Abstract
Extracellular matrix (ECM) refers to the non-cellular components of the tumour microenvironment, fundamentally providing a supportive scaffold for cellular anchorage and transducing signaling cues that orchestrate cellular behaviour and function. The ECM integrity is abrogated in several cases of cancer, ending in aberrant activation of a number of mechanotransduction pathways and induction of multiple tumorigenic events such as extended proliferation, cell death resistance, epithelial-mesenchymal transition and most importantly the development of chemoresistance. In this regard, the present study mainly aims to elucidate how the ECM-stiffening process may contribute to the development of chemoresistance during cancer progression and what pharmacological approaches are required for tackling this issue. Hence, the first section of this review explains the process of ECM stiffening and the ways it may affect biochemical pathways to induce chemoresistance in a clinic. In addition, the second part focuses on describing some of the most important pharmacological agents capable of targeting ECM components and underlying pathways for overcoming ECM-induced chemoresistance. Finally, the third part discusses the obtained results from the application of these agents in the clinic for overcoming chemoresistance.
Similar content being viewed by others
Introduction
Cancer chemotherapy is a common practice in the clinic aiming at improving the patient’s overall survival while reducing the rate of disease recurrence. In this context, “adjuvant” therapy is a specific module of chemotherapy that is performed after a successful surgery to eradicate the remaining malignant cells and non-detectable micro- and macro-metastases in secondary sites. Conversely, “neoadjuvant” therapy is a pre-operation intervention with the overall goal of restricting tumour outgrowth and improving the outcome of surgery by reducing cancer cell invasiveness and bolding the margins between healthy and malignant tissues [1]. However, in several cases, the efficacy of anti-anti-neoplastic therapy eventually reduces upon cancer progression and resistance develops, rendering the treatment schedule becoming either ineffective or negligibly effective in prolonging the overall survival of the patient [2, 3]. This usually takes place following the activation and/or suppression of a myriad of signaling cascades controlling cancer cell growth, differentiation, migration and invasiveness [4,5,6,7,8,9].
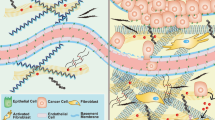
Extracellular matrix (ECM) refers to the non-cellular components of the tumour microenvironment (TME) which is mainly composed of proteins, proteoglycans and glycoproteins [10]. Regardless of preserving the intact structure of tissues and maintaining their functionality, the ECM structure and stiffness can affect different aspects of cellular responses, including adhesiveness, migration, proliferation, differentiation and apoptotic behaviour of epithelial cells through integrin-mediated interactions and local secretion of growth factors including epidermal growth factor (EGF), fibroblast growth factor (FGF), the transforming growth factor-β (TGF-β) and the like upon sequestration [10,11,12]. In physiologic conditions, cells can also rebuild and remodel their surrounding ECM by synthesising, reassembling and chemically modifying its components [10, 13]. Given that the physical and chemical properties of ECM mostly alter during cancer, the cellular behaviour of most cancer cells will change as well. Therefore, understanding the cancer cell-ECM negotiations is of utmost importance for establishing an effective cancer therapy.
Pathological progression of tumour stiffening
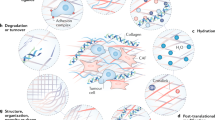
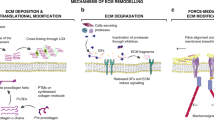
Change in tumour stiffness is the consequence of different ECM-remodeling processes, which can be categorised into three main subclasses. The aberrant activation of cancer-associated fibroblasts (CAFs) that alters the abundance and composition of ECM components, especially collagen, and ends in excessive ECM deposition. The other subclasses are the overexpression of lysyl oxidase (LOX) which enhances collagen cross-linkage in the tumour environment, and the enhanced level of matrix metalloproteinases (MMPs) and force-mediated remodeling in the TME which mediates the proteolytic degradation of ECM and parallel alignment of the collagen.
Excess ECM deposition due to the aberrantly activated CAFs
Even though the certain cause of ECM stiffening during tumour progression remains puzzling [14, 15], based on our present knowledge, ECM remodeling precedes tumour stiffening and resistance to chemotherapy [14, 16]. The aberrant interconnections between cancer cells, M2 macrophages and CAFs during the early stages of cancer development promote a myriad of destructive events including an excessive deposition of ECM components [17], production of a wide range of cytokines and growth factors [18, 19], production of various ECM-transforming enzymes [20, 21] and the alteration of the ECM-residing fiber alignment, culminating in the establishment of a remodeled ECM.
Based on this scenario, excessive secretion of growth factors during the early stages of cancer progression induces the activation and differentiation of fibroblasts while decreasing the speed of ECM degradation by reducing the expression of MMPs. Activated fibroblasts then differentiate into myofibroblasts (CAFs) and begin to secrete TGF-β, hepatocyte growth factor and FGF-2 that is the well-known stimulator of the epithelial-mesenchymal transition (EMT) process, the promoter of cellular growth and the main inducer of angiogenesis, respectively. CAFs can also promote remodeling and reinforcement of ECM through the upregulation of LOX activity and enhancement of the deposition and cross-linkage of types I, II, V and IX collagens. Similarly, stromal fibroblasts begin to secret different types of growth factors including insulin-like growth factor (IGF) and keratinocyte growth factor, which in turn, can accelerate cancer cell growth and TME remodeling. Additionally, radial alignment of thick collagen bundles by stromal cells provides a proper escaping route for metastatic cells to extravasate from the initial tumour site to the secondary compartment [22, 23].
Excessive proliferation of cancer cells and accelerated tumour growth can also induce hypoxia in the core region of the tumour, ending in the enhancement of vascular endothelial growth factor (VEGF) and connective tissue growth factor (CTGF) secretion (growth factors well-known for their supportive effect on angiogenesis and acceleration of cell recruitment into the tumour site, respectively) by cancer cells and promote ECM remodeling [24]. During the EMT process, cancer cells begin to secret the CSF-1 growth factor, which recruits macrophages to the tumour site and enhances the local release of macrophage-derived growth factors [25]. The recruited macrophages in the next place secret EGF and interleukin (IL)-33 which further promote metastasis [26]. Ultimately, invasive cancer cells lyse stiffened ECM by upregulating the expression of MMPs and a disintegrin and metalloprotease, induce ECM remodeling, and evade from the primary TME into lymph nodes and the bloodstream where they can spread into the distant secondary sites of metastasis [27].
Dysregulated ECM homeostasis by MMP overexpression
MMPs are a specific group of proteins responsible for the degradation of ECM components whose activity is tightly controlled by a specific group of enzymes, which are referred to as the tissue inhibitors of metalloproteinases (TIMPs) [28]. Different studies have outlined an enhanced MMP expression and activity for stiffened matrices. Accordingly, the increased amounts of MMPs lead to enhanced cellular invasion and chemoresistance development [29]. Using a pancreatic adenocarcinoma mouse model, Dangi-Garimella et al. also demonstrated that the increased collagen content of ECM accompanies enhanced MMP14 expression and development of resistance to gemcitabine while the co-administration of a selective MMP14 inhibitor with gemcitabine could reverse acquired resistance in mice. Investigating the underlying molecular mechanisms, they further found that MMP14 overexpression could promote the production of HMGA2, which is a non-histone binding protein and is capable of abrogating the anti-neoplastic effects of gemcitabine [30]. MMP14 can also activate autophagic responses in glioblastoma cell lines through the regulation of Bcl-2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) biosynthesis [31]. Moreover, MMP7 can promote chemoresistance by modulating apoptotic responses [32,33,34]. Experiments on colon cancer cell lines revealed that overtreatment with MMP7 alters the expression of the Fas death receptor which could induce oxaliplatin resistance [34]. Similarly, overtreating lung cancer cells with MMP7 promoted the expression of an anti-apoptotic protein (Bcl2), inducing resistance to cisplatin [35].
Dysregulated ECM homeostasis by LOX overexpression
The LOX family is a group of enzymes responsible for crosslinking collagen molecules and forming elastin networks [36]. LOXs can affect drug diffusion and delivery to their site of action by enhancing the accumulation of collagen fibers and/or densifying the existing collagen network. Furthermore, these enzymes are overexpressed in intrinsically chemoresistant breast, ovarian and colorectal cancers [37], and inhibiting LOX activity typically accompanies improved oxygenation [37, 38], reduced VEGF-A expression, angiogenesis and maturation of tumour vasculature [39]. They can also directly affect the expression of VEGF-A by oxidising the extracellular domain of the platelet-derived growth factor receptor [40]. Finally, endothelial invasion (i.e. the rate-limiting step of angiogenesis) is much more facilitated in LOX isoenzyme 2 (LOXL2) rich tumours compared to other tumours [41]. In this context, Schutze et al. concluded that LOXL2 overexpression strongly reduced doxorubicin diffusion into multicellular tumour spheroids [42].
Matrix stiffening induced chemoresistance
Increased matrix stiffness can directly stimulate a range of intracellular signaling pathways which can ultimately lead to the development of chemoresistance (Fig. 1). These pathways can be classified into three main subclasses of integrin-mediated signaling pathways, Discoidin domain receptor (DDR) mediated signaling pathway, and Rho GTPase/myocardin-related transcription factor (MRTF) mediated signaling pathway.

a Binding of a stiffened matrix to β1-integrin results in the activation of FAK, inducing the degradation of p53 and resistance to apoptosis. Furthermore, the interaction of integrin with stiffened ECM can promote the translocation of NF-kB to the nucleus and the development of resistance to cell death. b Stiffened matrix interaction with β1-integrin activates the ILK/PI3K/AKT pathway, promoting the expression of CSC-related markers including CD44, Nanog, CD49f and ALDH and inducing CSC behaviour in cancer cells. c Stiffened matrix can promote metastasis through the induction of the Wnt/YAP/TAZ pathway, the activation of myofibroblasts which secret activin/TGF-β and IGF that stimulates the EMT process, and finally, the promotion of MMP and ADAM secretion by cancer cells. d In addition to hypoxia-activated pathways and myofibroblast-mediated secretion of FGF-2, the ILK-mediated secretion of VEGF-A upon the interaction of stiffened ECM with β-integrins is highly responsible for the promotion of angiogenesis and development of acquired resistance to chemotherapy. e ECM stiffening promotes the FAK-mediated inhibition of p15 and p21 growth suppressors. f The ectopic expression of stromelysin and MMP3 promotes genome instability. Furthermore, a mutation in specific positions of alpha 5 and 6 chains of IV or VII collagen results in the development of smooth muscle tumours or skin malignancies. g ECM stiffening promotes the FAK-mediated activation of PI3K, promoting the expression of GLUT1 and GLUT4 and increasing the uptake of glucose by cancer cells. Furthermore, the ECM stiffening induced interaction of FAK with Ras and Myc promotes the conversion of glutamate to glutamine which guarantees cell survival through the induction of protein biosynthesis. h Finally, ECM stiffening promotes the FAK- and Src-mediated activation of PI3K/AKT AND MAPK/ERK regulated signaling pathways. ECM extracellular matrix, MMP matrix metalloproteinases, FAK focal adhesion kinase, PI3K phosphatidylinositol 3-kinase, AKT protein kinase B, CSC cancer stem cells, ALDH aldehyde dehydrogenase, TGF- β transforming growth factor-β, IGF insulin-like growth factor, ADAM a disintegrin and metalloprotease, FGF Fibroblast growth factor, VEGF vascular endothelial growth factor, EMT epithelial-mesenchymal transitions, ILK integrin-linked kinases, Wnt wingless-related integration sites. ECM stiffening influences the main hallmarks of acquired resistance to chemotherapy which can be broadly classified into eight categories.
Integrin-mediated signaling pathways
Integrins are a specific group of transmembrane proteins connecting ECM to the cell cytoskeleton and establishing a bidirectional interrelation between them [43]. These heterodimer proteins are composed of an α- and a β-chain subunit that is responsible for ligand specificity of the integrin and signal transduction, respectively. Upon ligand binding, integrins accumulate in specific regions of the membrane and recruit a range of coupling proteins to their intracellular region forming specific complexes known as focal adhesions. Among the recruited proteins, focal adhesion kinase (FAK), and proto-oncogene tyrosine-protein kinase Src, belonging to non-receptor tyrosine kinases, play a pivotal role in the transduction of the integrin-mediated signals from focal adhesions [44]. In this context, FAK and Src recruit specific adaptor proteins, including p130Cas and neural precursor cell expressed, developmentally downregulated 9 (NEDD9), which results in activation of a range of secondary signaling cascades including Rho GTPase, PI3k/protein kinase B (AKT), c-Jun N-terminal kinase, and extracellular signal-regulated protein kinase (ERK) regulated pathways, ending in distribution of integrin-mediated signals throughout the cell. [45].
Researches confirmed that the activation of β1-integrin-mediated signaling pathways can result in the development of resistance to radiotherapy in the head and neck and breast cancers [46,47,48,49]. Similarly, αvβ3/β5 integrins have been shown to take part in the development of radiotherapy resistance [50, 51]. β1-integrin is also responsible for the development of resistance to several chemotherapeutic agents [52, 53]. For instance, α5β1-integrin promotes temozolomide resistance in glioblastoma [54] through an IGFBP-2-dependent pathway [55] or α2β1-integrin activation is responsible for the development of doxorubicin-resistant leukemia [56]. The innate and/or acquired resistance mediated by integrin activation may also provoke tumour recurrence. In this regard, the inhibition of mutant BRAF by vemurafenib promotes the fabrication of an adhesion signaling network comprising α5β1-integrin, which is responsible for the development of resistance [57]. Moreover, the inhibition of BRAF in fibroblastic tumour stroma promotes the activation of a β1-integrin/FAK signaling pathway, which is responsible for enhancing tumour cell survival [58]. Furthermore, in breast cancer cells, fibronectin derived from CAFs activates β1-integrin-dependent signaling in neighboring cells, finally leading to the acquisition of resistance to tamoxifen therapy [59]. It has also been shown that taxol-resistant ovarian cancer cells express β1-integrin in higher amounts [60]. More importantly, numerous studies have confirmed integrin-GFR crosstalk and αvβ3 integrin/KRAS axis in the development of resistance to anti-epidermal growth factor receptor (EGFR) therapies [61,62,63]. Eventually, the crosstalk between β1-integrin and c-Met is highly responsible for the development of resistance to tyrosine kinase inhibitors in lung cancer [64].
DDR-mediated signaling pathway
The DDR family was first discovered in the early 1990s. It comprises DDR1 and DDR2 proteins, which are a specific group of RTKs with which collagens act as their specific ligands [65]. DDR-collagen interconnection distinctively results in the autophosphorylation of tyrosine moiety and the activation of DDRs, leading to the modulation of several genetic and cellular events and thus the regulation of cell-collagen interactions [66]. For instance, unsuccessful induction of cell death by DDR1-mediated pro-survival signaling pathways can result in the development of a chemoresistant phenotype [67]. Moreover, elevated expression levels of DDR1 have a direct association with a highly aggressive cancer phenotype and are considered to be an unfavourable disease prognosis marker [68]. Consequently, it is not surprising that a high level of DDR1 activity is related to the development of an intrinsic chemoresistant phenotype and a poor cancer outcome [65, 68].
Rho/MRTF-mediated signaling pathway
The RhoA member of the Rho guanosine triphosphatases family (Rho GTPases) is a specific signaling molecule that plays a pivotal role in the modulation of the actin cytoskeleton through the stimulation of G-actin polymerisation and the formation of F-actin stress fibers. Accordingly, the Rho GTPases modulate the activity of the MRTF/serum response factor and thus can modulate the transcription of a range of genes affecting cellular polarity, microtubular dynamics, membrane transportation pathways and the function of multiple transcription factors [69]. GTPases are usually aberrantly activated in cancer cells and are considered as one of the main driving forces of cancer progression. In this context, increased cellular contractility of fibroblasts occurs as a consequence of aberrant activation of the Rho/MRTF pathway [70]. Moreover, the same pathway is responsible for the generation of cancer cell migratory tracks by CAFs. Similarly, achieving amoeboid movement ability by cancer cells in response to ECM-remodeling action of CAFs is mediated by the above-mentioned pathway [71, 72]. Consequently, the application of Rho/MRTF pathway inhibitors during chemotherapy may be another way of tackling stiffened ECM-induced chemoresistance.
Therapeutic approaches targeting matrix stiffening induced chemoresistance
As comprehensively discussed in the previous section, matrix stiffening induced chemoresistance can be divided into two main sections. The first stage stems from a dysregulated synthesis and remodeling of the ECM which is the consequence of the aberrant activity of cancer-associated fibroblasts, M2 macrophages, and several enzymes including MMP2, LOXs, and tissue glutaminases resulting in the development of matrix stiffening in the second stage through the activation of integrins and specific underlying signaling pathways including FAK/Rho, FAK/Src and FAK/ERK (Fig. 2). Thus, therapeutic strategies can also be classified into two main subclasses. The first one consists of strategies that return back the homeostasis of ECM synthesis and the second group includes the modulators of signaling pathways connecting ECM stiffness to chemoresistance (Fig. 3). Names of a number of these agents have also been provided with detail in Table 1.

ECM extracellular matrix.

ECM extracellular matrix, FAK focal adhesion kinase, JNK c-Jun N-terminal kinase, PI3K phosphatidylinositol 3-kinase, ERK1/2 extracellular signal-regulated protein kinases 1 and 2, ROCK Rho-associated coiled-coil protein kinase. Extracellular signal-regulated protein kinase. Increased deposition of ECM components including collagen and fibronectin in the tumour microenvironment promotes the phosphorylation of FAK and subsequently, recruits Src which can together induce the activation of a number of signaling cascades including MAPKs (e.g. JNK, ERK1/2 and PI3K) and Rho/ROCK), ultimately resulting in an increased cancer cell proliferation and survival, as well as an enhancement in cell motility, invasion and metastasis through the re-organisation of cytoskeletal filaments and adhesion molecules.
Therapeutic strategies for targeting ECM homeostasis
2D-culture validated studies
Recent studies have outlined the great modulatory role of MMPs in the efficacy of different chemotherapeutic agents in cancer treatment. Based on the findings of Pratt et al., MMP14 is capable of activating autophagic responses and is an important mediator of resistance in glioblastoma cells that occurs through the regulation of the production of BNIP3 [31]. MMP7 is another enzyme that is involved in the development of chemoresistance through the modulation of apoptosis [32,33,34]. Experiments on colon cancer cell lines revealed that overtreatment with MMP7 alters the expression of the Fas death receptor, and thus inducing oxaliplatin resistance [34]. Similarly, the overtreatment of lung cancer cells with MMP7 promoted the expression of an anti-apoptotic protein Bcl2, increasing resistance to cisplatin in these cells [35]. Based on these data, MMP inhibitors could be an invaluable source of agents which may overcome chemoresistance upon co-administration with chemotherapeutic agents. In this respect, it was found that the application of (2 R)-2-((4-Biphenylsulfonyl) amino)-3 phenylpropionic acid (C21H19NO4S), as an inhibitor of MMP2 and MMP9, together with cisplatin demonstrates a synergistic effect in treating A2780cis (cisplatin-resistant) cancer cell lines [73]. According to these findings, MMP inhibitors could be an invaluable source of agents that may overcome chemoresistance relying on co-administration with chemotherapeutic agents.
Considering the putative role of aberrantly activated CAFs in the development of resistance during chemotherapy, using a Rho-associated kinase signaling pathway inhibitor (i.e. Y-27632), Calvo et al. could disrupt the feed-forward loop between matrix stiffening and activation of Yes-associated protein (YAP)-signaling pathway, leading to a long-lasting reversion of the CAF phenotype, and subsequently, inhibiting ECM remodeling and promotion of cancer cell invasion and metastasis [74]. These data were also confirmed in an engineered mouse model of breast cancer.
Three-dimensional (3D)-culture and in vivo validated studies
The role of MMP overexpression in the development of chemoresistance has also been confirmed in 3D-culture and in vivo studies. Based on the results of Dangi-Garimella et al., the increased collagen content in a pancreatic adenocarcinoma mouse model accompanied by the expression of MMP14, enhancing resistance to gemcitabine. Likewise, they found that the application of a specific inhibitor of MMP14 together with gemcitabine could reverse developed resistance. Examining the underlying mechanism for this event in 3D collagen I cultures, they revealed that the production of MMP14 induces the overproduction of non-histone binding protein (i.e. HMGA2), which can abrogate the anti-neoplastic effects of gemcitabine [30]. Similarly, AG3340, which is a nonpeptidic collagen-mimicking MMP inhibitor and is capable of targeting MMP2, 3, 9 and 13, demonstrated a synergistic effect in suppressing tumour growth and angiogenesis when concurrently administered with carboplatin and paclitaxel in chemoresistant MV522 human NSCLC cell line [75].
Considering the crucial role of myofibroblastic CAF (myCAF) in the development of chemoresistance, Özdemir et al. depleted the tumour environment from αSMA-positive myCAFs by applying a genetic approach and treated tumours with gemcitabine. Despite a significant reduction in the collagen content of the tumour, no significant improvement was observed in the anti-cancer potential of gemcitabine. Investigating the underlying mechanism, they proposed an increased tumour invasion in response to myCAFs depletion [76]. The results of this study, in addition to the heterogeneous nature of CAFs, suggested the application of a more selective inhibitor of CAF for reversing CAF-associated chemoresistance [77]. Similarly, Duluc et al. demonstrated that IL-6 secreted from CAFs is highly responsible for the development of resistance following chemotherapy. Using an inhibitor of the mTOR/4E‐BP1 pathway, pasireotide (SOM230 analogue), for reducing IL-6 expression together with gemcitabine, they reported a synergistic reduction in tumour growth and chemoresistance [78]. It is reported that IL-6 is responsible for the activation of Janus kinases (JAK)‐mediated signal transducer and activator of transcription 3 (STAT3) signaling pathway. Using AZD1480 (a JAK/STAT3 inhibitor) together with gemcitabine, Nagathihalli et al. found a significantly increased amount of drug delivery to the tumour site and enhanced anti-cancer effects without depleting stromal collagen or hyaluronan content [77].
Therapeutic strategies modulating signaling pathways involved in ECM-induced chemoresistance
2D-culture validated studies
Along with diverse groups of chemical and biotechnological products, a specific group of natural products can also target integrins and exhibit anti-cancer effects. For instance, the application of Ouabain, which is a cardioactive glycoside extracted from the seeds of Strophanthus gratus, in lung cancer resulted in a significant alteration in the expression pattern of integrins in cancer cells. In addition, curcumin has been shown to effectively reduce the motility of breast cancer cells by blocking the activity of α6β4 integrin. Finally, gambogic acid can strongly suppress the activation of VEGFR2 and induction of underlying protein kinases consisting of FAK, Src and AKT [79].
Numerous studies have delineated the crucial role of YAP/TAZ signaling in regulating cellular responses to chemotherapy. Nonetheless, no clinically viable drug has so far been developed or identified which can directly and specifically inhibit this pathway. Verteporfin was the first discovered agent which could directly prevent form YAP/transcriptional enhanced associate domain (TEAD) connection and suppress YAP-overexpression-induced organ overgrowth [80]. Further examinations confirmed that verteporfin is capable of mimicking YAP knockdown to reverse developed resistance to RAF inhibitors, tyrosine kinase inhibitors and chemotherapeutics [81,82,83]. Nevertheless, verteporfin can only block YAP/TEAD at extremely high concentrations and possess several YAP-independent effects. Thus, this agent cannot be considered as a clinically viable inhibitor of YAP [84, 85]. Different drugs have been identified to indirectly suppress the TAP/TAZ signaling pathway by regulating the upstream Hippo pathway [86]. In this regard, statins have shown to inhibit HMG-CoA reductase, suppress YAP nuclear translocation, and imitate genetic YAP ablation to sensitise cells against TK and MAPK inhibitors [87, 88]. Unfortunately, similar to verteporfin, high doses of statins are required for this action, making them improper for application in the clinic [87, 89].
Alternatively, there is a different group of drugs capable of targeting YAP/TAZ signaling by inhibiting its downstream effectors. AXL inhibitors including bemcentinib and TP-0903, and anti-CTGF monoclonal antibody pamrevlumab (FG-3019) are a few examples of this group which is currently under clinical examination in combination therapy with EGFR inhibitors [90, 91]. Nevertheless, considering multiple actions of YAP/TAZ downstream targets, it is highly improbable to completely block YAP/TAZ activity only by inhibiting one downstream effector. Finally, in recent studies, the application of the bromodomain and extraterminal domain family of protein BRD4 (BRD4) inhibitors could inhibit YAP-mediated transcription and reverse the acquired resistance to MAPK inhibitors in BRAF mutant melanoma and KRAS mutant lung cancer cells. From the mechanistic point of view, BRD4 is capable of inducing YAP expression, directly interacting with YAP/TAZ, and functioning as a cofactor for the YAP/TAZ/TEAD complex [92, 93]. Unfortunately, all BRD4 inhibitors can also inhibit BRD2 and BRD3, which possess different transcriptional targets and functionality [94]. Thus, the application of these agents in the clinic is accompanied by high toxicity, restricting the broad application of these agents in the clinic [95]. Additionally, BRD4 has several transcriptional partners other than YAP/TAZ [94], and numerous epigenetic regulators are also concurrently involved in the modulation of gene transcription, along with YAP/TAZ [96,97,98]. Therefore, the full functional interconnection among BRD4 and YAP/TAZ in different types of cancer may differ and require further investigations.
3D-culture and in vivo validated studies
The application of CNTO95, which is a fully humanised monoclonal antibody against αV-integrin, has been shown to effectively potentiate the anti-neoplastic effects of fractionated radiotherapy in different types of human xenograft tumour models in nude mice [99, 100]. Consistently, the application of ATN-161, as an α5β1-integrin antagonist, along with 5-fluorouracil could effectively suppress liver metastases and improve overall survival in a colon cancer model [101]. Volociximab is another chimeric monoclonal antibody targeting α5β1-integrin, which has gained much attention for application in cancer treatment due to its high anti-cancer effects and the absence of severe toxicities even at high doses in preclinical studies [102].
Further, the first developed small molecule capable of inhibiting FAK activity was TAE226, which could interact with the Adenosine-5’-triphosphate (ATP)-binding site and inhibit Tyr[397] and Tyr[861] phosphorylation in the FAK structure and cross-react with the VEGF signaling. This agent was accompanied by several beneficial outcomes in vitro and in vivo and could effectively suppress angiogenesis in a human colon cancer model [103]. This experimental success led to the development of other inhibitors from this prototype. For instance, PF-562,271 is another inhibitor of the ATP-binding site of FAK which is also capable of inhibiting the ATP-binding site of protein-rich tyrosine kinase 2, which is also involved in the activation of Src [104, 105]. This agent could effectively suppress cancer cell growth and metastasis development in breast, prostate, and lung cancers [104, 106, 107]. More importantly, a synergistic inhibitory effect on hepatocellular tumour growth was obtained upon the concurrent administration of this agent with sunitinib in vivo [108]. PF-573,228 is also an agent with similar activity to PF-562,271, which suppressed the proliferation of estrogen receptor-positive breast cancer cells upon co-administration with tamoxifen [106, 109]. Other FAK inhibitors include PF-04554878 and GSK2256098 which are in phases I and II the clinical trial for the treatment of advanced non-hematologic malignancies, respectively. PND-1186 is another agent with inhibitory effects on FAK activation, resulted from substituting the pyrimidine ring in other ATP-binding FAK inhibitors with the pyridine ring which has shown to enhance apoptosis and suppress the outgrowth and metastasis of the breast cancer cell. This agent could also inhibit ascites and peritoneal seeding in an ovarian cancer model [110,111,112]. Recently, a group of more specific and less toxic FAK inhibitors has been developed which are capable of targeting the Tyr[397] site in the FAK structure and inhibiting its autophosphorylation [113, 114]. For instance, 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) preferentially connects with Tyr[397], resulting in the inhibition of FAK rather than Pyk2. This agent could strongly suppress tumorigenesis and breast cancer cell adhesion in vivo and inhibit tumour growth in pancreatic and neuroblastoma models in vivo [114, 115]. Eventually, the application of this agent together with gemcitabine led to synergistic inhibitory effects against pancreatic cancer cell growth [115].
The translational gap between bench research and clinical application of ECM-targeting agents
Although different compounds capable of modulating ECM stiffness or its underlying pathways have so far been introduced, most of them were either non-applicable to clinical tests or extremely poorly effective in prolonging the survival of patients during the phase I/II clinical trials. For instance, many studies have demonstrated the critical role of CAFs in synthesising the protein components of ECM and conferring to tumour progression and metastasis. However, as mentioned earlier, CAFs are heterogeneous in nature and their targeting is a challenging process [116, 117]. Therefore, no viable approach currently exists in the clinical setting for targeting CAFs. Another potential approach for modulating ECM stiffening is to control its synthesis by targeting residing enzymes, most importantly, the LOX enzyme. Although this approach has shown to be somehow effective in the preclinical setting, clinical studies reported disappointing results, which may be due to the “network compensation” event occurring in response to single therapy. Additionally, MMP inhibitors have failed to demonstrate any meaningful effects on patients’ survival rates [118]. The application of marimastat after induction therapy in non-small cell lung cancer patients could not significantly improve survival, and surprisingly, even posed a negative effect on their quality of life [119]. This has mainly been attributed to their high toxicity profile and contradictory action on both enhancing and decreasing metastatic dissemination. Furthermore, MMP inhibitors have a nonspecific function, targeting multiple enzyme subtypes probably resulting in pro- and anti-tumorigenic effects. Accordingly, the application of more specifically acting MMPs and other ECM modulating proteases hold promise in overcoming resistance [120].
Similarly, the results of a limited number of clinical trials evaluating the efficacy of YAP/TAZ inhibitors in reversing resistance to chemotherapy completely contradicted their in vitro results. For example, the phosphorylation of YAP1 by Yes protein and its translocation to the nucleus could be prevented by dasatinib, which is a potent Src kinase inhibitor. Despite some excellent in vitro suppressive responses on the growth rate of triple-negative breast cancer cell lines, limited beneficial effects were achieved in a phase II clinical trial investigating the efficacy and safety profile of dasatinib in the management of these patients [121, 122]. Similarly, the results of another phase II clinical trial examining the efficacy of dasatinib against metastatic castration-resistant prostate cancer following disease progression on docetaxel demonstrated poor tolerability and limited therapeutic outcomes among the evaluated patients [123]. The findings of another phase II clinical trial investigating the potency of cediranib (i.e. a potent VEGFR2 inhibitor) and dasatinib regimen on patients with CRPC even represented worsening outcomes compared to cediranib therapy alone in these patients [124]. Finally, the administration of dasatinib was not practically associated with any significant beneficial outcomes in a clinical trial on patients with a specific type of lung adenocarcinoma, which is related to EGFR mutations and resistant to erlotinib or gefitinib [125]. The lack of a proper cell-ECM interaction model resembling the physiological condition of the human body is one of the main reasons behind the translational gap between bench research and clinical application of ECM stiffness targeting agents to overcome chemoresistance. Overall, preclinical studies for screening and testing drugs capable of affecting cell-ECM interaction are composed of three types of studies such as 2D in vitro cancer, 3D in vitro cancer, and small in vivo animal models [71]. Despite the observed advantages of these models, specific drawbacks associated with each model mostly restrict the translation of obtained data in these models to clinical settings.
Conclusion
As comprehensively discussed in this review, ECM and its aberrant stiffening play an important role in developing cancer and promoting chemoresistance induction. Thus, targeting ECM components may potentially be an effective approach in enhancing the delivery of chemotherapeutic drug efficacy and reversing developed chemoresistance. Nonetheless, the outcome of more than 10-year experience with the application of ECM-modifying agents or inhibitors of their downstream pathways in the clinic can be summarised in three lessons. Contrary to what was initially expected, targeting ECM is accompanied by unexpected high toxicity. One of the most important reasons is that ECM-modifying agents are also capable of disturbing homeostasis in normal organs. For instance, the enhanced deposition of collagen following the administration of MMPs in different tissues resulted in the development of severe musculoskeletal pain and inflammation which obligated the withdrawal of one-third of patients from treatment [126]. The other reason may be attributed to the suppression of specific cells or pathways that demonstrate opposing effects on cancer therapy. Furthermore, the application of MMP inhibitors has been shown to suppress specific subtypes of MMPs that are responsible for the synthesis of antiangiogenic peptides by sequestering responsible precursor proteins [126]. Moreover, resistance can develop against ECM-targeting agents. Contrary to the previously established idea, which stated that ECM modulators may not be subjected to genomic instability, development of resistance to Avastin (i.e., a monoclonal antibody targeting VEGF-A) as a result of redundancy to angiogenic signals proved the opposite statement [127]. In addition, agents targeting ECM stiffening may activate specific pathways in malignant cells, and thus promoting drug resistance or activation of bypass mechanisms. Finally, and perhaps most importantly, the potency of ECM-targeting agents in treating cancer should be considered in their optimal administered dosage. Unfortunately, most of the studied ECM-targeting agents in clinical trials have been applied at their maximal-tolerated dose (MTD) based on the common fact associated with chemotherapeutic agents indicating that further administration amount is better in this regard. Considering that ECM modulators are applied aiming at re-establishing a dysregulated homeostasis state in a “malignant organ” rather than completely reversing the direction of the equation, the application of MTD may not be always the optimum choice. Thus, determining the optimal biologic dose is more clinically valuable compared to the MTD.
Data availability
Not applicable.
References
Hayes DF, Schott AF. Neoadjuvant chemotherapy: what are the benefits for the patient and for the investigator? J Natl Cancer Inst Monogr. 2015;2015:36–39.
Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13.
Marquette C, Nabell L. Chemotherapy-resistant metastatic breast cancer. Curr Treat Options Oncol. 2012;13:263–75.
Farahmand L, Merikhian P, Jalili N, Darvishi B, Majidzadeh AK. Significant role of MUC1 in development of resistance to currently existing anti-cancer therapeutic agents. Curr Cancer Drug Targets. 2018;18:737–48.
Darvishi B, Majidzadeh AK, Ghadirian R, Mosayebzadeh M, Farahmand L. Recruited bone marrow derived cells, local stromal cells and IL-17 at the front line of resistance development to anti-VEGF targeted therapies. Life Sci. 2019;217:34–40.
Mahdi A, Darvishi B, Majidzadeh AK, Salehi M, Farahmand L. Challenges facing antiangiogenesis therapy: the significant role of hypoxia-inducible factor and MET in development of resistance to anti-vascular endothelial growth factor-targeted therapies. J Cell Physiol. 2019;234:5655–63.
Darvishi B, Farahmand L, Eslami SZ, Majidzadeh AK. NF-kappaB as the main node of resistance to receptor tyrosine kinase inhibitors in triple-negative breast cancer. Tumour Biol. 2017;39:1010428317706919.
Brasseur K, Gevry N, Asselin E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget. 2017;8:4008–42.
Salaritabar A, Berindan-Neagoe I, Darvish B, Hadjiakhoondi F, Manayi A, Devi KP, et al. Targeting Hedgehog signaling pathway: paving the road for cancer therapy. Pharmacol Res. 2019;141:466–80.
Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395–406.
Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia. 2004;9:325–42.
Kass L, Erler JT, Dembo M, Weaver VM. Mammary epithelial cell: influence of extracellular matrix composition and organization during development and tumorigenesis. Int J Biochem Cell Biol. 2007;39:1987–94.
Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4:165–78.
Mierke CT, Sauer F, Grosser S, Puder S, Fischer T, Kas JA. The two faces of enhanced stroma: Stroma acts as a tumor promoter and a steric obstacle. NMR Biomed. 2018;31:e3831.
Wei B, Zhou X, Liang C, Zheng X, Lei P, Fang J, et al. Human colorectal cancer progression correlates with LOX-induced ECM stiffening. Int J Biol Sci. 2017;13:1450–7.
Malik R, Lelkes PI, Cukierman E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015;33:230–6.
Farhood B, Najafi M, Mortezaee K. Cancer‐associated fibroblasts: secretions, interactions, and therapy. J Cell Biochem. 2019;120:2791–2800.
Witsch E, Sela M, Yarden Y. Roles for growth factors in cancer progression. Physiology (Bethesda). 2010;25:85–101.
de la Mare JA, Jurgens T, Edkins AL. Extracellular Hsp90 and TGFbeta regulate adhesion, migration and anchorage independent growth in a paired colon cancer cell line model. BMC Cancer. 2017;17:202.
Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801.
Lee J, Condello S, Yakubov B, Emerson R, Caperell-Grant A, Hitomi K, et al. Tissue transglutaminase mediated tumor-stroma interaction promotes pancreatic cancer progression. Clin Cancer Res. 2015;21:4482–93.
Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–32.
Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:1–15.
Kondo S, Kubota S, Shimo T, Nishida T, Yosimichi G, Eguchi T, et al. Connective tissue growth factor increased by hypoxia may initiate angiogenesis in collaboration with matrix metalloproteinases. Carcinogenesis. 2002;23:769–76.
Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005;65:5278–83.
Andersson P, Yang Y, Hosaka K, Zhang Y, Fischer C, Braun H, et al. Molecular mechanisms of IL-33–mediated stromal interactions in cancer metastasis. JCI Insight. 2018;3:e122375.
Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801.
Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015;44:247–54.
Nukuda A, Sasaki C, Ishihara S, Mizutani T, Nakamura K, Ayabe T, et al. Stiff substrates increase YAP-signaling-mediated matrix metalloproteinase-7 expression. Oncogenesis. 2015;4:e165–e165.
Dangi-Garimella S, Krantz SB, Barron MR, Shields MA, Heiferman MJ, Grippo PJ, et al. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP–mediated expression of HMGA2. Cancer Res. 2011;71:1019–28.
Pratt J, Annabi B. Induction of autophagy biomarker BNIP3 requires a JAK2/STAT3 and MT1-MMP signaling interplay in Concanavalin-A-activated U87 glioblastoma cells. Cell Signal. 2014;26:917–24.
Akers WJ, Xu B, Lee H, Sudlow GP, Fields GB, Achilefu S, et al. Detection of MMP-2 and MMP-9 activity in vivo with a triple-helical peptide optical probe. Bioconjugate Chem. 2012;23:656–63.
Huang Y, Yu H, Lei H, Xie C, Zhong Y. Matrix metalloproteinase 7 is a useful marker for 5-fluorouracil-based adjuvant chemotherapy in stage II and stage III colorectal cancer patients. Med Oncol. 2014;31:824.
Almendro V, Ametller E, García-Recio S, Collazo O, Casas I, Augé JM, et al. The role of MMP7 and its cross-talk with the FAS/FASL system during the acquisition of chemoresistance to oxaliplatin. PLoS ONE. 2009;4:e4728.
Liu H, Zhang T, Wu B, Huang J, Zhou Y, Zhu J. Chronic exposure to exogenous matrilysin induces chemoresistance and enhances Bcl-2 expression in A549 lung adenocarcinoma cells. Mol Biol Rep. 2009;36:2099.
Smith-Mungo LI, Kagan HM. Lysyl oxidase: properties, regulation and multiple functions in biology. Matrix Biol. 1998;16:387–98.
Rossow L, Veitl S, Vorlova S, Wax JK, Kuhn AE, Maltzahn V, et al. LOX-catalyzed collagen stabilization is a proximal cause for intrinsic resistance to chemotherapy. Oncogene. 2018;37:4921–40.
Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44.
Maxwell P, Dachs G, Gleadle J, Nicholls L, Harris A, Stratford I, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA. 1997;94:8104–9.
Baker A-M, Bird D, Welti JC, Gourlaouen M, Lang G, Murray GI, et al. Lysyl oxidase plays a critical role in endothelial cell stimulation to drive tumor angiogenesis. Cancer Res. 2013;73:583–94.
Zaffryar-Eilot S, Marshall D, Voloshin T, Bar-Zion A, Spangler R, Kessler O, et al. Lysyl oxidase-like-2 promotes tumour angiogenesis and is a potential therapeutic target in angiogenic tumours. Carcinogenesis. 2013;34:2370–9.
Schütze F, Röhrig F, Vorlová S, Gätzner S, Kuhn A, Ergün S, et al. Inhibition of lysyl oxidases improves drug diffusion and increases efficacy of cytotoxic treatment in 3D tumor models. Sci Rep. 2015;5:1–13.
Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22.
Berrier AL, Yamada KM. Cell–matrix adhesion. J Cell Physiol. 2007;213:565–73.
Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–26.
Eke I, Storch K, Krause M, Cordes N. Cetuximab attenuates its cytotoxic and radiosensitizing potential by inducing fibronectin biosynthesis. Cancer Res. 2013;73:5869–79.
Steglich A, Vehlow A, Eke I, Cordes N. α integrin targeting for radiosensitization of three-dimensionally grown human head and neck squamous cell carcinoma cells. Cancer Lett. 2015;357:542–8.
Nam J-M, Ahmed KM, Costes S, Zhang H, Onodera Y, Olshen AB, et al. 1-Integrin via NF-κB signaling is essential for acquisition of invasiveness in a model of radiation treated in situ breast cancer. Breast Cancer Res. 2013;15:R60.
Ahmed KM, Zhang H, Park CC. NF-κB regulates radioresistance mediated by β1-integrin in three-dimensional culture of breast cancer cells. Cancer Res. 2013;73:3737–48.
Ducassou A, Uro-Coste E, Verrelle P, Filleron T, Benouaich-Amiel A, Lubrano V, et al. vβ3 Integrin and Fibroblast growth factor receptor 1 (FGFR1): Prognostic factors in a phase I–II clinical trial associating continuous administration of Tipifarnib with radiotherapy for patients with newly diagnosed glioblastoma. Eur J Cancer. 2013;49:2161–9.
Lanvin O, Monferran S, Delmas C, Couderc B, Toulas C, Cohen-Jonathan-Moyal E. Radiation-induced mitotic cell death and glioblastoma radioresistance: a new regulating pathway controlled by integrin-linked kinase, hypoxia-inducible factor 1alpha and survivin in U87 cells. Eur J Cancer. 2013;49:2884–91.
Sørensen BH, Rasmussen LJH, Broberg BS, Klausen TK, Sauter DPR, Lambert IH, et al. Integrin β1, osmosensing, and chemoresistance in mouse ehrlich carcinoma cells. Cell Physiol Biochem. 2015;36:111–32.
Howe GA, Addison CL. β1 integrin: an emerging player in the modulation of tumorigenesis and response to therapy. Cell Adhes Migr. 2012;6:71–77.
Janouskova H, Ray A-M, Noulet F, Lelong-Rebel I, Choulier L, Schaffner F, et al. Activation of p53 pathway by Nutlin-3a inhibits the expression of the therapeutic target α5 integrin in colon cancer cells. Cancer Lett. 2013;336:307–18.
Han S, Li Z, Master L, Master Z, Wu A. Exogenous IGFBP-2 promotes proliferation, invasion, and chemoresistance to temozolomide in glioma cells via the integrin β1-ERK pathway. Br J Cancer. 2014;111:1400–9.
Naci D, Vuori K, Aoudjit, F. Alpha2beta1 integrin in cancer development and chemoresistance. Semin Cancer Biol. 2015;35:145–53.
Fedorenko IV, Abel EV, Koomen JM, Fang B, Wood ER, Chen YA, et al. Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene. 2016;35:1225–35.
Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, et al. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell. 2015;27:574–88.
Yuan J, Liu M, Yang L, Tu G, Zhu Q, Chen M, et al. Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: a new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and β1-integrin signaling pathway in tumor cells. Breast Cancer Res. 2015;17:69.
McGrail DJ, Khambhati NN, Qi MX, Patel KS, Ravikumar N, Brandenburg CP, et al. Alterations in ovarian cancer cell adhesion drive taxol resistance by increasing microtubule dynamics in a FAK-dependent manner. Sci Rep. 2015;5:9529.
Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25:234–40.
Eke I, Zscheppang K, Dickreuter E, Hickmann L, Mazzeo E, Unger K, et al. Simultaneous β1 integrin-EGFR targeting and radiosensitization of human head and neck cancer. J Natl Cancer Inst. 2015;107:dju419.
Kanda R, Kawahara A, Watari K, Murakami Y, Sonoda K, Maeda M, et al. Erlotinib resistance in lung cancer cells mediated by integrin β1/Src/Akt-driven bypass signaling. Cancer Res. 2013;73:6243–53.
Ju L, Zhou C. Association of integrin beta1 and c-MET in mediating EGFR TKI gefitinib resistance in non-small cell lung cancer. Cancer Cell Int. 2013;13:15.
Gadiya M, Chakraborty G. Signaling by discoidin domain receptor 1 in cancer metastasis. Cell Adhes Migr. 2018;12:315–23.
Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23.
Ambrogio C, Darbo E, Lee SW, Santamaría D. A putative role for Discoidin Domain Receptor 1 in cancer chemoresistance. Cell Adhes Migr. 2018;12:394–7.
Yang SH, Baek HA, Lee HJ, Park HS, Jang KY, Kang MJ, et al. Discoidin domain receptor 1 is associated with poor prognosis of non-small cell lung carcinomas. Oncol Rep. 2010;24:311–9.
Wang J, Hu K, Guo J, Cheng F, Lv J, Jiang W, et al. Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat Commun. 2016;7:1–13.
Leal AS, Misek SA, Lisabeth EM, Neubig RR, Liby KT. The Rho/MRTF pathway inhibitor CCG-222740 reduces stellate cell activation and modulates immune cell populations in Kras G12D; Pdx1-Cre (KC) mice. Sci Rep. 2019;9:1–12.
Vennin C, Chin VT, Warren SC, Lucas MC, Herrmann D, Magenau A, et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci Transl Med. 2017;9:eaai8504.
Haak AJ, Appleton KM, Lisabeth EM, Misek SA, Ji Y, Wade SM, et al. Pharmacological inhibition of myocardin-related transcription factor pathway blocks lung metastases of RhoC-overexpressing melanoma. Mol Cancer Ther. 2017;16:193–204.
Laios A, Mohamed BM, Kelly L, Flavin R, Finn S, McEvoy L, et al. Pre-treatment of platinum resistant ovarian cancer cells with an MMP-9/MMP-2 inhibitor prior to cisplatin enhances cytotoxicity as determined by high content screening. Int J Mol Sci. 2013;14:2085–103.
Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637–46.
Shalinsky D, Brekken J, Zou H, McDermott C, Forsyth P, Edwards D, et al. Broad antitumor and antiangiogenic activities of AG3340, a potent and selective MMP inhibitor undergoing advanced oncology clinical trials. Ann N Y Acad Sci. 1999;878:236–70.
Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–34.
Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R, et al. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology. 2015;149:1932–43. e1939.
Duluc C, Moatassim‐Billah S, Chalabi‐Dchar M, Perraud A, Samain R, Breibach F, et al. Pharmacological targeting of the protein synthesis mTOR/4E‐BP1 pathway in cancer‐associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol Med. 2015;7:735–53.
Aksorn N, Chanvorachote P. Integrin as a molecular target for anti-cancer approaches in lung cancer. Anticancer Res. 2019;39:541–8.
Fisher ML, Grun D, Adhikary G, Xu W, Eckert RL. Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells. Oncotarget. 2017;8:110257.
Scott LJ, Goa KL. Verteporfin. Drugs Aging. 2000;16:139–46.
Keam SJ, Scott LJ, Curran MP. Verteporfin. Drugs. 2003;63:2521–54.
Zhang H, Ramakrishnan SK, Triner D, Centofanti B, Maitra D, Győrffy B, et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci Signal. 2015;8:ra98–ra98.
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee S-J, Anders RA, et al. Genetic and pharmacological disruption of the TEAD–YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–5.
Dasari VR, Mazack V, Feng W, Nash J, Carey DJ, Gogoi R. Verteporfin exhibits YAP-independent anti-proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget. 2017;8:28628.
Tao Y, Cai F, Shan L, Jiang H, Ma L, Yu Y. The Hippo signaling pathway: an emerging anti-cancer drug target. Discov Med. 2017;24:7–18.
Oku Y, Nishiya N, Shito T, Yamamoto R, Yamamoto Y, Oyama C, et al. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Biol. 2015;5:542–9.
Xia H, Dai X, Yu H, Zhou S, Fan Z, Wei G, et al. EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis. 2018;9:1–12.
Matusewicz L, Meissner J, Toporkiewicz M, Sikorski AF. The effect of statins on cancer cells. Tumor Biol. 2015;36:4889–904.
Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer. 2017;116:415–23.
Heestand G, Pipas J, Valone F, McMullen A, Gadea P, Williams D, et al. A phase I trial of the monoclonal antibody FG-3019 to connective tissue growth factor (CTGF) in locally advanced or metastatic pancreatic cancer. J Clin Oncol. 2011;29:269–269.
Kitajima S, Asahina H, Chen T, Guo S, Quiceno LG, Cavanaugh JD, et al. Overcoming resistance to dual innate immune and MEK inhibition downstream of KRAS. Cancer Cell. 2018;34:439–52. e436.
Zanconato F, Battilana G, Forcato M, Filippi L, Azzolin L, Manfrin A, et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med. 2018;24:1599–610.
Xu Y, Vakoc CR. Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb Perspect Med. 2017;7:a026674.
Andrieu G, Belkina AC, Denis GV. Clinical trials for BET inhibitors run ahead of the science. Drug Discov Today Technol. 2016;19:45–50.
Elster D, Tollot M, Schlegelmilch K, Ori A, Rosenwald A, Sahai E, et al. TRPS1 shapes YAP/TEAD-dependent transcription in breast cancer cells. Nat Commun. 2018;9:1–16.
Kim M, Kim T, Johnson RL, Lim D-S. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep. 2015;11:270–82.
Oh H, Slattery M, Ma L, White KP, Mann RS, Irvine KD. Yorkie promotes transcription by recruiting a histone methyltransferase complex. Cell Rep. 2014;8:449–59.
Mullamitha SA, Ton NC, Parker GJ, Jackson A, Julyan PJ, Roberts C, et al. Phase I evaluation of a fully human anti–αv integrin monoclonal antibody (CNTO 95) in patients with advanced solid tumors. Clin Cancer Res. 2007;13:2128–35.
Ning S, Nemeth JA, Hanson RL, Forsythe K, Knox SJ. Anti-integrin monoclonal antibody CNTO 95 enhances the therapeutic efficacy of fractionated radiation therapy in vivo. Mol Cancer Ther. 2008;7:1569–78.
Doñate F, Parry GC, Shaked Y, Hensley H, Guan X, Beck I, et al. Pharmacology of the novel antiangiogenic peptide ATN-161 (Ac-PHSCN-NH2): observation of a U-shaped dose-response curve in several preclinical models of angiogenesis and tumor growth. Clin Cancer Res. 2008;14:2137–44.
Bhaskar V, Zhang D, Fox M, Seto P, Wong MH, Wales PE, et al. A function blocking anti-mouse integrin α5β1 antibody inhibits angiogenesis and impedes tumor growth in vivo. J Transl Med. 2007;5:61.
Schultze A, Decker S, Otten J, Horst AK, Vohwinkel G, Schuch G, et al. TAE226-mediated inhibition of focal adhesion kinase interferes with tumor angiogenesis and vasculogenesis. Invest N Drugs. 2010;28:825–33.
Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68:1935–44.
Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–50.
Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-β signaling and metastasis. Breast Cancer Res. 2009;11:R68.
Sun H, Pisle S, Gardner ER, Figg I. William D. Bioluminescent imaging study: FAK inhibitor, PF-562,271, preclinical study in PC3M-luc-C6 local implant and metastasis xenograft models. Cancer Biol Ther. 2010;10:38–43.
Bagi CM, Christensen J, Cohen DP, Roberts WG, Wilkie D, Swanson T, et al. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer Biol Ther. 2009;8:856–65.
Hiscox S, Barnfather P, Hayes E, Bramble P, Christensen J, Nicholson RI, et al. Inhibition of focal adhesion kinase suppresses the adverse phenotype of endocrine-resistant breast cancer cells and improves endocrine response in endocrine-sensitive cells. Breast Cancer Res Treat. 2011;125:659–69.
Tanjoni I, Walsh C, Uryu S, Tomar A, Nam J-O, Mielgo A, et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol Ther. 2010;9:764–77.
Schaller MD, Frisch SM. PND-1186 FAK inhibitor selectively promotes tumore cell apoptosis in three-dimensional environments. Cancer Biol Ther. 2010;9:791–3.
Walsh C, Tanjoni I, Uryu S, Tomar A, Nam J-O, Luo H, et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol Ther. 2010;9:778–90.
Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, et al. A small molecule inhibitor, 1, 2, 4, 5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J Med Chem. 2008;51:7405–16.
Zheng D, Golubovskaya V, Kurenova E, Wood C, Massoll NA, Ostrov D, et al. A novel strategy to inhibit FAK and IGF‐1R decreases growth of pancreatic cancer xenografts. Mol Carcinog. 2010;49:200–9.
Hochwald SN, Nyberg C, Zheng M, Zheng D, Wood C, Massoll NA, et al. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8:2435–43.
Wintzell M, Hjerpe E, Lundqvist EÅ, Shoshan M. Protein markers of cancer-associated fibroblasts and tumor-initiating cells reveal subpopulations in freshly isolated ovarian cancer ascites. BMC Cancer. 2012;12:359.
Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579–96.
Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer—trials and tribulations. Science. 2002;295:2387–92.
Shepherd FA, Giaccone G, Seymour L, Debruyne C, Bezjak A, Hirsh V, et al. Prospective, randomized, double-blind, placebo-controlled trial of marimastat after response to first-line chemotherapy in patients with small-cell lung cancer: a trial of the National Cancer Institute of Canada-Clinical Trials Group and the European Organization for Research and Treatment of Cancer. J Clin Oncol. 2002;20:4434–9.
Mack GS, Marshall A. Lost in migration. Nat Biotechnol. 2010;28:214–29.
Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13:63–79.
Finn RS, Bengala C, Ibrahim N, Roché H, Sparano J, Strauss LC, et al. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clin Cancer Res. 2011;17:6905–13.
Twardowski PW, Beumer JH, Chen C, Kraft AS, Chatta GS, Mitsuhashi M, et al. A phase II trial of dasatinib in patients with metastatic castration-resistant prostate cancer treated previously with chemotherapy. Anticancer Drugs. 2013;24:743.
Spreafico A, Chi KN, Sridhar SS, Smith DC, Carducci MA, Kavsak P, et al. A randomized phase II study of cediranib alone versus cediranib in combination with dasatinib in docetaxel resistant, castration resistant prostate cancer patients. Invest N Drugs. 2014;32:1005–16.
Johnson ML, Riely GJ, Rizvi NA, Azzoli CG, Kris MG, Sima CS, et al. Phase II trial of dasatinib for patients with acquired resistance to treatment with the epidermal growth factor receptor tyrosine kinase inhibitors erlotinib or gefitinib. J Thorac Oncol. 2011;6:1128–31.
Monia BP, Johnston JF, Geiger T, Muller M, Fabbro D. Antitumor activity of a phosphorothioate antisense oligodeoxynucleotide targeted against C-raf kinase. Nat Med. 1996;2:668–75.
Sun S, Irvine KD. Cellular organization and cytoskeletal regulation of the Hippo signaling network. Trends Cell Biol. 2016;26:694–704.
Guo W, Wei B, Chen T, Xu X, Ruan F, Xiang M. The Na+/K+ ATPase inhibitor ouabain attenuates stemness and chemoresistance of osteosarcoma cells. Med Sci Monit Int Med J Exp Clin Res. 2019;25:9426.
Javadi S, Rostamizadeh K, Hejazi J, Parsa M, Fathi M. Curcumin mediated down‐regulation of αVβ3 integrin and up‐regulation of pyruvate dehydrogenase kinase 4 (PDK4) in Erlotinib resistant SW480 colon cancer cells. Phytother Res. 2018;32:355–64.
Chen P, Huang H-P, Wang Y, Jin J, Long W-G, Chen K, et al. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J Exp Clin Cancer Res. 2019;38:1–17.
Hehlgans S, Lange I, Eke I, Cordes N. 3D cell cultures of human head and neck squamous cell carcinoma cells are radiosensitized by the focal adhesion kinase inhibitor TAE226. Radiother Oncol. 2009;92:371–8.
Bagi CM, Roberts GW, Andresen CJ. Dual focal adhesion kinase/Pyk2 inhibitor has positive effects on bone tumors: implications for bone metastases. Cancer. 2008;112:2313–21.
Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–60.
Das S, Ongusaha PP, Yang YS, Park J-M, Aaronson SA, Lee SW. Discoidin domain receptor 1 receptor tyrosine kinase induces cyclooxygenase-2 and promotes chemoresistance through nuclear factor-κB pathway activation. Cancer Res. 2006;66:8123–30.
Morimatsu M, Yamashita E, Seno S, Sudo T, Kikuta J, Mizuno H, et al. Migration arrest of chemoresistant leukemia cells mediated by MRTF-SRF pathway. Inflamm Regen. 2020;40:1–9.
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
BD, MRE, KMA and LF wrote the manuscript. BD and MRE collected and organised the data, and designed the figures. Further, KMA and LF edited the manuscript for scientific errors. All authors read and approved the manuscript in its final format.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent for participation
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Darvishi, B., Eisavand, M.R., Majidzadeh-A, K. et al. Matrix stiffening and acquired resistance to chemotherapy: concepts and clinical significance. Br J Cancer 126, 1253–1263 (2022). https://doi.org/10.1038/s41416-021-01680-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01680-8
