
Abstract
This study utilized Next-Generation Sequencing (NGS) to explore genetic determinants of survival duration in Glioblastoma Multiforme (GBM) patients. We categorized 30 primary GBM patients into two groups based on their survival periods: extended survival (over two years, N = 17) and abbreviated survival (under two years, N = 13). For identifying pathogenic or likely pathogenic variants, we leveraged the ClinVar database. The cohort, aged 23 to 66 (median: 53), included 17 patients in Group A (survival >2 years, 10 males, 7 females), and 13 patients in Group B (survival <2 years, 8 males, 5 females), with a 60% to 40% male-to-female ratio. Identified mutations included CHEK2 (c.1477 G > A, p.E493K), IDH1 (c.395 G > A, p.R132H), and TP53 mutations. Non-coding regions exhibited variants in the TERT promoter (c.-146C > T, c.-124C > T) and TP53 RNA splicing site (c.376-2 A > C, c.376-2 A > G). While Group A had more mutations, statistical significance wasn’t reached, likely due to sample size. Notably, TP53, and ATR displayed a trend toward significance. Surprisingly, TP53 mutations were more prevalent in Group A, contradicting Western findings on poorer GBM prognosis. In Taiwanese GBM patients, bevacizumab usage is linked to improved survival rates, affirming its safety and effectiveness. EGFR mutations are infrequent, suggesting potential distinctions in carcinogenic pathways. Further research on EGFR mutations and amplifications is essential for refining therapeutic approaches. TP53 mutations are associated with enhanced survival, but their functional implications necessitate detailed exploration. This study pioneers genetic analysis in Taiwanese GBM patients using NGS, advancing our understanding of their genetic landscape.
Similar content being viewed by others

Introduction
Glioblastoma multiforme (GBM), a notoriously aggressive primary brain tumor, poses significant challenges in management and treatment due to its rapid progression and dismal prognosis. Over the years, intensive research has sought to unravel its complex pathogenesis, focusing on genetic mutations and exploring novel therapeutic avenues. The latest WHO classification’s fifth edition underscores the pivotal role of genetic alterations in shaping patient outcomes, highlighting the need for personalized treatment strategies in the ongoing battle against this formidable disease [1].
Previously, GBM classification relied primarily on histological characteristics. However, advancements in molecular biology have unveiled a more intricate scenario. We now understand that GBM’s genetic profile involves a dynamic interplay of mutations, epigenetic factors, and disruptions in cellular signaling pathways. This molecular perspective has revolutionized our approach to understanding GBM’s biology, offering deeper insights into its complex pathophysiology [2]. The Cancer Genome Atlas (TCGA) project’s comprehensive genomic analysis of GBM marked a significant milestone in understanding the disease. This project identified key genetic alterations, including mutations in genes such as TP53, PTEN, EGFR, and NF1. These findings have been instrumental in elucidating the molecular mechanisms underpinning GBM, paving the way for targeted therapies and personalized medicine approaches in treating this challenging brain tumor [3].
While the majority of research has focused on GBM in Western populations, there’s an increasing emphasis on understanding its genetic profile in non-Western groups. For instance, a notable study on the Chinese population revealed unique genetic characteristics. This research uncovered a comparatively lower incidence of EGFR amplifications and a higher frequency of TP53 mutations than typically observed in Western populations. This highlights the importance of exploring ethnic and regional variations in GBM genetics, which could lead to more tailored and effective treatment strategies globally [4]. These findings underscore the importance of considering ethnic and regional genetic variations in GBM. Such diversity can significantly influence diagnostic accuracy, prognostic assessments, and the effectiveness of therapeutic interventions. This perspective encourages a more personalized approach to managing GBM, taking into account the unique genetic makeup of diverse populations.
The conventional approach to treating GBM typically involves a sequence of surgical resection, followed by radiotherapy and chemotherapy using temozolomide (TMZ). Despite this regimen, the prognosis for GBM patients remains challenging, with median survival rates hovering around just 15 months. This reality underscores the urgency for more effective therapeutic strategies in the battle against this aggressive brain tumor [5]. The discovery of MGMT promoter methylation has become a key factor in predicting the efficacy of TMZ treatment in GBM patients. Research indicates that tumors with methylated MGMT promoters tend to respond more favorably to TMZ, leading to improved survival outcomes. This insight has been pivotal in guiding personalized treatment plans and enhancing the therapeutic approach for GBM patients [6]. Explorations into targeted therapies for GBM, particularly EGFR inhibitors for cases with EGFR amplification, have been conducted. However, the effectiveness of these treatments is often limited due to the tumor’s inherent heterogeneity and its capacity to develop resistance mechanisms. This complexity poses a significant challenge in achieving consistent success with targeted therapeutic strategies in GBM treatment [7]. In summary, current research underscores the genetic intricacies of GBM and the associated treatment challenges. While there has been notable advancement in understanding GBM’s molecular underpinnings, the journey from these insights to effective, practical treatments remains a significant and complex challenge. This highlights the ongoing need for innovative research and therapeutic strategies in the fight against this formidable brain cancer.
A retrospective study at Taichung Veterans General Hospital, spanning from 2010 to 2022, analyzed primary GBM patients and found that the average overall survival rate post-standard treatment was 18.7 months. This duration, slightly longer than the general average, provides valuable insights into patient outcomes within this specific population and timeframe [8]. This duration is significantly longer than the 14.6 months reported by Wang F. et al. [9] in 2018. Given the hypothesis that GBM patients from different ethnic backgrounds may exhibit distinct genetic profiles, we conducted a study involving 30 patients diagnosed with malignant brain tumors between 2009 and 2023. These tumors were classified as grade IV according to traditional WHO histopathological grading. These patients were divided into two groups based on a two-year survival threshold: those who survived for more than two years (Group A) and those who survived for less than two years (Group B). Using advanced Next-Generation Sequencing (NGS) molecular biology techniques, we performed gene sequencing and compared the results with the TCGA database. Our primary goal was to identify differences in genetic mutation sites between Eastern and Western populations. Furthermore, we aimed to identify factors that influence prognosis within these two patient groups.
Material and methods
Study population and tumor samples
Our study, a retrospective analysis at Taichung Veterans General Hospital in Taichung, Taiwan, evaluated 30 patients diagnosed with GBM between 2009 and 2022. Selection was based on the availability of comprehensive data and high-quality tumor samples. All patients underwent standardized therapy protocols, including surgical intervention, radiotherapy, and temozolomide-based chemotherapy, with bevacizumab for progressive GBM cases.
A limitation of our study is the absence of an initial power analysis, owing to its retrospective design. Our primary objective was to ensure a representative GBM patient sample, aiming to provide insightful findings despite this methodological constraint. This focus highlights the significance of a predetermined power analysis in future prospective studies to determine an adequate sample size that can reliably detect significant effects.
Inclusion and exclusion criteria
Our inclusion criteria encompassed: (1) a confirmed diagnosis of GBM; (2) adherence to the standard therapy protocol; (3) availability of follow-up data and samples; (4) survival beyond one-month post-operation; and (5) average necrosis below 40% on top and bottom slides. The exclusion criteria were patients with incomplete follow-up data or inadequate sample quality for analysis. A validation cohort of 30 cases was carefully chosen from the primary group based on these criteria.
The rationale for blinding or lack thereof
In our retrospective analysis, the nature of data collection and patient selection precluded the implementation of blinding. As a retrospective study, we relied on existing medical records and samples, which means the investigators were inherently aware of patient outcomes and treatment protocols during data analysis. However, to mitigate potential bias from this non-blinded approach, we employed strict inclusion and exclusion criteria and systematic data analysis methods. This methodology aimed to ensure the integrity and objectivity of our findings. Future studies, particularly prospective ones, should consider incorporating blinding techniques to further reduce bias and enhance the validity of results.
Sample processing and data analysis
Post-surgery, tumor samples were immediately frozen for subsequent analysis. Overall survival (OS) was calculated from the surgery date to the date of death or the last follow-up examination.
Ethical compliance
The Medical Ethics Committee of Taichung Veterans General Hospital approved our study protocols (Approval number: CF17263B-4), with the study concluding on March 1, 2023. All specimens were collected in compliance with institutional review board-approved protocols and anonymized to maintain patient confidentiality. Informed written consent was obtained from all participants, adhering to the principles of the Declaration of Helsinki. For a detailed overview of patient characteristics, please refer to Table 1.
DNA extraction and quality control
DNA was extracted from frozen samples using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The quantity and purity of the genomic DNA (gDNA) were assessed using the Qubit® 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and NanoDrop ND-1000 (Thermo Scientific, Wilmington, DE, USA). The fragmentation status of the gDNA was evaluated by the Agilent 2200 TapeStation system using the Genomic DNA ScreenTape assay (Agilent Technologies, Santa Clara, CA, USA), which generates a DNA Integrity Number (DIN). Additional quality control (QC) steps were performed to assess gDNA integrity using a multiplex Polymerase Chain Reaction (PCR) approach. In this approach, 30 ng of gDNA were amplified using three different-sized sets of primers targeting the Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) gene (200-300-400 base pair), and the concentration of PCR products was determined using the Agilent 2100 Bioanalyzer instrument (Agilent Technologies). To estimate gDNA fragmentation, an Average Yield Ratio (AYR) value was calculated by comparing the yield ratio of each amplicon with a reference DNA.
Targeted sequencing:
Each subject’s GBM tumor sample was collected for genomic DNA extraction. Genomic DNA was extracted from leukocytes using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) for subsequent next-generation sequencing analysis. Targeted sequencing was employed to sequence the specific regions of interest associated with GBM, including the complete exons of the IDH1, TP53, and TERT genes. Custom-designed probes and primers were used for these genes. The targeted panel used was the Carcinogens Gene Test Assay, utilized in clinical genetic trials at the Precision Medicine Laboratory of Taichung Veterans General Hospital.
Polymerase chain reaction (PCR) was performed to amplify and sequence the targeted DNA fragments. Library construction was carried out using the Qiagen Target Panel Kit (Qiagen, CDHS-15658z-227, Hilden, Germany), followed by quantification. The prepared library was loaded onto the Illumina Sequencing System (iSeq 100/MiniSeq, San Diego, CA, USA). FastQ files generated from the targeted DNA libraries were stored in CLC Genomics Workbench 12 (QIAGEN, Denmark), and variant calling was performed using QIAGEN Panel analyses. The pathogenicity assessment of variants was conducted using the Illumina Basespace Variant Interpreter. The pathogenic or likely pathogenic variants were further confirmed using the ClinVar database, a public archive providing information on human genomic variants and their associations with diseases, supported by clinical or functional evidence.
DNA libraries were generated using the QIAseq Human Comprehensive Cancer Panel, covering 275 genes (0.8 Mbp). Each sample utilized 40 ng of tumor genomic DNA following the manufacturer’s instructions. The prepared library underwent paired-end sequencing on the NovaSeq 6000 sequencer. DNA reads were aligned to the human reference genome GRCh37, and variant calling was performed using the integrated workflow in CLC Genomics Workbench 21. Somatic mutation annotation filtering and characterization were performed via QIAGEN Clinical Insight (QCI). Variants with a frequency below 3% were excluded, and pathogenic and likely pathogenic variants were identified based on the ACMG variant interpretation guidelines.
Statistical analysis
Our analysis encompassed demographic data, which were presented as frequencies for categorical variables and examined using the chi-squared test or Fisher’s exact test, as appropriate. We assessed Overall Survival (OS) employing the Kaplan-Meier method, complemented by the log-rank test to discern survival differences. In our Cox proportional hazards regression analysis, we meticulously adjusted for key variables, including age, sex, and bevacizumab treatment. Crucially, we implemented Levene’s Test to evaluate the homogeneity of variance both within and across groups, thereby solidifying the validity of our statistical underpinnings. All statistical analyses were conducted using IBM SPSS, version 22.0. We considered p values less than 0.05 to be statistically significant. Moreover, the normal distribution of our data was confirmed through the Shapiro-Wilk test (p > 0.05), further bolstering the credibility of our results.
Results
Glioblastoma patient characteristics in the Taiwanese population
000These 30 patients were enrolled between February 2009 and September 2022, with ages spanning from 23 to 66 years, calculated from the date of surgery. The median age was 53 years. Within this cohort, Group A consisted of 17 patients who exhibited a survival of over two years, comprising 10 males and 7 females. Meanwhile, Group B comprised 13 patients who survived less than two years, consisting of 8 males and 5 females. The male-to-female ratio was 60% to 40%.
The maximum diameter and number of tumors were evaluated using preoperative Brain MRI scans with contrast. Furthermore, patients were stratified based on their received treatments, which encompassed TMZ + CCRT (Concurrent Chemoradiotherapy) and TMZ + CCRT+Bevacizumab. All treatments were in accordance with the current GBM treatment guidelines.
Furthermore, the impact of diabetes and hypertension on survival was evaluated at the time of diagnosis using Fisher’s exact test (Table 1). No statistically significant disparities were noted in terms of age, gender, tumor size, or number between the two groups. However, the utilization of Bevacizumab demonstrated a statistically significant correlation with prolonged survival (p = 0.030), indicating a positive association between Bevacizumab use and extended survival [8].
The frequently mutated genes in glioblastoma within the Taiwanese population
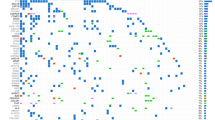
We employed NGS target panel techniques to analyze 30 glioblastoma samples. Utilizing the tertiary analysis system, QIAGEN Clinical Insight (QCI), we selected variants categorized as pathogenic or likely pathogenic, and filtered out those with a frequency of less than 3%. The samples were then stratified into two groups based on survival period. The results revealed a spectrum of mutations, encompassing missense mutations, nonsense mutations, frameshift mutations, and indels spanning the promoter, exon, and intron regions. Additionally, we conducted a quantification of the number of patients and the proportion of patients with each mutated gene. A visual representation of gene mutations was generated using Comut (Fig. 1) [10].

This figure illustrates the genetic profiles of glioblastoma in the Taiwanese population, coupled with an investigation into the survival characteristics of patients. It provides a comprehensive analysis of genetic variations and their potential impact on patient survival outcomes, highlighting the significance of personalized medicine in the treatment of glioblastoma.
Upon closer examination of the heat map (Fig. 1), it is evident that the total mutation count in Group A patients surpasses that in Group B. We conducted Fisher’s exact test to scrutinize each mutated gene in both groups, excluding genes with zero mutations in both groups (such as ATRX, MUTYH, PIK3R1, etc.). Due to the limited sample size, our analysis results indicate that none of the mutations reached statistically significant levels (Table 2). However, there is a noteworthy trend towards a p value of 0.05 for TP53.
During the course of this experiment, we observed a higher prevalence of TP53 mutations in Group A. This observation contradicts current Western research, which suggests an association between TP53 mutations and a worsened prognosis in GBM [11].
Characteristics of glioblastoma-associated variants in the genes CHEK2, IDH1, TP53, and TERT promoter in the Taiwanese population
We utilized lollipop plots to visually represent glioblastoma-associated gene mutation sites specific to the Taiwanese population. These mutation sites were categorized into coding and non-coding regions. Within the coding region, a single mutation in the CHEK2 gene was identified as (c.1477 G > A, p.E493K) (Fig. 2A). In the IDH1 gene, a solitary mutation was identified as (c.395 G > A, p.R132H) (Fig. 2B). In the TP53 gene, seven mutations were identified as (c.326 T > C, p.F109S), (c.473 G > A, p.R158H), (c.578 A > G, p.H193R), (c.718 A > G, p.S240G), (c.743 G > A, p.R248Q), (c.817 C > T, p.R273C), and (c.833 C > T, p.P278L) (Fig. 2C). The coding region diagrams were generated using Mutation Mapper [12].

This figure illustrates amino acid substitutions in the A CHEK2, B IDH1, and C TP53 genes. The gray bar denotes the location of amino acids (aa). The circular lollipop marker indicates the specific site of amino acid substitution, with the height representing the variant count at those positions. Colored boxes represent distinct functional domains. D Schematic diagram of the TERT promoter. The bar marks the upstream regulatory region. The circular lollipop marker shows the specific site of nucleotide substitution. E Schematic diagram of the TP53 RNA splicing site. The bar denotes the TP53 gene in the chromosome 17 region using the GRCh37 reference genome. The circular lollipop marker indicates the specific site of nucleotide substitution.
In the non-coding region, two variants were detected in the TERT promoter regulatory region: (c.-146C > T) and (c.-124C > T) (Fig. 2D). In the TP53 gene, two variants were identified at the RNA splicing site: (c.376-2 A > C) and (c.376-2 A > G) (Fig. 2E). The non-coding region diagrams were prepared using track Viewer [13].
The disparities in glioblastoma patient gene mutations between the Taiwanese and Western populations
This study endeavors to elucidate the distinctions in the genetic mutation profiles of glioblastoma between the Taiwanese population and Western populations. We curated data from the TCGA database and various publications to pinpoint the top 10 mutated genes in glioblastoma. Our observations indicate that mutations in the ATR, KMT2C, TERT, RAD50, and CHEK2 genes are more prevalent in the Taiwanese population, whereas the mutation frequencies of TP53, IDH1, ATRX, NF1, and PIK3R1 are more akin to those in Western populations (Table 3). Additional details regarding these six TCGA projects can be found in the Supplementary Materials.
Statistical analysis of the effects of IDH1 and TP53 mutations on survival rate and age distribution of patients
Following the NGS results, we conducted a thorough statistical analysis. Due to the limited sample size, none of the mutations yielded statistically significant results. However, we identified the genes IDH1 and TP53 as having potential statistical significance. Regarding IDH1, we observed six patients in Group A and two patients in Group B (Fig. 3A). The age distribution analysis for IDH1 mutations indicated one patient below the age of 55 and seven patients aged 55 or above (Fig. 3B). These findings suggest that mutations in IDH1 among glioblastoma patients in Taiwan are linked with a more favorable prognosis, and the majority of patients with IDH1 mutations are under the age of 55.

A Count of patients with IDH1 mutation in the two-year survival rate. B Count of patients with IDH1 mutation aged 55 years and below. C Count of patients with TP53 mutation in the two-year survival rate. OS: overall survival.
As for TP53, our investigation revealed seven patients in Group A and one patient in Group B (Fig. 3C). This outcome suggests that mutations in TP53 among glioblastoma patients in Taiwan are associated with a better prognosis.
Based on our NGS analysis data, Table 3 illustrates the heat map. ATR stands out as the most frequently mutated gene, accounting for 83% of all mutations. Following KMT2C, TERT, and CHEK2 mutations are the next most prevalent. For further specifics, please refer to the gene mutation heat map and Table 3.
In our study analyzing the top 10 genetic mutations in the Taiwanese population, we utilized a Cox proportional hazards regression model. This model was adjusted for age, gender, and bevacizumab treatment, as detailed in Table 4. Our findings indicate a notable association between the IDH1 mutation and patient prognosis. Specifically, compared to the wild type, the IDH1 mutation shows a hazard ratio (HR) of 0.31 (95% CI: 0.11–0.83) with a p value of 0.020 in the multivariate analysis. This suggests a significantly lower risk ratio (p < 0.05), indicating that glioblastoma patients with the IDH1 mutation may have a higher survival rate. Our study’s findings reveal that patients harboring IDH1 mutations demonstrate a notably prolonged survival rate. According to the World Health Organization’s 2021 glioma classification protocol, which emphasizes the significance of IDH1 mutations in its hierarchical categorization and integrates histopathological features, including microvascular proliferation and/or necrosis, these patients are classified as ‘Astrocytoma, IDH-mutant, CNS WHO Grade 4’ [14]. This classification demonstrates a comparatively favorable prognosis within the CNS WHO Grade 4 spectrum, aligning with our study’s results. This correlation echoes the trends observed in recent literature [14, 15]. In contrast, the TP53 mutation did not demonstrate statistical significance in the risk ratio. After adjustments, the HR for the mutation group was 0.49 (95% CI: 0.21–1.17), with a p value of 0.107. Despite the decreased HR value, the lack of statistical significance underscores the need for further investigation with larger sample sizes. Other genetic mutations analyzed did not show significant effects in this multivariate analysis.
Discussion
EGFR mutations in the Taiwanese population in our study
The data from the TCGA database, accessed through cBioPortal and outlined in Table 3 [16,17,18,19] shows that prevalent mutations, especially in PTEN and EGFR, are observed less frequently in our patient cohort compared to others. In particular, EGFR mutations are completely absent, as demonstrated in the mutation heat maps for both Group A and Group B. To corroborate these observations, we employed first-generation SANGER sequencing and verified the nonexistence of EGFR mutations in all 30 patients in our study. This contrasts with the findings in Asian populations; for instance, Fukushima and Favereaux [16] observed a 2% mutation rate in EGFR kinase regions of Japanese glioblastomas using SSCP and DNA sequencing. This rate is markedly different from the polymorphic allele frequency in Swiss glioblastomas, which show a lower mutation rate in the Japanese group. Our results are consistent with these findings, indicating a similar trend in mutation rates across different populations.
In the classic subtype of The Cancer Genome Atlas (TCGA), a high frequency of EGFR amplification is typically noted [20]. However, this trend is not commonly seen in Chinese patients [4]. Similarly, Nayuta HIGA et al. [21], utilizing NGS, reported a lower rate of EGFR amplification in GBM in Asian patients compared to those in other regions. Our research confirms a low frequency of EGFR gene mutations in both Groups A and B of our study. It is important to note that our study did not evaluate the extent of EGFR amplifications. A more focused investigation into EGFR amplifications could reveal significant differences in the oncogenic pathways and treatment approaches for GBM between Eastern and Western populations. This differentiation could be crucial for developing region-specific treatment strategies.
TP53 mutation analysis
In our study, both Group A and Group B exhibit a p-value close to 0.05 in association with TP53 mutations, hinting at a potential link to enhanced survival rates. This observation aligns with the findings of Noor H, Briggs NE, et al. [22], who reported that TP53 mutations significantly improved overall survival in astrocytoma patients. Their research highlighted the presence of hotspot mutations in TP53, particularly at codon 273, in 33% (17 out of 51) of astrocytoma samples. Retrospective analysis indicated markedly better clinical outcomes in patients who received chemotherapy, suggesting that specific mutations in TP53, especially at codon 273, could be critical in determining the effectiveness of therapy in astrocytomas and, consequently, in affecting survival rates. In our study, we also detected mutations at codon 273 (specifically R273C (c.817 C > T)) in two patients, labeled G71 and G35. However, these patients were part of Groups A and B, respectively, and thus, we could not establish statistical significance for this observation.
The research by Lauren R. Olafson et al. [23] revealed that the TP53 gene in G53 tumors (classified as secondary GBM) harbors a c.818 G > A (p.R273H) mutation at codon 273 of exon 8. This particular mutation leads to a gain of function (GOF) in the p53 protein, which could be responsible for increased tumor aggression, proliferation, invasiveness, and metastatic potential. Given these findings, it becomes crucial to further investigate how missense mutations at various locations within the TP53 gene contribute to functional changes and influence tumor progression. Additionally, identifying any differences in mutation sites between Eastern and Western populations calls for a more comprehensive dataset. This would help in understanding regional variations in tumor genetics and could guide targeted treatment strategies.
Bevacizumab treatment in GBM recurrence
In a study conducted by our institution focusing on the use of bevacizumab in recurrent GBM, we employed the Polymerase Chain Reaction (PCR) - Restriction Fragment Length Polymorphism (RFLP) technique to investigate the CDKN1A (p21) c.93 C > A gene polymorphism [8]. This research included 139 glioblastoma patients and explored the prevalence of different CDKN1A c.93 C > A genotypes. Although we found no direct link between these genotypes and the overall survival rate in glioblastoma patients, our findings did reveal a notable survival benefit in patients possessing certain genotypes (Arg/Arg and Arg/Ser). This advantage was observed in those who received a combination of concurrent chemoradiotherapy and bevacizumab monoclonal antibody treatment, as opposed to those treated with chemoradiotherapy alone. Additionally, our study suggests a positive association between bevacizumab use and prolonged survival in these patients.
In Japan, significant research has been conducted on the use of bevacizumab in GBM patients. These studies conclude that bevacizumab monoclonal antibody treatment can be particularly beneficial for tumors in patients who lack MGMT methylation [24]. Additionally, a post-market surveillance study in Japan examined the safety and effectiveness of bevacizumab for treating malignant gliomas [24]. While this study did not pinpoint specific genetic variations that might predict a positive response to bevacizumab, it did confirm that the use of bevacizumab in GBM patients is associated with extended survival. Furthermore, it underscored that bevacizumab is a safe and effective treatment option for these patients.
Conclusion
In Taiwan’s GBM patient cohort, bevacizumab use correlates positively with increased survival rates, underscoring its safety and effectiveness. Notably, the prevalence of EGFR mutations in this group is lower than in Western counterparts, hinting at distinct carcinogenesis pathways. This calls for further detailed research on EGFR mutations and amplifications to deepen our understanding and improve treatment strategies. Additionally, our study links TP53 mutations to better survival outcomes, although the specific impacts of missense mutations require further investigation. This pioneering study utilizing NGS technology sheds new light on the genetic variations in Taiwanese GBM patients, significantly enriching our knowledge of their genetic profile.
Data availability
Our research data, housed at Taichung Veterans General Hospital, is subject to specific licensing restrictions that limit its public availability. This data was primarily utilized for our study. Requests for data access can be made to the corresponding authors and are subject to hospital approval. For detailed insights into our Next-Generation Sequencing (NGS) methods and results, please refer to https://www.ncbi.nlm.nih.gov/sra/. Note: The accession number mentioned must match the raw data provided in the Supplementary Materials.
In our commitment to rigorous data-sharing standards, we are implementing the following measures:
Accession Codes: We will obtain and publish unique accession codes for each dataset from established public repositories.
Regulatory Compliance: We are committed to adhering to all relevant licensing, ethical standards, and confidentiality agreements. In cases where public data sharing is restricted, we will outline alternative access methods.
Collaborative Compliance: Our team will collaborate closely with co-authors and affiliated institutions to secure all necessary approvals and ethical permissions for data distribution.
References
Park YW, Vollmuth P, Foltyn-Dumitru M, Sahm F, Ahn SS, Chang JH, et al. The 2021 WHO Classification for Gliomas and Implications on Imaging Diagnosis: Part 1-Key Points of the Fifth Edition and Summary of Imaging Findings on Adult-Type Diffuse Gliomas. J Magn Reson Imaging. 2023;58:677–89.
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66.
Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8.
Yan W, Zhang W, You G, Zhang J, Han L, Bao Z, et al. Molecular classification of gliomas based on whole genome gene expression: a systematic report of 225 samples from the Chinese Glioma Cooperative Group. Neuro Oncol. 2012;14:1432–40.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003.
Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–24.
Cheng WY, Shen CC, Liang YJ, Chiao MT, Yang YC, Hsieh WY, et al. Polymorphism at codon 31 of CDKN1A (p21) as a predictive factor for bevacizumab therapy in glioblastoma multiforme. BMC Cancer. 2023;23:886.
Wang F, Zheng Z, Guan J, Qi D, Zhou S, Shen X, et al. Identification of a panel of genes as a prognostic biomarker for glioblastoma. EBioMedicine. 2018;37:68–77.
Crowdis J, He MX, Reardon B, Van Allen EM. CoMut: visualizing integrated molecular information with comutation plots. Bioinformatics. 2020;36:4348–9.
Dolma L, Muller PAJ. GOF Mutant p53 in Cancers: A Therapeutic Challenge. Cancers (Basel). 2022; 14.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Ou J, Zhu LJ. trackViewer: a Bioconductor package for interactive and integrative visualization of multi-omics data. Nat Methods. 2019;16:453–4.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23:1231–51.
Wang K, Wang Y, Fan X, Wang J, Li G, Ma J, et al. Radiological features combined with IDH1 status for predicting the survival outcome of glioblastoma patients. Neuro Oncol. 2016;18:589–97.
Network TC. Corrigendum: Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2013;494:506.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25:462–9.
Wang LB, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell. 2021;39:509–528.e520.
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110.
Higa N, Akahane T, Yokoyama S, Yonezawa H, Uchida H, Fujio S, et al. Molecular Genetic Profile of 300 Japanese Patients with Diffuse Gliomas Using a Glioma-tailored Gene Panel. Neurol Med Chir (Tokyo). 2022;62:391–9.
Noor H, Briggs NE, McDonald KL, Holst J, Vittorio O. TP53 Mutation Is a Prognostic Factor in Lower Grade Glioma and May Influence Chemotherapy Efficacy. Cancers (Basel). 2021;13:5362.
Obrecht D, Mynarek M, Hagel C, Kwiecien R, Spohn M, Bockmayr M, et al. Clinical and molecular characterization of isolated M1 disease in pediatric medulloblastoma: experience from the German HIT-MED studies. J Neurooncol. 2022;157:37–48.
Nagane M, Ichimura K, Onuki R, Narushima D, Honda-Kitahara M, Satomi K, et al. Bevacizumab beyond Progression for Newly Diagnosed Glioblastoma (BIOMARK): Phase II Safety, Efficacy and Biomarker Study. Cancers (Basel). 2022;14:5522.
Acknowledgements
We extend our deepest gratitude to the Taichung Veterans General Hospital for their invaluable support throughout this study. Their unwavering assistance and resources have been fundamental in enabling us to conduct our research on the genetic mutation patterns among glioblastoma patients in the Taiwanese population. This research would not have been possible without their generous support and commitment to advancing medical science and patient care.
Funding
This work received partial support from Taichung Veterans General Hospital, under grant numbers TCVGH-111733E, TCVGH-1107333E, TCVGH-11097333E, and TCVGH-11087333E in Taiwan. The funders played no role in the study’s design, data collection, analysis, decision to publish, or manuscript preparation. None of the authors received a salary from the aforementioned funding sources.
Author information
Authors and Affiliations
Contributions
Study design: Y.F.H., T.H.H., C.C.S., M.Y.Y.; data acquisition: M.T.C., Y.X.Z., T.Y.C., C.H.L., S.Y.L., C.H.L., W.Y.C., C.M.Y., C.M.L.; statistical analysis: J.P.C.; data interpretation: Y.F.H., T.H.H.; manuscript preparation: Y.H.F., M.T.C.; manuscript editing: Y.F.H., C.C.S., M.Y.Y.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and informed consent
The study protocols received approval from the Medical Ethics Committee of Taichung Veterans General Hospital (Approval number: CF17263B-4). Informed consent was obtained from all subjects or their legal guardians prior to surgery, and the collected samples were promptly frozen. The validation cohort consisted of 254 cases selected from the primary cohort based on the following criteria: (1) availability of follow-up data and samples, and (2) postoperative survival time exceeding 1 month. Between January 2010 and March 2023, tissue specimens were collected from 30 patients diagnosed with GBM. Exclusion criteria included: 1. GBM patients with unconfirmed pathology, 2. GBM patients with spinal involvement, and 3. GBM patients with incomplete data records. All methods strictly adhered to the guidelines and regulations of Taichung Veterans General Hospital and were conducted in accordance with the Declaration of Helsinki.
Consent for Publication
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, YF., Chiao, MT., Hsiao, TH. et al. Genetic mutation patterns among glioblastoma patients in the Taiwanese population – insights from a single institution retrospective study. Cancer Gene Ther (2024). https://doi.org/10.1038/s41417-024-00746-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41417-024-00746-y
