Wu F, Liu Y, Anderson DM, McAnally J, Lin W, et al. Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+release and contractility. Proc Natl Acad Sci USA. 2013;110:11881–6.
Polster A, Nelson BR, Olson EN, Beam KG. Stac3 has a direct role in skeletal muscle-type excitation-contraction coupling that is disrupted by a myopathy-causing mutation. Proc Natl Acad Sci USA. 2016;113:10986–91.
Huh JE, Shin JH, Jang ES, Park SJ, Park DR, Ko R, et al. Sirtuin 3 (SIRT3) maintains bone homeostasis by regulating AMPK-PGC-1beta axis in mice. Sci Rep. 2016;6:22511.
Lin J, Lee D, Choi Y, Lee SY. The scaffold protein RACK1 mediates the RANKL-dependent activation of p38 MAPK in osteoclast precursors. Sci Signal. 2015;8:ra54.
Ko R, Park JH, Ha H, Choi Y, Lee SY. Glycogen synthase kinase 3beta ubiquitination by TRAF6 regulates TLR3-mediated pro-inflammatory cytokine production. Nat Commun. 2015;6:6765.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MSIP) (No. 2016R1A2B3010699; No. 2012R1A5A1048236).
Author information
Authors and Affiliations
Contributions
Study design: EJ, HKC, and SYL; Study conduct: EJ, HKC, and JHP; Data analysis and interpretation: EJ, HKC, and SYL; Drafting manuscript: EJ and SYL; All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Jeong, E., Choi, H.K., Park, J.H. et al. STAC2 negatively regulates osteoclast formation by targeting the RANK signaling complex. Cell Death Differ 25, 1364–1374 (2018). https://doi.org/10.1038/s41418-017-0048-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0048-5
This article is cited by
-
Modulation of fracture healing by senescence-associated secretory phenotype (SASP): a narrative review of the current literature
European Journal of Medical Research (2024)
-
The histone methyltransferase ASH1L protects against bone loss by inhibiting osteoclastogenesis
Cell Death & Differentiation (2024)
-
Brevilin A inhibits RANKL-induced osteoclast differentiation and bone resorption
In Vitro Cellular & Developmental Biology - Animal (2023)
-
Spi-C positively regulates RANKL-mediated osteoclast differentiation and function
Experimental & Molecular Medicine (2020)
-
RANKL signaling in bone marrow mesenchymal stem cells negatively regulates osteoblastic bone formation
Bone Research (2018)
