
Abstract
Caffeine induces multiple vascular effects. In this study we investigated the angiogenic effect of physiological concentrations of caffeine with focus on endothelial cell behaviors (migration and proliferation) during angiogenesis and its mitochondrial and bioenergetic mechanisms. We showed that caffeine (10–50 μM) significantly enhanced angiogenesis in vitro, evidenced by concentration-dependent increases in tube formation, and migration of human umbilical vein endothelial cells (HUVECs) without affecting cell proliferation. Caffeine (50 μM) enhanced endothelial migration via activation of cAMP/PKA/AMPK signaling pathway, which was mimicked by cAMP analog 8-Br-cAMP, and blocked by PKA inhibitor H89, adenylate cyclase inhibitor SQ22536 or AMPK inhibitor compound C. Furthermore, caffeine (50 μM) induced significant mitochondrial shortening through the increased phosphorylation of mitochondrial fission protein dynamin-related protein 1 (Drp1) in HUVECs, which increased its activity to regulate mitochondrial fission. Pharmacological blockade of Drp1 by Mdivi-1 (10 μM) or disturbance of mitochondrial fission by Drp1 silencing markedly suppressed caffeine-induced lamellipodia formation and endothelial cell migration. Moreover, we showed that caffeine-induced mitochondrial fission led to accumulation of more mitochondria in lamellipodia regions and augmentation of mitochondrial energetics, both of which were necessary for cell migration. In a mouse model of hindlimb ischemia, administration of caffeine (0.05% in 200 mL drinking water daily, for 14 days) significantly promoted angiogenesis and perfusion as well as activation of endothelial AMPK signaling in the ischemic hindlimb. Taken together, caffeine induces mitochondrial fission through cAMP/PKA/AMPK signaling pathway. Mitochondrial fission is an integral process in caffeine-induced endothelial cell migration by altering mitochondrial distribution and energetics.
Similar content being viewed by others


Introduction
Ischemic diseases, a group of disorders caused by the obstruction or stenosis of arterioles and capillaries that is not compensated for by collateral circulation or vessel dilatation, are a leading cause of disability and death around the world [1]. These diseases are characterized by a reduction in blood supply that results in limited oxygen transfer and nutrient uptake for cellular metabolism. Angiogenesis, a crucial process in new blood vessel formation and subsequent expansion of the vascular network, is triggered by ischemic conditions [2]. Thus, as a compensatory mechanism in response to ischemia, angiogenesis is important for physiological recovery and the functional protection of ischemic tissues [3, 4].
During angiogenesis, coordinated endothelial cell behaviors, including cellular migration, polarity, proliferation, differentiation, and communication, play a critical role in functional blood vessel morphogenesis [5]. Among these behaviors, endothelial cell migration and proliferation initiate the formation of capillary networks, which provide the primitive vascular plexus for further vascular maturation [6].
Mitochondria are the primary energy-generating organelles in most eukaryotic cells and have been widely reported to play a pivotal role in modulating angiogenesis by regulating the migration, proliferation, and survival of endothelial cells, which form the inner lining of blood vessels [7, 8]. Mitochondria exist in dynamic networks that frequently change shape and subcellular distribution; these dynamics are precisely controlled by two opposite processes: fusion and fission, which are regulated by mitofusins (Mfns) and dynamin-related protein 1 (Drp1), respectively [9]. A previous study reported that increased Drp1-dependent mitochondrial fission plays an important role in breast cancer cell migration and invasion [10] as well as vascular smooth muscle cell migration [11]. It was proposed that mitochondrial fission regulated by adenosine monophosphate-activated protein kinase (AMPK)/mitochondrial fission factor (MFF)/Drp1 signaling also serves to eliminate damaged organelles from the mitochondrial network, which presumably serves as an organellar quality control mechanism, and maintains bioenergetic capacity [12]. However, whether alterations in mitochondrial dynamics and energetics contribute to angiogenesis is unknown.
Caffeine is the most widely consumed bioactive substance in the world as it is a component of coffee, tea, carbonated beverages, and a wide variety of medications. Caffeine intake induces several physiological and pharmacological effects, including stimulation of the nervous and musculoskeletal systems [13] as well as the relaxation of vascular and bronchial smooth muscle [14]. However, very little is known about its role in angiogenesis. Previous studies demonstrated that caffeine increased mitochondrial function in Alzheimer’s model mice and cells [15] and improved the functional capacity of endothelial cells in a mitochondria-dependent process [16], suggesting that caffeine may regulate mitochondrial function. Nevertheless, whether caffeine plays a critical role in angiogenesis by modulating mitochondrial dynamics is not understood.
Therefore, we hypothesized the existence of a molecular link among caffeine, mitochondrial dynamics, and angiogenesis. In this study, we showed that (1) caffeine at low concentrations induced mitochondrial fission in endothelial cells through cAMP/PKA/AMPK/MFF signaling, (2) increased fission enhanced mitochondrial energetics and mediated mitochondrial redistribution in the leading edge to facilitate lamellipodia formation, (3) caffeine at low concentrations exerted angiogenic effects in endothelial cells in a mitochondrial fission-dependent manner, and (4) finally, treatment of mice with caffeine promoted angiogenesis and perfusion in the ischemic hindlimb. Our study provides new insight into the therapeutic effect of caffeine in ischemic diseases.
Materials and methods
Cell culture and treatments
Primary human umbilical vein endothelial cells (HUVECs) were harvested from umbilical cords using standard procedures according to established guidelines. HUVECs at passages 3–7 were cultured in endothelial cell medium (ECM) (PromoCell, Heidelberg, Baden-Württemberg, Germany) supplemented with 5% fetal bovine serum (FBS), 1% endothelial cell growth supplement, and the antibiotics penicillin and streptomycin at 1%. In some experiments, 10, 50, or 100 μM caffeine (Sigma, St. Louis, MO, USA); 200 μM SQ22536 (Selleck Chemicals, Houston, TX, USA); 200 μM 8-Br-cAMP (Biovision, Milpitas, CA, USA), 10 μM H89 (Meilun Bio, Dalian, Liaoning, China); 5 μM compound C (Sigma); 10 μM Mdivi-1 (Enzo, Farmingdale, NY, USA); 10 μM FCCP (Sigma); or 1 μg/mL oligomycin A (Sigma) was added to the culture medium as indicated.
Scratch wound healing assay
HUVECs administered the indicated treatment were seeded in 6-well plates and cultured overnight. When the cells reached ~100% confluence, a wound was formed by scraping the cells with a 200 μl pipette tip, and the cells were washed with PBS. Migration of the wounded cells was evaluated at 0, 6, and 12 h after scraping by recording photographic images using an inverted microscope, and the percentage of wound healing was analyzed at the indicated timepoints using ImageJ software.
Boyden chamber migration assay
A Boyden chamber migration assay was performed using a Boyden chamber (8 μm pore size; Corning, Acton, MA, USA). Briefly, HUVECs suspended in ECM with 1% FBS at a concentration of 5 × 104 cells/mL were added to the upper chambers, and ECM containing 1% FBS with the indicated chemicals was added to the lower chambers. After 12 h of incubation at 37 °C, the cells on the upper surfaces were removed with cotton swabs, and migrated cells on the lower surfaces were fixed and stained with a 1% crystal violet solution. For each Boyden chamber, the total cell numbers were determined from six random fields and analyzed.
WST-1 proliferation assay and cell viability assay
The proliferation of HUVECs and cytotoxic effects of the indicated activators and inhibitors on the HUVECs were investigated using a water-soluble tetrazolium salt (WST-1) assay (Sigma). HUVECs were seeded at 1 × 104 cells per well in 96-well plates. After treatment with the indicated chemicals, a 1/10 volume of WST-1 solution was added to the cells and incubated at 37 °C for 2 h. Cell proliferation and viability were determined by measuring the absorbance at 450 nm using an iMark microplate reader (Bio-Rad, Hercules, CA, USA).
Cell counting analysis
For cell counting, briefly, HUVECs were seeded in 6-well plates in triplicate at an equal density, and the cells were manually counted with a hemocytometer at the indicated timepoints.
Western blot analysis
For protein analysis, cells were lysed in ice-cold RIPA buffer (Sigma) with 1% PMSF. Following lysis for 30 min, the protein was obtained by centrifugation at 12,000 × g for 10 min at 4 °C, and the protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA). Samples containing the same amount of protein (20–30 μg) were separated by SDS-PAGE, and the proteins were then transferred to a 0.45 μm PVDF membrane by electrophoretic transfer. After blocking with 5% BSA for 1 h, the membranes were immunoblotted with primary antibody at 4 °C overnight and then incubated with HRP-conjugated secondary antibody for 1 h at room temperature. After the washing steps, immunoreactive bands were visualized with the ChemiDoc MP system (Bio-Rad), and the band densities were analyzed with Image Lab software (Bio-Rad). Primary antibodies targeting the following proteins were used in this study: p-AMPK (rabbit, 1:2000; Cell Signaling Technology, Danvers, MA, USA, Cat# 2531), AMPK (mouse, 1:2000; Abcam, Cambridge, MA, USA, Cat# ab80039), pS616-Drp1 (rabbit, 1:1000; Cell Signaling Technology, Cat# 3455), Drp1 (rabbit, 1:1000; Cell Signaling Technology, Cat# 8570), Mfn1 (rabbit, 1:1000; Proteintech, Chicago, IL, USA, Cat# 13798-1-AP), Mfn2 (rabbit, 1:2000; Proteintech, Cat# 12186-1-AP), p-MFF (rabbit, 1:2000; Affinity Biosciences, Cincinnati, OH, USA, Cat# AF2365), MFF (rabbit, 1:2000; Proteintech, Cat# 17090-1-AP), and β-actin (mouse, 1:5000; Santa Cruz Biotechnology, Santa Cruz, CA, USA, Cat# sc-81178).
Tube formation assay
For the tube formation assay, growth factor-reduced Matrigel (BD Bioscience, San Jose, CA, USA) was added to 96-well plates (60 μL/well) and placed at 37 °C for over 30 min to form a gel. HUVECs administered the indicated pretreatment were suspended at a concentration of 1 × 105 cells/mL. Then, 100 μL of the cell suspension was added to each well and incubated at 37 °C for 12 h. Tube formation was observed with an inverted fluorescence microscope and analyzed with ImageJ software.
Measurement of intracellular cAMP levels
HUVECs were treated with or without caffeine for different durations. Samples were prepared, and cAMP concentrations were determined using a cyclic AMP ELISA kit (Cayman, Ann Arbor, MI, USA) according to the manufacturer’s protocol. Briefly, after culture, HUVECs were lysed with 0.1 mol/L HCl (1 mL for every 35 cm2 surface area) and the supernatants were diluted at least 1:2 in ELISA buffer after centrifugation at 1000 × g for 10 min. After the addition of samples and reagents according to the protocol and 18 h of incubation at 4 °C, followed by five washes with wash buffer, Ellman’s reagent was added to the assay plate and incubated using an orbital shaker at room temperature for 2 h in the dark. The plate absorbance was read at wavelengths from 405 to 420 nm, and the cAMP concentration of the sample was calculated with a cAMP standard curve.
Measurement of cellular energetics
For metabolic measurements, HUVECs were seeded at 5 × 103 cells per well in Seahorse XF96 polystyrene tissue culture plates (Seahorse Bioscience, North Billerica, MA, USA) and then incubated at 37 °C overnight, followed by treatment with the indicated chemicals for the indicated duration. The following day, the medium was changed to XF basal medium supplemented with 25 mM glucose, 2 mM glutamine, and 1 mM pyruvate for measurement of the oxygen consumption rate (OCR), after which the plate was incubated in a non-CO2 incubator at 37 °C for 1 h. The OCR was analyzed using an XF96 extracellular flux analyzer (Seahorse Bioscience), and the following activators and inhibitors were used in the experiment: oligomycin (1 µM), FCCP (2 µM), antimycin A (0.5 µM), and rotenone (0.5 µM). The extracellular acidification rate (ECAR) for glycolysis in the HUVECs was measured under the same conditions.
RNA interference
HUVECs at 50%–70% confluence were transiently transfected with control siRNA or siRNA targeting human Drp1 (siDrp1, GenePharma, Shanghai, China) using Lipofectamine RNAiMAX reagent according to the manufacturer’s protocol. The cells were used for Western blot analysis, the scratch wound healing assay, the Boyden chamber migration assay, the tube formation assay, and immunofluorescence at 24 h after transfection.
Mouse hindlimb ischemia model
All animal studies were approved by the Institutional Animal Care and Use Committee of Guangzhou Medical University and in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 2011). Eight-week-old male C57BL/6 mice were purchased from the Guangdong Medical Laboratory Animal Center (Guangzhou, Guangdong, China). As described in a previous study [17], the femoral arteries of 8-week-old male C57BL/6 mice that were untreated (control) or had received caffeine (0.05% in 200 mL of drinking water daily) were ligated at two positions spaced 5 mm apart, and the arteries between the ligatures were excised. Then, tissue perfusion was measured preoperatively; immediately; and 3, 7, and 14 days after surgery. Images of blood flow in the lower limbs were obtained using laser speckle contrast imaging (Pericam PSI system, Perimed AB, Järfalla, Sweden) at 37 ± 0.2 °C under isoflurane anesthesia. Additionally, PIMsoft was used to analyze the data, which are presented as the ratio of blood flow in the ischemic/nonischemic hindlimb.
H&E staining
The gastrocnemius muscles of the model mice were dissected, fixed in 4% paraformaldehyde for 24 h, dehydrated and used to prepare frozen sections at a 10 μm thickness, which were then stained with hematoxylin and eosin (H&E) (Beyotime, Shanghai, China) on the 7th day after femoral artery ligation. Areas of necrotic muscle were identified according to morphology, differential eosin staining, and leukocyte infiltration of the section observed at ×200.
Immunofluorescence
For mitochondria (MitoTracker Red; Invitrogen, Carlsbad, CA, USA) and lamellipodia (fluorescein phalloidin; Invitrogen) or Drp1 (Cell Signaling Technology) immunostaining, pretreated cell slides were incubated with MitoTracker Red for 35 min at 37 °C after being washed three times. Then, the cell slides were fixed with 4% paraformaldehyde for 30 min, permeabilized in PBS containing 0.2% Triton X-100 for 20 min, blocked with 10% goat serum for 1 h, and then incubated with fluorescein phalloidin and DAPI (Invitrogen) for 20 min or primary antibodies against Drp1 overnight in the dark. For CD31 (PECAM-1; Cell Signaling Technology) and p-AMPK (Cell Signaling Technology) immunostaining of the gastrocnemius muscles, the frozen sections were fixed with 4% paraformaldehyde for 30 min, permeabilized in PBS with 0.2% Triton X-100 for 20 min, and then blocked with 10% goat serum for 1 h. Then, the sections were incubated with primary antibodies at 4 °C overnight, followed by incubation with FITC-labeled secondary antibodies for 1 h at room temperature in the dark. The CD31+ cells and CD31+/p-AMPK+ cells in four mice from each group were counted. All images were obtained using an upright confocal microscope.
Determination of mtDNA copy number
Total cellular DNA was isolated using a TIANamp Genomic DNA Kit (TIANGEN Biotech, Beijing, China) as recommended by the manufacturer. The relative mtDNA copy numbers in HUVECs were examined using real-time polymerase chain reaction and calibrated by simultaneously measuring nuclear DNA. The forward and reverse primers complementary to β-actin used for nuclear gene assessment were 5′-TCACCCACACTGTGCCCATCTACGA-3′ and 5′-CAGCGGAACCGCTCATTGCCAATGG-3′, respectively. The forward and reverse primers specific for mtDNA, which were complementary to the ND1 gene, were 5′-TGGGTACAATGAGGAGTAGG-3′ and 5′-GGAGTAATCCAGGTCGGT-3′, respectively. PCR was performed on a Life Technologies QuantStudio 5 real-time PCR system (Thermo Fisher Scientific) using the SYBR Green PCR Master Mix kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. The relative expression of mtND1 to β-actin was determined using the ΔΔCt method.
Statistical analyses
All data are presented as the mean ± SEM of three or more independent experiments. After confirming the normal distribution of the data using the Shapiro–Wilk test, two-tailed unpaired Student’s t test or one-way analysis of variance was used for statistical comparisons using GraphPad Prism 8.0 (La Jolla, CA). A two-sided P value was calculated, and a P < 0.05 indicated a statistically significant difference. Statistical significance was defined as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Results
Caffeine enhanced angiogenesis in vitro
To investigate whether caffeine affects angiogenesis in vitro, HUVECs were incubated with caffeine at doses ranging from 10 to 100 μM. We observed that caffeine at each concentration promoted tube formation, as manifested by the increased number of branch points and increased tube length (Fig. 1a, b). The scratch wound healing assay (Fig. 1c, d) and Boyden chamber migration assay (Fig. 1e, f) showed that caffeine at concentrations of 10–100 μM enhanced the migration of HUVECs, especially at a concentration of 50 μM. In accordance with increased migration of the HUVECs, caffeine treatment at 50 μM also augmented the formation of lamellipodia and flat, sheet-like F-actin-mediated membrane protrusions at the leading edge of migrating cells, which generated the driving force for cell migration (Fig. 1g). To evaluate the effect of caffeine on HUVEC proliferation, we performed WST-1 proliferation and cell number counting assays. These assays consistently showed that caffeine at doses ranging from 10 to 100 μM did not significantly affect the proliferation of HUVECs (Supplementary Fig. S1). Collectively, these data imply that the proangiogenic capacity of caffeine is due to changes in cell migration.

a–f Representative images and quantification of tube formation (a, b; Scale bar, 100 μm), wound closure (c, d; Scale bar, 200 μm) and migrated cells (e, f; Scale bar, 100 μm) after the administration of caffeine at the indicated concentration for 12 h (n = 6). g Representative images and quantification of lamellipodia stained with phalloidin dye in HUVECs incubated with vehicle or caffeine (50 μM) for 12 h. The lamellipodia extent at cell edges was quantified as a percentage of the cell circumference (n = 10). Scale bar, 25 μm. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 vs. the control group.
Caffeine promotes endothelial cell motility via cAMP/PKA/AMPK signaling
Since the physiological concentration of caffeine is ~30–70 μM and the proangiogenic effect of caffeine peaks at 50 μM, we chose 50 μM caffeine for all the following in vitro studies. Several studies have demonstrated that the activation of AMPK differentially regulates endothelial proliferation and migration [18, 19], mimicking the effect of caffeine on endothelial cells. This prompted us to test whether the promigratory effect exerted by caffeine is dependent on the activation of AMPK. Compared with that in the control group, the phosphorylation of AMPK was significantly enhanced (Supplementary Fig. S2a) and peaked at 60 min after caffeine treatment in HUVECs (Fig. 2a). As a nonselective phosphodiesterase (PDE) inhibitor, caffeine is known to increase intracellular cAMP levels and PKA activity. Thus, we attempted to dissect whether cAMP/PKA signaling was responsible for the activation of AMPK and the promigratory effect induced by caffeine. As expected, caffeine treatment markedly elevated intracellular cAMP levels by ~50%, and this effect was blocked by an inhibitor of adenylate cyclase, SQ22536 (Fig. 2b). In addition, pharmacological activation of PKA with the cAMP analog 8-Br-cAMP at a dose that had no effect on cell viability (Supplementary Fig. S2b) mimicked the effect of caffeine in inducing AMPK phosphorylation (Fig. 2c), while the PKA blocker H89 or adenylate cyclase inhibitor SQ22536 blocked the increased AMPK phosphorylation induced by caffeine (Fig. 2c, d), suggesting that cAMP/PKA signaling acts as an upstream activator of AMPK in HUVECs upon caffeine treatment. Furthermore, 8-Br-cAMP treatment markedly enhanced the mobility of HUVECs, as shown by the scratch wound healing assay (Fig. 2e, g) and Boyden chamber migration assay (Fig. 2f, h), while treatment with H89, SQ22536, and the AMPK inhibitor compound C at relatively safe doses (Supplementary Fig. S2b) dramatically compromised the enhanced endothelial cell migration induced by caffeine (Fig. 2e–h). Altogether, these data demonstrate that caffeine enhances endothelial migration via a cAMP-PKA-AMPK-dependent mechanism.

a Western blot analysis of p-AMPK protein levels in HUVECs incubated with caffeine (50 μM) at the indicated timepoints (n = 3). b The cAMP levels in cell lysates after caffeine treatment (50 μM) for 2 h (n = 4) were measured using a commercial kit. c, d. Western blot analysis of p-AMPK protein levels in HUVECs incubated with vehicle or caffeine (50 μM) in the presence of 8-Br-cAMP (200 μM), H89 (10 μM), compound C (5 μM) or SQ22536 (200 μM, d) for 1 h (n = 3). e–h Representative images and quantification of wound closure (e, g) and migrated cells (f, h) among HUVECs incubated with vehicle or caffeine (50 μM) in the presence of SQ22536 (200 μM), 8-Br-cAMP (200 μM), H89 (10 μM) or compound C (5 μM) for 12 h (n = 6). Scale bar, 200 μm. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 vs. the control or caffeine-treated group.
Caffeine increased mitochondrial fission in endothelial cells
In cells in which migration represents an essential physiological function, such as tumoral metastatic cells and T cells, mitochondria exhibit excess fragmentation and are recruited to specific subcellular regions to enable cell movement [20]. We therefore assessed whether caffeine enhances endothelial migration by regulating mitochondrial dynamics. We found that the mitochondria in vehicle-treated HUVECs exhibited filamentous structures, whereas those in caffeine-treated HUVECs became short and fragmented, characteristics of mitochondrial fission (Fig. 3a, b).Western blot assays showed that the levels of Drp1 and the mitofusin proteins Mfn1 and Mfn2 remained unchanged in HUVECs treated with caffeine compared to those in control HUVECs (Fig. 3c), whereas caffeine treatment significantly increased Drp1 phosphorylation at serine 616 [21], increasing its regulation of mitochondrial fission (Fig. 3c). The cellular localization of Drp1 was further examined by immunofluorescent staining. Drp1 in control HUVECs diffused throughout the cytoplasm with little colocalization with mitochondria; in caffeine-treated cells, many Drp1 puncta existed in the short mitochondria, whereas cytosolic Drp1 was decreased (Fig. 3d). Collectively, these data indicate that caffeine treatment induced mitochondrial fission in endothelial cells.

a Representative mitochondria from HUVECs incubated with vehicle or caffeine (50 μM) for 6 h. Scale bars: left panels, 40 μm; right panels, 20 μm. b Mitochondrial length was measured and scored as follows: fragmented (globular): mainly small and round (<2 μm in diameter) and filamentous: long and more highly interconnected (>2 μm in length). The percentage of cells with the indicated mitochondrial morphology was determined as a percentage of the total number of stained cells counted in each group (n = 3). c Western blot analysis of p-Drp1, Drp1, Mfn1, and Mfn2 protein levels in HUVECs incubated with vehicle or caffeine (50 μM) for 2 h (n = 3). d Drp1 colocalization with mitochondria in HUVECs after caffeine treatment for 2 h. Scale bar, 10 μm. ***P < 0.001; ****P < 0.0001 vs. the control group.
Mitochondrial fission is essential for caffeine-induced lamellipodia formation and migration in endothelial cells
To examine the role of mitochondrial fission in caffeine-induced endothelial cell migration, we pretreated HUVECs with Mdivi-1, a specific Drp1 inhibitor, at a dose that did not affect the cell viability, as demonstrated by the MTT assay (data not shown). As expected, while the elongated mitochondrial morphology was maintained (Supplementary Fig. S3a, b), pharmacological blockade of Drp1 by Mdivi-1 significantly suppressed caffeine-induced tube formation (Fig. 4a, b) and cell migration (Fig. 4c, Supplementary Fig. S3c, d). In addition, phalloidin staining indicated that caffeine-induced lamellipodia formation was also impaired by inhibition of Drp1 (Fig. 4d). In an alternative approach to pharmacological blockade of Drp1, genetic silencing of endogenous Drp1 was achieved by two siRNAs (siDrp1#1 and siDrp1#2), with siDrp1#1 found to be more efficient (Supplementary Fig. S3e–g). As expected, the mitochondria became tubular and elongated in Drp1-silenced HUVECs compared to those in control HUVECs (Supplementary Fig. S3h). Furthermore, knockdown of Drp1 significantly reduced caffeine-induced migration (.Fig. 4e), tube formation (Fig. 4f), and lamellipodia formation (Fig. 4g) in HUVECs.

a–c Representative images and quantification of tube formation (a, b; 100 μm) and migrated cells (c; 200 μm) in HUVECs incubated with vehicle or caffeine (50 μM) in the presence of Mdivi-1 (10 μM) for 12 h (n = 6). d Representative images and quantification of lamellipodia in HUVECs treated for 12 h (n = 10). e–g HUVECs were transfected with scrambled siRNA or Drp1 siRNA for 24 h and then incubated with vehicle or caffeine (50 μM) for 12 h. Representative images and quantification of migrated cells (e; n = 6; 200 μm), tube formation (f; n = 6; 100 μm) and lamellipodia extent (g; n = 10; 20 μm) are shown. h The lamellipodia region was defined as the area from the leading edge of a cell to half of the distance to the nucleus, as indicated by the dashed line. The relative fluorescence intensity of mitochondria in the lamellipodia region is shown (n = 5; 20 μm). **P < 0.01; ***P < 0.001; ****P < 0.0001 vs. the control, caffeine-treated or caffeine-scramble group.
Previous studies have shown that fragmented mitochondria may be transported to the region with energy demands [10]. In this regard, we observed that during lamellipodia formation in endothelial cells induced by caffeine, the mitochondria switched from an elongated and perinuclear accumulated state to a fragmented and dispersed state. MitoTracker Red and phalloidin staining indicated that nearly threefold more mitochondria were distributed to the lamellipodia region at the leading edge of caffeine-treated HUVECs compared to that of control HUVECs (Fig. 4h). Inhibition of mitochondrial fission by silencing Drp1 significantly reduced mitochondrial accumulation in the lamellipodia region (Fig. 4h).
Collectively, these data demonstrate that Drp1-mediated mitochondrial fission and trafficking to the lamellipodia region are essential for caffeine-induced lamellipodia formation and endothelial cell migration.
Functional importance of mitochondrial energetics in caffeine-induced lamellipodia formation and endothelial cell migration
The formation of lamellipodia relies on actin cytoskeleton remodeling and requires the hydrolysis of large amounts of ATP [22, 23]. Since mitochondria are organelles that provide the majority of the energy in most cells due to their synthesis of ATP by oxidative phosphorylation, we examined the mitochondrial energetic function in caffeine-treated endothelial cells. Using Seahorse flux analysis, we observed that basal cellular oxygen consumption, ATP production, and the maximal respiratory capacity were significantly higher in HUVECs incubated with 50 or 100 µM caffeine for 12 or 24 h than in untreated cells (Supplementary Fig. S4a), while the ECAR remained unchanged (Supplementary Fig. S4b), suggesting that mitochondrial energetic function but not glycolysis is enhanced in response to caffeine stimulation. Furthermore, we also found that, while it had no effect on the ECAR (Supplementary Fig. S4c, d), inhibition of mitochondrial fission via pharmacological and genetic blockade of Drp1 compromised the caffeine-induced enhancement of mitochondrial energetics (Fig. 5a, b), which is consistent with findings observed in other cells in which mitochondrial fission deficiency decreased the efficiency of mitochondrial ATP synthesis. To determine whether increased mitochondrial energetics play a role in caffeine-induced endothelial cell motility, caffeine-induced lamellipodia formation and endothelial cell migration were assessed in the presence of uncoupler carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP), which disrupts ATP synthesis by interfering with the proton gradient [24]. FCCP completely blocked caffeine-induced lamellipodia formation (Fig. 5c, d) and the migrative ability of HUVECs (Fig. 5e–h). In addition, we examined the effect of oligomycin A, which inhibits mitochondrial ATP synthase activity. Oligomycin A almost completely suppressed lamellipodia formation (Fig. 5c, d) and the promigratory effects exerted by caffeine (Fig. 5e–h). Altogether, these data suggest that mitochondrial energy production plays an important role in lamellipodia formation and the migration of endothelial cells.

a HUVECs were incubated with vehicle or caffeine (50 μM or 100 μM) in the presence or absence of Mdivi-1 (10 μM) for 12 h. Quantified mitochondrial function parameters are from the upper panel for each group (n = 3). *P < 0.05; **P < 0.01; ****P < 0.0001 vs. the control, 50 μM caffeine or 100 μM caffeine groups. b HUVECs were transfected with scrambled siRNA or Drp1 siRNA for 24 h and then incubated with vehicle or caffeine (50 μM) for 12 h. Quantified mitochondrial function parameters are from the upper panel for each group (n = 4). *P < 0.05; ****P < 0.0001 vs. the control-scramble or caffeine-scramble group. c–h HUVECs were pretreated with FCCP (10 μM) or oligomycin A (1 μg/ml) for 30 min and then incubated with vehicle or caffeine (50 μM) in the presence or absence of FCCP or oligomycin A for 12 h. Representative images and quantification of lamellipodia (c, d; n = 10; 25 μm), wound closure (e, f; n = 6; 200 μm), and migrated cells (g, h; n = 6; 200 μm) among HUVECs. ***P < 0.001; ****P < 0.0001 vs. the control or caffeine-treated group.
The cAMP/PKA/AMPK signaling pathway contributes to caffeine-induced mitochondrial fission and mitochondrial distribution to lamellipodia region
Our previous data demonstrated that caffeine enhances endothelial migration via a cAMP-PKA-AMPK-dependent mechanism. We further determined whether the activation of this signaling pathway is related to mitochondrial fission induced by caffeine. As shown in Fig. 6a, b, the PKA blocker H89 and the AMPK inhibitor compound C inhibited caffeine-induced mitochondrial fission by ~71% and 74%, respectively. Moreover, H89 and compound C almost completely abolished lamellipodia formation and mitochondrial distribution to the lamellipodia region induced by caffeine (Fig. 6c–e). Previous studies have shown that pharmacological activation of AMPK promotes mitochondrial fragmentation through the phosphorylation of MFF, a mitochondrial outer membrane receptor for Drp1 [12]. In our study, we found that phosphorylation of MFF at Ser172 was significantly increased by caffeine treatment, and this effect was mimicked by the activation of PKA via 8-Br-cAMP (Fig. 6f, g). In contrast, pharmacological blockade of PKA and AMPK significantly inhibited the phosphorylation of MFF induced by caffeine (Fig. 6f, g). Taken together, these data suggest a causal role of cAMP/PKA/AMPK signaling pathway activation in mitochondrial fission and its distribution to the lamellipodia region.

HUVECs were incubated with vehicle or caffeine (50 μM) in the presence or absence of H89 (10 μM) or compound C (5 μM) for the indicated durations. a, b Representative mitochondria from HUVECs cultured for 6 h (n = 3). Scale bar, 25 μm. c–e Representative images and quantification of the lamellipodia extent (d; n = 10) and relative fluorescent intensity of mitochondria in the lamellipodia region (e; n = 5) in HUVECs cultured for 12 h. Scale bar, 25 μm. f, g Western blot analysis of p-MFF protein levels in HUVECs incubated with vehicle or caffeine (50 μM) in the presence of 8-Br-cAMP (200 μM), H89 (10 μM), or compound C (5 μM) for 2 h (n = 3). *P < 0.05; ****P < 0.0001 vs. the control or caffeine-treated group.
Caffeine enhanced ischemia-induced angiogenesis in vivo
The impaired angiogenic response to limb ischemia in diabetes is accompanied with poor clinical outcome [25]. We next evaluated the impact of caffeine on limb perfusion after ischemic injury in vivo by introducing a mouse hindlimb ischemic model. Male C57BL/6 mice that received drinking water supplemented with caffeine (0.05%) or vehicle were subjected to femoral artery ligation. Blood flow was restored to ~45% and 65% at days 7 and 14 after ischemic surgery, respectively; however, caffeine-treated mice showed ~70% and almost complete blood flow restoration 7 and 14 days after ischemia, respectively (Fig. 7a, b). In addition, histological assessment showed that the injured and necrotic areas in the ischemic gastrocnemius muscles were significantly decreased in caffeine-fed mice (Fig. 7c). In line with accelerated blood flow recovery in response to caffeine treatment, the number of CD31-positive endothelial cells (Fig. 7d) was significantly higher in the ischemic sections of caffeine-treated mice than in those of control mice. Immunostaining revealed that phosphorylated AMPK (p-AMPK) in the ischemic muscles was localized mainly to CD31-positive endothelial cells, and the level of p-AMPK was also significantly enhanced in caffeine-treated mice compared with control mice (Fig. 7d). Taken together, our results demonstrate that caffeine treatment promoted angiogenesis and was associated with significantly improved perfusion as well as the activation of endothelial AMPK signaling after hindlimb ischemia.

a Representative laser Doppler perfusion images of mice that received vehicle or caffeine (0.05% in 200 mL of drinking water daily) following unilateral hindlimb ischemia on various days after hindlimb ischemia (n = 5). Scale bar, 5 mm. b The blood perfusion rate over time was examined with computer-assisted analysis of the laser Doppler program at the indicated timepoints (n = 5). c Representative images and quantification of histologically assessed necrotic areas drawn out using red coils in the gastrocnemius (GC) muscle at day 7 after ischemia (n = 4). Scale bar, 1 mm. d Representative images and quantification of immunofluorescence staining of CD31 and p-AMPK in sections of GC muscle at day 7 after ischemia (n = 4). Scale bar, 100 μm. *P < 0.05; **P < 0.01; ***P < 0.001 vs. the control group.
Discussion
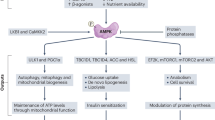
The data presented in the current study indicate that caffeine stimulation of endothelial cells increased mitochondrial fission, accompanied by enhanced mitochondrial energetics, and endothelial cell lamellipodial formation (Fig. 8). Importantly, we showed that inhibition of mitochondrial fission reduced caffeine-induced endothelial cell migration and tube formation. Our studies further provided the mechanisms by which caffeine stimulation activates mitochondrial fission and how mitochondrial fission induces endothelial cell lamellipodial formation and migration.

Caffeine activates protein kinase A (PKA, a cAMP-dependent protein kinase) by increasing the intracellular cyclic adenosine monophosphate (cAMP) content. Subsequently, phosphorylated AMP-activated protein kinase (AMPK), activated by PKA, facilitates mitochondrial fission by phosphorylating mitochondrial fission factor (MFF), which recruits dynamin-related protein 1 (Drp1) from the cytosol to the mitochondrial outer membrane. Together, the increased mitochondrial distribution to the lamellipodia region of endothelial cells (ECs) and elevated adenosine triphosphate (ATP) content induced by caffeine enhance the angiogenesis of ECs, which can be impaired by inhibiting cAMP, PKA, AMPK, and Drp1 activation using SQ22536, H89, compound C and Mdivi-1, respectively.
Studies regarding the roles of caffeine in angiogenesis are controversial [26]. Although our study, indicates the angiogenic effect of caffeine for the first time, previous data demonstrated that caffeine at concentrations of 250 μM or higher attenuates angiogenesis, and the inhibitory effect of caffeine on angiogenesis is associated, at least in part, with its induction of endothelial cell apoptosis, probably mediated by a caspase-3-dependent mechanism [27]. Additionally, Yeh et al. assessed caffeine-induced toxicity on embryonic vascular development using zebrafish and showed that caffeine-treated (250–350 ppm) embryos revealed developmental defects in the dorsal longitudinal anastomotic vessels, the intersegmental vessels, and subintestinal vein sprouting [28]. Considering that the average weight of the zebrafish embryo is ~1.2 mg, 250–350 ppm of caffeine is estimated to correspond to 4200–5800 mg/kg [29], which is much higher than the acceptable daily intake of caffeine in adults (150–300 mg per day) and close to the toxic dosage [30]. However, several reports support the proangiogenic effects of caffeine. Caffeinated coffee was demonstrated to increase the serum concentration of caffeine from 2 to 23 μM in patients with coronary artery disease, accompanied by a significant increase in the migratory ability of patient-derived endothelial progenitor cells. In a mouse model after denudation of the carotid artery, caffeine at physiologically relevant concentrations (50–100 μM) was proven to enhance reendothelialization. Therefore, the effect of caffeine on angiogenesis appears to be dose dependent, and the mechanisms explaining the proangiogenic effects of caffeine observed in our study should be attributed to the use of concentrations of less than 100 μM. Moreover, the controversial role of caffeine in angiogenesis may be attributed to the distinct tissue-specific or pathophysiological context, since caffeine at low (micromolar) and relatively nontoxic concentrations has been shown to inhibit tumor angiogenesis by antagonizing adenosine receptors [31], while in our study, we demonstrated that caffeine at physiologically relevant concentrations protected mice against hindlimb ischemic injury by promoting angiogenesis in the hindlimb.
While mainly quiescent in healthy adult tissues, endothelial cells are flexible, undergoing rapid migration and proliferation during angiogenesis, thus increasing their biosynthetic and bioenergetic requirements. Recent findings indicate that metabolism is a critical regulator of endothelial cell function during angiogenesis. We and others previously found that endothelial cells are highly glycolytic, and glycolysis is necessary for endothelial cell proliferation and migration during angiogenesis [32,33,34]. A new and recent finding demonstrated that mitochondrial respiratory chain-linked metabolism is critical for the proliferation of endothelial cells during angiogenesis [35]. However, the role of mitochondrial energy metabolism in endothelial cell migration has long been grossly underestimated. Cell migration relies on reorganization of the actin cytoskeleton to form filopodia and lamellipodia, which requires ATP [36]. Mitochondria are too bulky to fit into thin filopodia and lamellipodia. In addition, mitochondria are mostly contained in perinuclear clusters, distant from the lamellipodia and filopodia regions. The diffusion of ATP from distant perinuclear mitochondria may thus not be sufficient to support the high and rapidly changing ATP demands for lamellipodial formation during migration [33]. However, previous studies have also shown that altering mitochondrial morphology affects cell migration. Decreased cell migration was observed with mitochondrial elongation, whereas mitochondrial fragmentation was found to increase cell migration in both metastatic breast cancer cells and chemokine-stimulated lymphocytes [10, 37]. Here, in our study, we observed that mitochondria in migrated endothelial cells upon caffeine treatment switched from an elongated and perinuclear accumulated state to a fragmented and scatted state, suggesting a correlation between mitochondrial fission and cell migration. In addition, mitochondrial fission was shown to be necessary for the redistribution of mitochondria to the leading edge to facilitate lamellipodia formation [10]. This phenomenon has not been reported in endothelial cells or other cell types with a high capacity for glycolysis. Nevertheless, our findings in this study suggest that mitochondrial fragmentation may make it easier for mitochondria to access the lamellipodial region in caffeine-treated endothelial cells.
Our study demonstrates that caffeine increased mitochondrial energetics and that inhibition of mitochondrial ATP synthesis by oligomycin A suppressed caffeine-induced cell migration. Additionally, the proton ionophore FCCP, which disperses the proton gradient across the inner mitochondrial membrane and causes the uncoupling of mitochondrial respiration from ATP synthesis, completely interfered with caffeine-induced lamellipodia formation. These data suggest that mitochondrial ATP is an important local energy source that is required for endothelial cell migration. Our study also showed that inhibition of mitochondrial fission prevented caffeine-induced endothelial cell mitochondrial energetics and migration. The limitation of cell migration upon fission inhibition might be due to an increase in respiration uncoupling and a decrease in mitochondrial respiratory capacity. This is consistent with previous findings that chemotactic stimulation of vascular smooth muscle cells enhanced mitochondrial energetics with the formation of short, small mitochondria, whereas the long, large mitochondria in fission-deficient cells exhibited increased respiration uncoupling [11]. Increased fission may facilitate the uptake of substrate into the mitochondria by increasing the overall mitochondrial surface area in the cell. In addition, the alteration of mitochondrial shape through fission may reorganize respiratory chain complexes to optimize electron flux for energetic metabolism. However, an assessment of subcellular ATP concentrations will be required to further understand the role and mechanisms of mitochondria in lamellipodia formation in caffeine-treated endothelial cells.
Over decades, the effects of caffeine have been proven to be mediated in the following different ways: (1) by antagonizing adenosine receptors, (2) by inhibiting PDEs, and (3) by elevating intracellular calcium levels [38]. Our previous studies have demonstrated that endothelial cells predominantly express adenosine receptor A2A or A2B [39]. Activation of these receptors promoted angiogenesis, whereas their blockade reduced angiogenesis and endothelial cell migration [40]. Thus, the promigratory effect of caffeine seems to be independent of adenosine receptor antagonization. Our study showed that caffeine elevated the intracellular cAMP level in endothelial cells. This indicated the activation of PKA as a potential signaling pathway involved in the effects induced by caffeine. As expected, the activation of PKA mimicked the promigratory activity of caffeine, while inhibition of PKA blocked caffeine-induced mitochondrial fragmentation and redistribution, lamellipodia formation, and migration in endothelial cells. AMPK has been shown to play an important role in endothelial migration [18]. As reported in other cells, our study showed that caffeine treatment activated AMPK in endothelial cells. Several studies have demonstrated that PKA and AMPK interact to regulate mitochondrial function in cardiac and endothelial cells [41]. Studies regarding the role of AMPK activation in mitochondrial fission are controversial. The activation of AMPK attenuated palmitate-induced mitochondrial fragmentation in pancreatic β cells [42], whereas a recent publication reported that activated AMPK orchestrates MFF phosphorylation in response to cellular stress, thereby recruiting Drp1 from the cytosol to the mitochondrial outer membrane to initiate mitochondrial fission [12, 43]. Our current study found that Drp1 localization in mitochondria was sensitized by caffeine treatment and that a PKA inhibitor suppressed caffeine-mediated AMPK activation and MFF phosphorylation, suggesting that the cAMP/PKA/AMPK/MFF signaling pathway activates mitochondrial fission. However, endothelial cell-specific AMPK-knockout mice will be required to provide a more specific experimental model for the functional investigation of AMPK in caffeine-induced angiogenesis and mitochondrial fission in endothelial cells.
Caffeine treatment might also improve endothelial cell migration via other mechanisms. Previous studies have demonstrated that caffeine induces ryanodine receptor activity in the endothelial endoplasmic reticulum, thereby stimulating the release of Ca2+ from the reticulum to increase the cytosolic Ca2+ concentration [26]. Increased intracellular Ca2+ has been proven to promote mitochondrial fission [44]. Additionally, Ca2+ in mitochondria activates dehydrogenases of the TCA cycle as well as ATP synthase [45,46,47,48,49]. Thus, it is possible that the caffeine-evoked Ca2+ increase activated mitochondrial fission through AMPK in the cytosol and, concomitantly, elevated oxidative phosphorylation in mitochondria. Since activation of AMPK has been shown to regulate mitochondrial biogenesis, which controls mitochondrial quality and quantity [50], it is also possible that mitochondrial biogenesis serves to fuel endothelial cell migration. Indeed, caffeine treatment increased the mitochondrial DNA content in endothelial cells (Supplementary Fig. S5). In addition, as a critical cAMP-dependent signaling pathway mediator, the exchange protein activated by cAMP 1 (EPAC1) has been shown to positively regulate endothelial cell migration [51], raising the possibility that caffeine exerts the promigratory effect on endothelial cells through EPAC1.
In conclusion, we demonstrate for the first time that caffeine markedly stimulates mitochondrial fission and that increased fission mediates endothelial cell migration and angiogenesis, likely due to reduced spatial impediments to repositioning brought by the long-intertwined tubules. Since therapeutic angiogenesis is of major importance for treating ischemic diseases, coffee consumption or caffeine per se could be considered as an additional protective dietary factor for patients with critical limb ischemia. Moreover, since the effects of caffeine are linked to increased mitochondrial fission and thus improved mitochondrial function, enhancing mitochondrial fission could serve as a potential therapeutic strategy in ischemic vascular diseases.
References
Bian X, Ma K, Zhang C, Fu X. Therapeutic angiogenesis using stem cell-derived extracellular vesicles: an emerging approach for treatment of ischemic diseases. Stem Cell Res Ther. 2019;10:158.
Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49:507–21.
Losordo DW, Dimmeler S. Therapeutic angiogenesis and vasculogenesis for ischemic disease: part II: cell-based therapies. Circulation. 2004;109:2692–7.
Losordo DW, Dimmeler S. Therapeutic angiogenesis and vasculogenesis for ischemic disease. Part I: angiogenic cytokines. Circulation. 2004;109:2487–91.
Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12:551–64.
Eilken HM, Adams RH. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol. 2010;22:617–25.
Takahashi T, Kalka C, Masuda H, Chen D, Silver M, Kearney M, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–8.
Gale NW, Yancopoulos GD. Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development. Genes Dev. 1999;13:1055–66.
Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–84.
Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32:4814–24.
Wang L, Yu T, Lee H, O’Brien DK, Sesaki H, Yoon Y. Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc Res. 2015;106:272–83.
Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Loson OC, Hellberg K, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission response energy stress. Science. 2016;351:275–81.
Poleszak E, Szopa A, Wyska E, Kukula-Koch W, Serefko A, Wosko S, et al. Caffeine augments the antidepressant-like activity of mianserin and agomelatine in forced swim and tail suspension tests in mice. Pharmacol Rep. 2016;68:56–61.
Mitchell DC, Knight CA, Hockenberry J, Teplansky R, Hartman TJ. Beverage caffeine intakes in the U.S. Food Chem Toxicol. 2014;63:136–42.
Dragicevic N, Delic V, Cao C, Copes N, Lin X, Mamcarz M, et al. Caffeine increases mitochondrial function and blocks melatonin signaling to mitochondria in Alzheimer’s mice and cells. Neuropharmacology. 2012;63:1368–79.
Ale-Agha N, Goy C, Jakobs P, Spyridopoulos I, Gonnissen S, Dyballa-Rukes N, et al. CDKN1B/p27 is localized in mitochondria and improves respiration-dependent processes in the cardiovascular system-new mode of action for caffeine. PLoS Biol. 2018;16:e2004408.
Xu Y, Wang Y, Yan S, Zhou Y, Yang Q, Pan Y, et al. Intracellular adenosine regulates epigenetic programming in endothelial cells to promote angiogenesis. EMBO Mol Med. 2017;9:1263–78.
Levine YC, Li GK, Michel T. Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK -> Rac1 -> Akt -> endothelial nitric-oxide synthase pathway. J Biol Chem. 2007;282:20351–64.
Peyton KJ, Liu XM, Yu Y, Yates B, Durante W. Activation of AMP-activated protein kinase inhibits the proliferation of human endothelial cells. J Pharmacol Exp Ther. 2012;342:827–34.
da Silva AF, Mariotti FR, Maximo V, Campello S. Mitochondria dynamism: of shape, transport and cell migration. Cell Mol Life Sci. 2014;71:2313–24.
Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–9.
Goveia J, Stapor P, Carmeliet P. Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol Med. 2014;6:1105–20.
Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100:782–94.
Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-Doulcier AM, Bouillaud F, et al. The biology of mitochondrial uncoupling proteins. Diabetes. 2004;53 Suppl 1:S130–5.
Sawada N, Jiang A, Takizawa F, Safdar A, Manika A, Tesmenitsky Y, et al. Endothelial PGC-1alpha mediates vascular dysfunction in diabetes. Cell Metab. 2014;19:246–58.
Echeverri D, Montes FR, Cabrera M, Galan A, Prieto A. Caffeine’s vascular mechanisms of action. Int J Vasc Med. 2010;2010:834060.
Li H, Jin SY, Son HJ, Seo JH, Jeong GB. Caffeine-induced endothelial cell death and the inhibition of angiogenesis. Anat Cell Biol. 2013;46:57–67.
Yeh CH, Liao YF, Chang CY, Tsai JN, Wang YH, Cheng CC, et al. Caffeine treatment disturbs the angiogenesis of zebrafish embryos. Drug Chem Toxicol. 2012;35:361–5.
Chen YH, Huang YH, Wen CC, Wang YH, Chen WL, Chen LC, et al. Movement disorder and neuromuscular change in zebrafish embryos after exposure to caffeine. Neurotoxicol Teratol. 2008;30:440–7.
Christian MS, Brent RL. Teratogen update: evaluation of the reproductive and developmental risks of caffeine. Teratology. 2001;64:51–78.
Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Simioni C, et al. Caffeine inhibits adenosine-induced accumulation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and interleukin-8 expression in hypoxic human colon cancer cells. Mol Pharmacol. 2007;72:395–406.
Xu Y, An X, Guo X, Habtetsion TG, Wang Y, Xu X, et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler Thromb Vasc Biol. 2014;34:1231–9.
De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–63.
Eelen G, Cruys B, Welti J, De Bock K, Carmeliet P. Control of vessel sprouting by genetic and metabolic determinants. Trends Endocrinol Metab. 2013;24:589–96.
Diebold LP, Gil HJ, Gao P, Martinez CA, Weinberg SE, Chandel NS. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat Metab. 2019;1:158–71.
Xue F, Janzen DM, Knecht DA. Contribution of filopodia to cell migration: a mechanical link between protrusion and contraction. Int J Cell Biol. 2010;2010:507821.
Campello S, Lacalle RA, Bettella M, Manes S, Scorrano L, Viola A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med. 2006;203:2879–86.
Grosso G, Godos J, Galvano F, Giovannucci EL. Coffee, caffeine, and health outcomes: an umbrella review. Annu Rev Nutr. 2017;37:131–56.
Xu Y, Wang Y, Yan S, Yang Q, Zhou Y, Zeng X, et al. Regulation of endothelial intracellular adenosine via adenosine kinase epigenetically modulates vascular inflammation. Nat Commun. 2017;8:943.
Headrick JP, Ashton KJ, Rose’meyer RB, Peart JN. Cardiovascular adenosine receptors: expression, actions and interactions. Pharmacol Ther. 2013;140:92–111.
Zhang J, Wang Y, Liu X, Dagda RK, Zhang Y. How AMPK and PKA interplay to regulate mitochondrial function and survival in models of ischemia and diabetes. Oxid Med Cell Longev. 2017;2017:4353510.
Wikstrom JD, Israeli T, Bachar-Wikstrom E, Swisa A, Ariav Y, Waiss M, et al. AMPK regulates ER morphology and function in stressed pancreatic beta-cells via phosphorylation of DRP1. Mol Endocrinol. 2013;27:1706–23.
Zhang CS, Lin SC. AMPK promotes autophagy by facilitating mitochondrial fission. Cell Metab. 2016;23:399–401.
Hom J, Yu T, Yoon Y, Porter G, Sheu SS. Regulation of mitochondrial fission by intracellular Ca2+ in rat ventricular myocytes. Biochim Biophys Acta. 2010;1797:913–21.
Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J. 1978;176:899–906.
Denton RM, McCormack JG, Edgell NJ. Role of calcium ions in the regulation of intramitochondrial metabolism. Effects of Na+, Mg2+ and ruthenium red on the Ca2+-stimulated oxidation of oxoglutarate and on pyruvate dehydrogenase activity in intact rat heart mitochondria. Biochem J. 1980;190:107–17.
McCormack JG, Denton RM. The role of Ca2+ ions in the regulation of intramitochondrial metabolism and energy production in rat heart. Mol Cell Biochem. 1989;89:121–5.
Das AM. Regulation of the mitochondrial ATP-synthase in health and disease. Mol Genet Metab. 2003;79:71–82.
De Marchi U, Thevenet J, Hermant A, Dioum E, Wiederkehr A. Calcium co-regulates oxidative metabolism and ATP synthase-dependent respiration in pancreatic beta cells. J Biol Chem. 2014;289:9182–94.
Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–35.
Baljinnyam E, Umemura M, Chuang C, De Lorenzo MS, Iwatsubo M, Chen S, et al. Epac1 increases migration of endothelial cells and melanoma cells via FGF2-mediated paracrine signaling. Pigment Cell Melanoma Res. 2014;27:611–20.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant numbers: 81700395 and 81870217); the Key Project of Department of Education of Guangdong Province (Grant number: 2018KZDXM053); the Science and Technology Planning Project of Guangzhou (Grant numbers: 201903010005 and 202002030190) and “Yangcheng Scholars” Research Project by Guangzhou Education Bureau (Grant number: 201831843).
Author information
Authors and Affiliations
Contributions
YMX contributed to the conception and design; LTW, PCH, AQL, KXC, JWY, SG, LJ, LY, and XYD contributed to the acquisition of data or analysis and interpretation of data; LTW and YMX drafted the paper; YMX, DF, and NT supervised the experiments. All authors read and approved the final version to be published.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, Lt., He, Pc., Li, Aq. et al. Caffeine promotes angiogenesis through modulating endothelial mitochondrial dynamics. Acta Pharmacol Sin 42, 2033–2045 (2021). https://doi.org/10.1038/s41401-021-00623-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00623-6
